

Abstract
The halophile environment has a number of compelling aspects with regard to the origin of structured polypeptides (i.e., proteogenesis) and, instead of a curious niche that living systems adapted into, the halophile environment is emerging as a candidate “cradle” for proteogenesis. In this viewpoint, a subsequent halophile-to-mesophile transition was a key step in early evolution. Several lines of evidence indicate that aromatic amino acids were a late addition to the codon table and not part of the original “prebiotic” set comprising the earliest polypeptides. We test the hypothesis that the availability of aromatic amino acids could facilitate a halophile-to-mesophile transition by hydrophobic core-packing enhancement. The effects of aromatic amino acid substitutions were evaluated in the core of a “primitive” designed protein enriched for the 10 prebiotic amino acids (A,D,E,G,I,L,P,S,T,V)—having an exclusively prebiotic core and requiring halophilic conditions for folding. The results indicate that a single aromatic amino acid substitution is capable of eliminating the requirement of halophile conditions for folding of a “primitive” polypeptide. Thus, the availability of aromatic amino acids could have facilitated a critical halophile-to-mesophile protein folding adaptation—identifying a selective advantage for the incorporation of aromatic amino acids into the codon table.
Keywords: protein evolution, proteogenesis, prebiotic, protein design, protein folding
Introduction
Abiogenesis (the origin of living systems) is hypothesized to have used the simple chemical building blocks that were freely available in the prebiotic environment (the Oparin–Haldane “heterotroph hypothesis”). A number of abiotic processes have been proposed to generate the critical organic compounds required for life to develop, including spark discharge chemistry,1 hydrothermal vent chemistry,2 high energy particle synthesis,3 and deep space chemistry with subsequent delivery to the Earth's surface by comets and meteorites.4,5 Strikingly, these processes produce a consistent set of 10 of the 20 common α-amino acids (termed the “prebiotic set”) comprised of A, D, E, G, I, L, P, S, T, and V.6,7 Notably, this set has also been confirmed in recently reanalyzed original spark discharge samples of Miller.8
The prebiotic set of α-amino acids has several remarkable and compelling features as regards potential fitness for proteogenesis. For example, while the set resides at the theoretical minimum of complexity required for foldability9,10 it contains amino acids having among the highest propensity values for formation of all three types of common protein 2° structure (i.e., α-helix, β-strand, reverse turn). With regard to hydrophobic/hydrophilic patterning essential for folding of soluble globular proteins,11 the prebiotic set contains five hydrophobic and five hydrophilic amino acids. The prebiotic set is also U.V. transparent—indicating the potential for persistence and accumulation in a high-U.V. flux environment, as would be present prior to development of oxygen/ozone in the atmosphere,12 (for a detailed discussion of such properties see Ref.6). Given the above, and the ubiquity of proteins as the molecular workhorses in all extant life, it is likely that polypeptides were incorporated early in the abiogenic process; that is, proteogenesis (the origin of polypeptides) was a key event in the overall process of abiogenesis.
Consistent with the proteogenic hypothesis is recent experimental evidence that the prebiotic set of amino acids likely defines a “foldable set” (i.e., is supportive of protein folding) within a halophilic (high salt) environment.13 The compatibility of prebiotic protein folding and the halophilic environment is due to the unique composition of the prebiotic amino acid alphabet, which is devoid of both basic and aromatic amino acids and is also—a distinctive hallmark of halophile proteomes.7,14–16 High salt serves to stabilize protein structures having reduced hydrophobic packing volume, shields surface acidic charges, and promotes solubility through carboxylate binding of hydrated Na+ cations. Salt-induced peptide formation (SIPF) also promotes favorable condensation reactions of peptide bonds in aqueous solution.17 Thus, rather than being a curious niche that life adapted into, the halophile environment has been proposed as the likely site of origin of both proteogenesis and abiogenesis.13,17,18
The general consensus that aromatic amino acids (both canonical and noncanonical) were essentially absent when life first emerged is supported by several observations. The aromatic amino acids are the largest and most complex of the common α-amino acids19 and prebiotic aromatic amino acid synthesis appears highly inefficient (with most abiotic chemical syntheses failing to yield aromatics altogether).6,7 Furthermore, due to an essential lack of ozone in the atmosphere, abiotically generated aromatic compounds (i.e., aromatic amino acids and nucleic acid bases having absorption wavelengths falling within the U.V. range) would have been highly susceptible to photodegradation. As such, the concentrations of aromatic amino acids in unprotected surface environments on the early Earth are expected to have been marginal and accumulation unlikely.12 Attempts at reconstructing the order of amino acid incorporation into the genetic code are in agreement: coevolution theory identifies the aromatics as being part of the “Phase 2” amino acids (that is, those amino acids incorporated well after establishment of the genetic code).20 A detailed multifactorial analysis by Trifonov identifies the three aromatic amino acids (F, Y, and W) as being the last amino acids to be incorporated into the genetic code (along with aliphatic M).21 Evolutionary analysis of W biosynthesis strongly suggests that biosynthetic pathways for this aromatic amino acid evolved only once and spread between species by horizontal gene transfer, sometime after the last universal common ancestor (LUCA).22 An evolutionary analysis of F and Y biosynthesis was unable to establish whether the LUCA could synthesize these amino acids, although synthesis of chorismate (a key metabolic precursor to the aromatic amino acids) was possible.23 Taken together, the above data identify aromatic amino acid biosynthesis as a key adaptation acquired sometime after the emergence of life, separate from the initial proteogenesis/abiogenesis event, and concurrent with, or preceding, the LUCA.
A reasonable assumption is that aromatic amino acids provided a selective advantage, initially as metabolites, and subsequently upon incorporation into polypeptides. As metabolites, aromatic amino acids could have provided protection from damaging U.V. radiation (as in the aqueous humor of avian eyes)24 enabling nascent living systems to move from protective environments (i.e., physically shielded from U.V. radiation) to more open expanses. Subsequent incorporation of aromatic amino acids into polypeptides may have provided a selective advantage by their ability to stabilize proteins via improved core packing (through increased hydrophobic volume and combinatorial packing efficiency). Increased stability would enable broader functional mutations (which typically occur at the expense of stability)25,26 or to adapt to novel (destabilizing) environments that would otherwise be inaccessible. To date, however, there has been no formalism or testable hypothesis of how aromatic amino acids might have affected protein evolution.
In this study, we test the hypothesis that the availability of aromatic amino acids could have played a major role in protein evolution by enabling a halophile-to-mesophile adaptive transition in protein folding. This hypothesis is evaluated by testing the effects of incorporating aromatic amino acids into a “primitive” designed protein that is highly enriched for the prebiotic amino acids, devoid of aromatics, and is an obligate halophile with regard to foldability.13 Previous studies of the primitive protein model system showed that a mesophile/halophile folding transition occurred concomitant with six simultaneous substitutions of buried aromatic amino acids. The requirement of multiple simultaneous substitutions is a steep barrier to evolutionary change in comparison to a single substitution. In this study, we test whether incorporation of only a single aromatic amino acid can obviate the need for halophile conditions for the efficient folding of a “primitive” obligate halophile protein. The results show this to be the case, supporting the hypothesis that incorporation of a Shikimate-like pathway into the genome of early HaloArchea could relax the requirement of salt for protein foldability, thereby facilitating expansion into a low-salt mesophile environment, and demonstrating a plausible biophysical basis for the evolutionary selection of the “Phase 2” aromatic α-amino acids.
Results
Mutant sequence characteristics
The design of the “primitive” protein (“PV2”) utilized in this study has previously been described.13 PV2 is a small β-trefoil protein made up of three identical repeats of 42-amino acids, comprising an amino acid alphabet of only 12 letters. PV2 is highly enriched (∼80%) for the prebiotic set of amino acids,6 is devoid of aromatic amino acids, and has an acidic pI = 4.36. Importantly, the hydrophobic core of PV2 (21 of 126 residues total, or 17% of amino acid positions) is entirely prebiotic (i.e., comprised of only L, I, and V). PV2 was designed by Top-Down Symmetric Deconstruction27 and, as a consequence, the identical sequences of the three 42-amino acid structural subdomains that form the β-trefoil architecture define a threefold rotational symmetry that substantially simplifies mutational design (Fig. 1).28
Figure 1.

The 1° structure of the PV2 protein (using single letter amino acid code) and aligned to reflect the threefold symmetric architecture of the overall β-trefoil fold. The boxed positions comprise the hydrophobic core packing group. The shaded positions are the locations where aromatic amino acids were substituted as point mutations or in various combinations (see Table I).
The aromatic amino acids F, Y, or W were incorporated at two buried locations within the PV2 scaffold known to exhibit a high statistical preference for aromatic amino acids in the β-trefoil fold29: symmetry-related positions 22, 64, and 108 (comprising three independent hydrophobic “mini-core” regions) and symmetry-related positions 44, 85, and 132 (participating in a cooperatively packing central hydrophobic core). L → V mutation at the mini-core and central core positions resulted in markedly reduced expression and solubility and were not pursued further. L→I mutation was tolerated in both the mini-core and central core, but was less stable than L in both cases (data not shown). Constructs denoted “6xAro” incorporate the indicated aromatic amino acid at all six positions (e.g., 6xF indicates a combined F mutation at positions 22, 64, 108, 44, 85, 132 in the PV2 protein). Other constructs are named according to the number of aromatics, the type of aromatic amino acid incorporated, and the positions of incorporation (e.g., 2xF(22,108) has an F residue incorporated at positions 22 and 108 in PV2).
Differential scanning calorimetry
Incorporation of aromatic residues F, Y, or W in the mini-core region of PV2 (as 3xAro(22,64,108)) is stabilizing in each case. Compared to PV2, the Aro3x(22,64,108) mutants (2.4% aromatic amino acid incorporation) display an increase in Tm (i.e., ΔTm) ranging from +27.8 (W) to +33.0°C (Y). This increase is essentially equivalent to the increase in Tm exhibited by PV2 in response to a high salt environment (ΔTm = +30.3°C in 2.0M vs. 0.1M NaCl) (Fig. 2, Table I). Likewise, all 6xAro constructs are more thermostable than PV2, with increases in Tm ranging from +14.7 (Y) to +40.4 (W) °C (Fig. 2, Table I). Comparisons between the 3xAro(22,64,108) and the 6xAro series indicate that while F or W incorporation into the central core is stabilizing, the Y mutation is destabilizing, and the melting temperature of 6xY is lowered by 18.3°C relative to 3xY(22,64,108).
Figure 2.
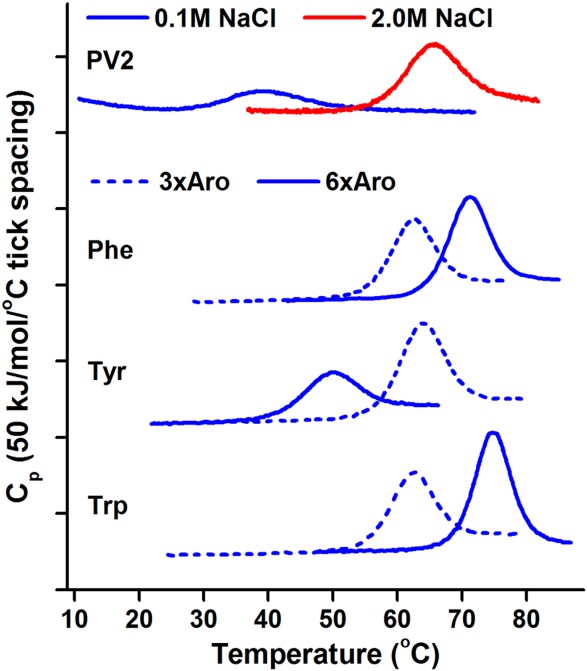
DSC of PV2 derivatives with aromatic amino acids at mini-core and central core positions. 3xAro constructs have the stated aromatic amino acid at positions 22, 64, 108. All data was collected under low salt conditions (0.1M NaCl) except where noted (red curve).
Table I.
DSC data for the thermal denaturation of PV2 and mutant proteins
| ΔH(Tm) (kJ mol−1) | Tm (°C) | ΔHvan't Hoff/ΔHcal | ΔTm (°C) | |
|---|---|---|---|---|
| Proteina | ||||
| PV2b | 157 ± 5 | 34.2 ± 0.2 | 0.87 ± 0.19 | – |
| PV2 (2.0M NaCl)b | 357 ± 2 | 64.5 ± 0.1 | 0.84 ± 0.04 | 30.3 |
| Mini-core mutants | ||||
| 1xF(22) | 306 ± 3 | 48.5 ± 0.1 | 0.71 ± 0.01 | 14.3 |
| 1xF(64) | 302 ± 3 | 48.3 ± 0.2 | 0.74 ± 0.02 | 14.1 |
| 1xF(108) | 300 ± 2 | 48.2 ± 0.1 | 0.75 ± 0.01 | 14.0 |
| 2xF(22,108) | 401 ± 2 | 56.9 ± 0.1 | 0.71 ± 0.01 | 22.7 |
| 3xF(22,64,108) | 446 ± 3 | 63.2 ± 0.1 | 0.81 ± 0.02 | 29.0 |
| 3xY(22,64,108) | 437 ± 1 | 67.2 ± 0.1 | 1.12 ± 0.01 | 33.0 |
| 3xW(22,64,108) | 414 ± 3 | 62.0 ± 0.1 | 1.03 ± 0.02 | 27.8 |
| Central core and mini-core mutants | ||||
| 6xFb | 490 ± 3 | 70.7 ± 0.1 | 0.96 ± 0.08 | 36.5 |
| 6xY | 301 ± 1 | 48.9 ± 0.1 | 1.04 ± 0.02 | 14.7 |
| 6xW | 544 ± 1 | 74.6 ± 0.1 | 0.99 ± 0.01 | 40.4 |
Buffer contains 0.1M NaCl unless otherwise noted.
From Ref.13
To determine how many aromatics are necessary to achieve essentially complete fractional folding (i.e., ≥0.99) of PV2, 1xF, and 2xF constructs were evaluated. F was selected for further study because it is less complex and more resistant to photodegradation than either Y or W; additionally, F is considered the earliest aromatic amino acid acquisition in Trifonov's analysis (discussed above). Each of the three mini-core positions 22, 64, and 108 was mutated independently to probe for differential effects on stability. The melting temperatures and enthalpies of unfolding of 1xF(22), 1xF(64), and 1xF(108) are essentially indistinguishable, indicating that all three of the mini-core positions are structurally equivalent in the native and unfolded states. Likewise, a plot of the number of F residues in the mini-core versus ΔGunf is linear (Supporting Information Fig. S1), as expected if the mini-core sites are noninteracting. Melting temperature, however, is nonlinear with respect to the number of incorporated F residues (Supporting Information Fig. S2) and it is the first F mutation that results in the greatest increase in Tm, with subsequent F mutations having diminished effects. At its temperature of maximum stability and in a low (i.e., mesophile) salt condition, PV2 is only 0.81 fractionally folded; in contrast, the 1xF mini-core variants achieve fractional folding of ≥0.99 at their respective temperatures of maximum stability in low salt (Fig. 3). A comparison of 3xY and 3xW mini-core mutant stability with 3xF shows that the Y mutation is more stable, while W is essentially isoenergetic with F. Thus, with incorporation of just a single aromatic amino acid (that is, at 0.8% of positions–an ∼11-fold reduction in the typical percentage of aromatic residues found in extant, mesophile proteins30,31), involving either F, Y, or W, the requirement of high salt concentrations for essentially complete folding is eliminated.
Figure 3.
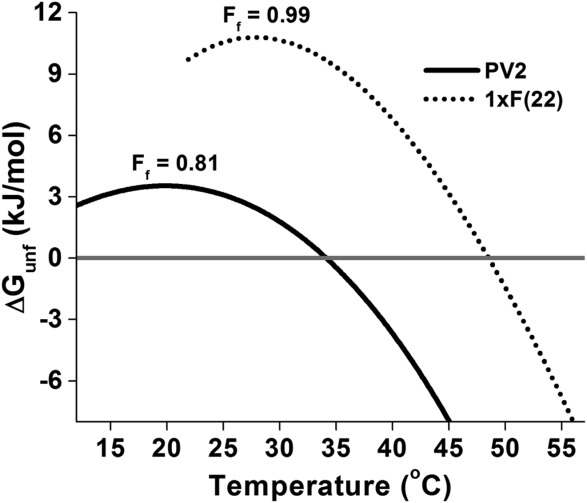
Stability of PV2 and 1xF(22) as a function of temperature. Stability curves were generated from fitted DSC parameters. Ff is the fraction folded at the temperature of maximum stability (i.e., ΔS = 0).
X-ray crystallography
Crystal structures for 6xY and 6xW were solved to a resolution of 1.70–1.75 Å (Table II); crystal structures of PV2 and 6xF have been previously reported.13 Each mutant demonstrates the predicted β-trefoil architecture and, despite a difference of 30 buried carbons between PV2 and 6xW, there is no evidence of any significant global structural expansion or collapse. Indeed, the main chain RMSD values for the 6xAro constructs range from 0.48 (6xF) to 0.56 Å (6xY) in comparison to PV2.
Table II.
Crystallographic data collection and refinement statistics
| 6xWa | 6xYb | |
|---|---|---|
| Space group | P212121 | P212121 |
| Cell constants (Å) | a = 47.1 | a = 34.8 |
| b = 48.5 | b = 46.8 | |
| c = 69.7 | c = 67.6 | |
| α = 90° | α = 90° | |
| β = 90° | β = 90° | |
| γ = 90° | γ = 90° | |
| Max resolution (Å) | 1.70 | 1.75 |
| Highest shell (Å) | 1.74–1.70 | 1.81–1.75 |
| Mosaicity (°) | 0.76 | 0.63 |
| Redundancy | 7.5 | 7.3 |
| Mol/ASU | 1 | 1 |
| Matthews coef. (Å3/Da) | 2.68 | 1.87 |
| Total reflections | 135,200 | 85,201 |
| Unique reflections | 17,987 | 11,690 |
| I/σ (overall) | 58.0 | 54.3 |
| I/σ (highest shell) | 4.1 | 4.3 |
| Completion overall (%) | 98.8 | 98.3 |
| Completion highest shell (%) | 99.9 | 99.6 |
| Rmerge overall (%) | 7.6 | 5.2 |
| Rmerge highest shell (%) | 40.2 | 31.1 |
| Nonhydrogen protein atoms | 1005 | 1023 |
| Solvent molecules/ion | 136/2 | 116/1 |
| Rcryst (%) | 19.5 | 18.7 |
| Rfree (%) | 23.2 | 21.6 |
| RMSD bond length (Å) | 0.007 | 0.007 |
| RMSD bond angle (°) | 1.04 | 1.10 |
| Ramachandran plot: | ||
| favored (%) | 97.5 | 100.0 |
| allowed (%) | 2.5 | 0.0 |
| outlier (%) | 0.0 | 0.0 |
| PDB accession | 4QKS | 4QKR |
1.4M (NH4)2SO4, 0.1M Tris pH 7.0, 0.07M Li2SO4.
30% PEG 8000, 0.1M Imidazole HCl pH 8.0, 0.2M NaCl.
Position 22 mutations (“mini-cores”)
Residue positions L13 and I42, along with the aliphatic chains of R15 and R37, form a hydrophobic environment around residue position 22 (Fig. 4). This hydrophobic “mini-core” is a distinct packing environment from the central hydrophobic core-packing group, and is replicated by the threefold symmetry of the β-trefoil structure at equivalent positions 22, 64, and 108. The introduced F, W, and Y aromatic residues at position 22 are accommodated with remarkably minimal structural perturbation. Each aromatic residue adopts an identical χ1 angle as the parental L22 residue in PV2. In response to the presence of the bulkier aromatic rings at position 22, the adjacent Arg15 side chain adopts an alternative rotamer in each case to avoid a close contact (Fig. 4); all other neighbor residues are unchanged. The mutant Y hydroxyl extends into partial solvent accessibility, and its hydrogen bonding requirement is satisfied by two novel water molecules (Sol77 and Sol60, Fig. 4). Similarly, the mutant W Nε1 nitrogen of the pyrrole ring achieves partial solvent accessibility, and its hydrogen bonding requirement is satisfied by a novel water molecule (Sol33, Fig. 4).
Figure 4.
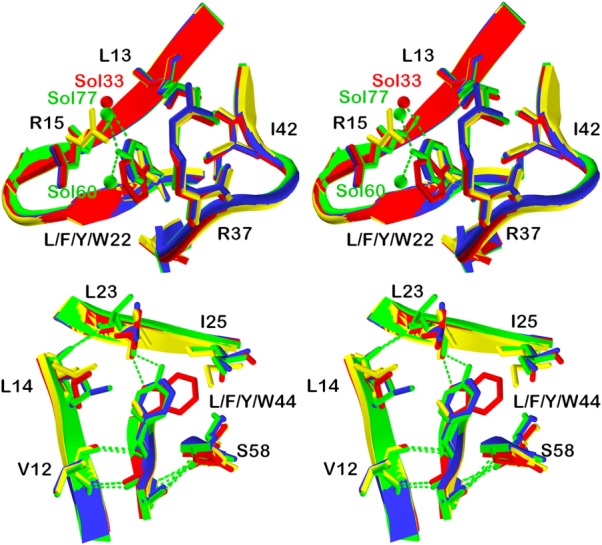
X-ray crystal structure overlays of aromatic substitutions in the PV2 protein. Upper panel: relaxed stereo diagram overlay of PV2 (yellow) with the 6xF (blue), 6xW (red), and 6xY (green) X-ray structures in the region of the position 22 mutations. Residue positions I42, L13 and the aliphatic side chains of R15 and R37 form a hydrophobic “mini-core” region around position 22. Lower panel: a similar overlay in the region of position 44 mutations. Residue positions L23, I25, V12, and L14 form a hydrophobic region around position 44—comprising part of the central hydrophobic core.
Position 44 mutations (central core)
Residue positions V12, L14, L23, and I25 form a hydrophobic environment around residue position 44 (Fig. 4). This region comprises part of the main central hydrophobic packing group, and is replicated by the threefold symmetry of the β-trefoil structure at equivalent positions 44, 85, and 132. The F, W, and Y aromatic residues introduced at position 44 are accommodated with minimal positional changes, or alternate rotamer conformations, of the adjacent residues. The introduced aromatic side chains, in each case, adopt the same χ1 angle as the parental L44 residue in PV2. The substitution of L44 by aromatic amino acids eliminates the L44 Cδ1 atom (the mutant aromatic rings are coplanar with the Cδ2 atom in each case). In response to the loss of the Leu44 Cδ1 atom the adjacent Leu14 adopts an alternative rotamer to effectively fill this space (Fig. 4). The bulkier F Cζ carbon and W Cε2 carbon introduce a close contact with L23, which is relieved by adoption of an alternate χ2 angle rotamer of L23. In the case of mutant Y44, the much longer OH group results in an alternative χ1 angle rotamer of L23 to avoid a close contact. The aromatic rings also result in a positional shift (∼1.0 Å) of adjacent I25 Cε1. In response to the bulkier indole ring of the introduced W44, the adjacent I25 residue adopts an alternative χ1 angle rotamer to avoid a steric clash. The hydrogen-bonding requirement of the mutant Y OH hydroxyl is provided by the main chain carbonyl of L23 with minimal (0.5 Å) positional shift (Fig. 4). Similarly, the hydrogen-bonding requirement of the mutant W Nε1 is also provided by the main chain carbonyl of Leu23 with minimal (0.3 Å) positional shift.
Refined coordinates and structure factors for the 6xW and 6xY mutants have been deposited in the Protein Databank (accession numbers 4QKS and 4QKR, respectively).
Empirical phase diagrams
Circular dichroism (CD), differential scanning calorimetry (DSC), and optical density at 360 nm were used to construct a temperature versus [NaCl] empirical phase diagram for PV2 and 1xF(64) (Fig. 5; raw data given in Supporting Information Fig. S3). Taken together, these probes provide a comprehensive view of the conformational state occupied by the protein, in which CD monitors secondary structure, DSC is sensitive to the heat capacity change associated with a conformational phase transition, and OD360 monitors protein aggregation. The OD360 data show that neither PV2 nor 1xF(64) aggregate, even at high temperatures and in the presence of 2.0M NaCl. Based on both DSC and CD, 1xF(64) is more thermostable than PV2, and differences in Tm as a function of salt concentration are greatest at low concentrations of NaCl: ΔTm (0.1M) = +14.3°C and ΔTm (2.0M NaCl) = +7.0°C (Fig. 5, panel c).
Figure 5.
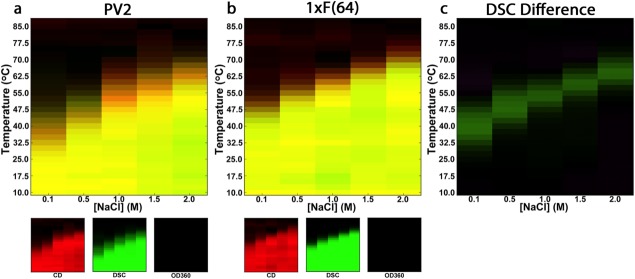
Temperature vs. salt concentration empirical phase diagrams of PV2 and 1xF(64). Empirical phase diagrams were generated using CD (a measure of secondary structure formation; red color indicates native-like structure), DSC (monitors heat associated with unfolding; intensity of green color corresponds integrated DSC signal, in which green color is assigned to the pretransition signal) and OD360 (turbidity; blue color). Phase diagrams are generated with an additive color scheme; thus, yellow color indicates that both CD and DSC report native-like interactions whereas black color indicates an unfolded protein that does not aggregate. A difference DSC EPD (c) was generated by subtracting the interpolated, integrated DSC signals of PV2 from that of 1xF(64); green indicates regions where 1xF(64) has a greater population of folded molecules than PV2.
Discussion
Although abiogenesis is one of the great unsolved problems in biochemistry, practical hypotheses are notoriously difficult to formulate and test. Among the challenges is the evaluation of key physical or chemical processes that took place over geological time scales, as well as assumptions regarding uncertain conditions. Furthermore, it is highly improbable that a single experiment will arrive at a solution; as with other major scientific problems, elucidating abiogenesis will be achieved through a series of individual advances—identifying what is possible, plausible, or implausible for key aspects of the overall abiogenic process. The Miller–Urey gas discharge experiments, along with recent related studies, have identified a consensus set of 10 of the common α-amino acids (the “prebiotic set”) that were plausibly available in the prebiotic soup as raw material for the very first peptides.6,32,33 A testable hypothesis is whether this restricted “abiotic” set comprises a foldable set that is able to support complex, stably folded polypeptide architecture.6 Basic and aromatic amino acids are notably absent from the prebiotic set, thus, salt bridges and aromatic core packing interactions are not feasible structural features to promote foldability in the earliest polypeptides. Successful studies of simplified protein design have been reported whereby foldable proteins have been constructed from a reduced α-amino acid alphabet,34,35 and relevance for proteogenesis have been described. However, such studies have focused exclusively on achieving minimization of the alphabet size, without regard to the prebiotic relevance of the included amino acid alphabet. Thus, without exception, such minimal foldable proteins have depended on critical aromatic amino acids within the core, as well as stabilizing salt bridges (dependent on basic amino acids), to achieve a stable structure. Thus, more work is needed to elucidate the critical question of whether the prebiotic amino acids form a foldable set; however, there is compelling evidence to support the prebiotic foldable set hypothesis, with studies indicating a dependency of such foldability on a halophile environment.6,7,13
Use of the β-trefoil architecture as a model of early folded proteins is motivated by several factors: first, the β-trefoil fold is comprised of β-strands and β-turns, which organize into β-hairpins and a β-barrel. Both the architecture itself and the structural motifs contained within it are common to every domain of life. Second, the structural evolution of the β-trefoil is well characterized, including an experimental demonstration of structural emergence by homo-oligomeric self-assembly (from a much simpler 42-mer peptide subdomain). Such data include a detailed experimentally validated path through stable, foldable sequence space linking an evolved β-trefoil protein (human fibroblast growth factor-1) to a simple 42 residue peptide “building block” (Monofoil-4P).27,36 Furthermore, sequence simplification (a recognized feature of ancient proteins) was accomplished with the development of the PV2 protein, comprised of an alphabet of only 12 different amino acids types. Although a number of protein simplification studies have reported stable folded structure using reduced amino acid alphabets, such simplified proteins fail to achieve prebiotic relevance because they depend upon non-prebiotic amino acids for structure and stability—notably involving aromatic or basic amino acids. As such, the observation that such simplified proteins can fold within a mesophile environment does not contradict the present results. Given that the β-trefoil is a common architecture with a unique robustness to sequence simplification, we conclude that PV2—which is entirely devoid of aromatic amino acids, with a purely prebiotic protein core—represents one of the best model systems currently available for studies of the folding potential of the prebiotic set of amino acids.
The positions selected for evaluating the effects of introducing aromatic residues in PV2 (22, 44, 64, 85, 108, and 132) have the property of residing within buried hydrophobic environments and being positions statistically preferred by aromatics in consensus sequence analyses of the β-trefoil fold.37–39
The large hydrophobic aromatic amino acids have long been known as major contributors to efficiently packed protein cores, providing substantial stabilizing Gibbs energy.40–42 There are two structural challenges to aromatic amino acid accommodation within a protein core: first, the adjacent residues must be able to adjust in response to the larger bulk of the aromatic amino acids, otherwise unfavorable strain (“overstuffing”) among core residues will result.43 The choice of the core and mini-core positions used in this study minimizes the potential for overstuffing as it is already known that these sites can accommodate an aromatic amino acid. Second, the protein must provide an appropriate hydrogen-bonding partner to the polar groups of Y (OH) and W (Nε1). In this regard, Y has both donor and acceptor requirements, while W requires only an acceptor. The X-ray structure analysis of the aromatic mutants shows that these positions in the β-trefoil have a plasticity that facilitates ready accommodation of essentially any of the aromatic amino acids.
As expected, the added bulk of the aromatic amino acids are accommodated with minor adjacent side chain rotamer adjustments and no substantial main chain perturbations. At positions 22, 64, 108 the hydrogen-bonding requirements of Y and W are achieved by solvent—two solvent molecules (one apparent donor, one apparent acceptor) in the case of Y and one (an acceptor) in the case of W. At positions 44, 85, 132 the protein architecture itself provides an appropriate acceptor in the main chain carbonyl 23O (as a second donor interaction, 23O has an H-bond donor partner in an adjacent buried solvent). No donor is observed interacting with the Y OH; thus, while the hydrogen-bonding requirements of the introduced W may be fully satisfied, those of the introduced Y appear to be incomplete. Water/hydrophobic solvent transfer free energy values, as well as experimental values for A → S and V → T polar substitutions at hydrophobic (i.e., buried) positions in proteins, indicate an upper value of ΔG ∼+12 kJ/mol for effective desolvation of such polar groups with no corresponding novel H-bond partner.44,45 The derived ΔΔG value for an F → Y point mutation at symmetry-related positions 44, 85, and 132 is ∼+10 kJ/mol per mutation, in agreement with the expected destabilization of an unsatisfied H-bonding requirement. The stability data is consistent with the structural data: the added hydrophobic bulk of the buried aromatics provide substantial increased stability regardless of type of aromatic amino acid, with the exception of Y at positions 22, 44, 85. Thus, at the two buried environments evaluated, the protein achieves significant stability gains with a general introduction of aromatic amino acids (i.e., with 15 of 18 possible aromatic substitutions). The stability increase in response to aromatic substitution is not due to π-stacking or π-cation interactions as the prebiotic design is devoid of basic and aromatic amino acids within the core. The stability increase is due to a combination of hydrophobic effect (solvent entropy gain on aromatic burial) and more extensive van der Waals interactions within the core, combined with a structural ability of the basic β-trefoil architecture to satisfy H-bond requirements of the aromatics Y and W. Subsequently, the aromatic substitutions—potentially as point mutations—have the ability to move the folding properties of the PV2 protein from halophilic to mesophilic conditions. This ability of the β-trefoil architecture to accommodate and thermodynamically benefit from aromatic substitutions (primarily involving a main chain architecture able to provide necessary hydrogen-bonding interactions without perturbation) suggests a plausible selective advantage for this fold upon the evolutionary availability of aromatic amino acids.
Protein design studies suggest that a halophilic environment may have been involved in supporting protein folding early in abiogenesis (i.e., before the incorporation of amino acid biosynthesis).6,13 Consistent with this view is the observation that peptide bond formation is promoted by high NaCl concentrations.17,46 This demonstrates that polymerization (an otherwise thermodynamically unfavorable condensation reaction in water) of a key class of biopolymer is achievable under plausible prebiotic conditions. These studies suggest that the cradle of life may have resided within evaporative salt ponds, within which nonvolatile metabolites—in this case, amino acids—would have been concentrated and undergone chemical condensation to form polypeptides. Thus far, NaCl has been assumed to be the most appropriate salt of the halophile environment. Other salts are of interest to study, both as potential cosalts in halophile environments and as probes to understand the biophysical basis of enhanced stability in more detail (e.g., effects of the Hoffmeister series); such studies are currently in progress. Although it is known that copolymers (e.g., PEG, Dextran, and Ficoll) as well as various sugars can stabilize proteins, these additives (unlike simple salts) lack prebiotic relevance and their accumulation in the environment to concentrations that would significantly affect folding appears improbable.
If high salt conditions are a requirement for stable folding of the earliest polypeptides, then a key question is how life could have adapted out of such halophilic environments. Previously, it was shown that a construct with a combined total of six F residues can shift folding requirements from the halophile to mesophile environment. However, if six F substitutions are simultaneously required for a halophile-mesophile shift in folding, it would be evolutionarily implausible. We show here that incorporation of a single aromatic amino acid can effectively convert a foldable “prebiotic” polypeptide from an obligate halophile to a stable mesophile (i.e., with fractional folding of ≥0.99 in the absence of high concentrations of salt). Notably, the stability data demonstrate that potentially any of the aromatic amino acids (F, Y, or W) substituted into PV2 (involving the mini-core position) could enable this folding transition, and that the first aromatic amino acid yields the greatest increase in melting temperature. These results are consistent with the observation that aromatic amino acids are significantly more common in the proteomes of mesophiles than in halophiles,7,14–16 perhaps due to the alleviated need for optimized core packing in a halophile context. Therefore, incorporation of aromatic amino acids into early proteins may have facilitated a critical halophile-to-mesophile transition. Subsequent incorporation of multiple aromatic groups within protein core regions can provided additional stability gains, enabling further adaptation into more demanding (i.e., extremophile) environments for protein folding, such as physical extremes of temperature or pH.
Materials and Methods
Protein expression and purification
Synthetic genes and mutagenesis primers were ordered from integrated DNA technologies. The 1xF and 2xF mutant proteins were constructed via site-directed mutagenesis following the Quikchange (Agilent Technologies, Santa Clara, CA) protocol. DNA sequences were verified before expression in E. coli BL21(DE3) competent cells. Transformed cells were grown in M9 media cultures, induced with 1 mM IPTG, and expressed for 8–10 h at 27°C. Cells were harvested via centrifugation at 5400g for 15 min at 4°C using and stored at −20°C. Cell pellets were resuspended in 5 mM imidazole, 50 mM NaPi, 500 mM NaCl, 0.01% Tween-80, pH 7.5. The cell suspension was lysed by passage through a French pressure cell at 1000 psi and the cell lysate was clarified by centrifugation at 29,600g for 60 min at 4°C. Expressed proteins contained an N-terminal (His)6x tag which has shown no influence upon stability or folding properties.38 The supernatant was loaded onto a packed nickel affinity (Ni-NTA) chromatography column and the protein was eluted with 100 mM Imidazole, 500 mM NaCl, 50 mM NaPi, pH 7.5. Samples were further purified by gel filtration on a Superdex 75 column (GE Healthcare, Buckinghamshire, United Kingdom). The extinction coefficients for mutants containing W or Y residues were obtained by the Gill and von Hippel method.47 Concentrations for all other mutant proteins were obtained using a bicinchoninic acid (BCA) assay using a standard curve generated against known concentrations of Symfoil-1. The purified protein was dialyzed against either phosphate buffer (100 mM NaCl, 10 mM (NH4)2SO4, 50 mM NaPi pH 7.5) for crystallization studies or ADA buffer (20 mM N-(2-acetamido) iminodiacetic acid solution, 100 mM NaCl, pH 6.6.) for biophysical characterization and empirical phase diagram preparation.
X-ray crystallography
Purified protein was concentrated to 10–15 mg/mL in phosphate buffer. Crystal conditions were screened by the hanging drop vapor diffusion method at 25°C. 6xW crystals grew in 1.4M (NH4)2SO4, 0.1M Tris, 0.07M Li2SO4, pH 7.0; 6xY crystals grew in 30% (w/v) PEG 8,000, 0.1M Imidazole HCl, 0.2M NaCl, pH 8.0. Both crystals exhibited the same space group (P212121), however, the crystal cell dimensions differ. 6xLeu (PV2; PDB ID code 4D8H) and 6xF (PDB ID code 3QYX) crystal structures have been previously reported.13 Crystals were mounted using Hampton Research nylon cryo-loops and were cryo-cooled to 100 K by gaseous N2 using an Oxford cryo-system (Oxford, UK). Crystals were diffracted in-house with a Rigaku RU-H3R rotating anode X-ray source (Rigaku, Tokyo, Japan) equipped with Osmic confocal mirrors (Osmic, Troy, MI) and a MarCCD165 detector (Rayonix, Evanston, IL). Data sets were analyzed with the DENZO software package to integrate, index, and scale all reflections. Molecular replacement for 6xW and 6xY was conducted using PV1 (PDB ID code 3QYX) as a search model with the PHENIX software program.48
Differential scanning calorimetry
DSC data was collected using a VP-DSC calorimeter (GE Healthcare, Buckinghamshire, United Kingdom). Three buffer–buffer scans were collected prior to protein loads to establish proper thermal history. 40 µM protein samples in ADA Buffer were scanned from 10 to 95°C at a rate of 0.25°C/min under 34 psi. Analysis of the resulting endotherms was performed using the DSCfit software package.49
Empirical phase diagrams
CD was performed using a Chirascan-plus CD spectrometer (Applied Photophysics, Leatherhead, UK) equipped with a four-cuvette position Pelletier temperature controller (Quantum Northwest, Liberty Lake, WA) and a solid-state detector. The lamp, monochromator, and sample chamber were continually purged with N2. Far-U.V. CD spectra of triplicate samples at 0.75 mg/mL were collected in the range of 260–200 nm in 1 nm steps and a 0.5 s sampling time using a quartz cuvette (0.1 cm path length) sealed with a Teflon stopper (Starna Cells Inc., Atascadero, CA). The CD signal at 230 nm was monitored as a function of temperature from 10 to 87.5°C at 2.5°C intervals. The heating rate was 1°C/min, and the equilibration time at each temperature was 1 min. The ellipticity of the buffer was subtracted from all measurements. All data were subjected to a three-point Savitzky–Golay smoothing filter using the Chirascan software (Applied Photophysics).
To quantify turbidity, the optical density at 360 nm was measured as a function of temperature (10.0–87.5°C) using a Cary-100 U.V.-Vis spectrophotometer equipped with a 12 cell-temperature controlled Pelletier (Agilent Technologies, Santa Clara CA). A 1°C/min heating rate, a 2 s integration time, and a 2 min equilibration time at each temperature were used. Samples were diluted in ADA Buffer to 0.2 mg/mL using a 1 cm path length quartz cuvette. The optical density of buffer alone was subtracted from all measurements.
DSC was performed using an Auto-VP capillary differential scanning calorimeter (MicroCal/GE Health Sciences) equipped with Tantalum sample and reference cells. Two water-water scans were taken prior to the reference and sample scans. Scans were completed from 10–90°C using a scanning rate of 15°C/h and a concentration of 1 mg/mL. Reference subtraction and concentration normalization were performed using the instrument software.
Three-index EPDs were constructed as described50 using the MiddaughSuite software. The DSC data was interpolated from 10.0–87.5°C at 2.5°C increments and integrated prior to EPD construction.
Acknowledgments
The X-ray Facility at Florida State University is acknowledged for assistance with data collection and processing. The authors declare no conflicts of interests.
Glossary
- ADA
N-(2-acetamido) iminodiacetic acid
- Aro
aromatic
- BCA
bicinchoninic acid
- CD
circular dichroism
- DSC
differential scanning calorimetry
- EPD
empirical phase diagram
- ΔGunf
unfolding Gibbs energy
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- LUCA
last universal common ancestor
- OD
optical density
- NaPi
sodium phosphate
- Ni-NTA
Nickel-nitrilotriacetic acid
- OH
hydroxyl
- PDB
protein databank
- PEG
polyethylene glycol
- PV1
designed primitive β-trefoil protein version 1
- PV2
designed primitive β-trefoil protein version 2
- RMSD
root mean square deviation
- Sol
water solvent
- Tm
melting temperature
- Tween
polysorbate
- U.V.
ultraviolet.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- 1.Miller SL. A production of amino acids under possible primitive earth conditions. Science. 1953;117:528–529. doi: 10.1126/science.117.3046.528. [DOI] [PubMed] [Google Scholar]
- 2.Hennet RJ-C, Holm NG, Engel MH. Abiotic synthesis of amino acids under hydrothermal conditions and the origin of life: a perpetual phenomenon? Naturwissenschaften. 1992;79:361–365. doi: 10.1007/BF01140180. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi K, Kaneko T, Saito T, Oshima T. Amino acid formation in gas mixtures by high energy particle irradiation. Orig Life Evol Biosph. 1998;28:155–165. doi: 10.1023/a:1006561217063. [DOI] [PubMed] [Google Scholar]
- 4.Wolman Y, Haverland WJ, Miller SL. Nonprotein amino acids from spark discharges and their comparison with the Murchison meteorite amino acids. Proc Natl Acad Sci USA. 1972;69:809–811. doi: 10.1073/pnas.69.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chyba CF, Thomas PJ, Brookshaw L, Sagan C. Cometary delivery of organic molecules to the early Earth. Science. 1990;249:366–373. doi: 10.1126/science.11538074. [DOI] [PubMed] [Google Scholar]
- 6.Longo LM, Blaber M. Protein design at the interface of the pre-biotic and biotic worlds. Arch Biochem Biophys. 2012;526:16–21. doi: 10.1016/j.abb.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 7.Longo LM, Blaber M. Prebiotic protein design supports a halophile origin of foldable proteins. Front Microbiol. 2014;4:418. doi: 10.3389/fmicb.2013.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parker ET, Zhou M, Burton AS, Glavin DP, Dworkin JP, Krishnamurthy R, Fernandez FM, Bada JL. A plausible simultaneous synthesis of amino acids and simple peptides on the primordial Earth. Angew Chem. 2014;126:8270–8274. doi: 10.1002/anie.201403683. [DOI] [PubMed] [Google Scholar]
- 9.Romero P, Obradovic Z, Dunker AK. Folding minimal sequences: the lower bound for sequence complexity of globular proteins. FEBS Lett. 1999;462:363–367. doi: 10.1016/s0014-5793(99)01557-4. [DOI] [PubMed] [Google Scholar]
- 10.Murphy LR, Wallqvist A, Levy RM. Simplified amino acid alphabets for protein fold recognition and implications for folding. Protein Eng. 2000;13:149–152. doi: 10.1093/protein/13.3.149. [DOI] [PubMed] [Google Scholar]
- 11.Kamtekar S, Schiffer JM, Xiong H, Babik JM, Hecht MH. Protein design by binary patterning of polar and nonpolar amino acids. Science. 1993;262:1680–1685. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 12.Cockell CS, Airo A. On the plausibility of a UV transparent biochemistry. Origins Life Evol Biosphere. 2002;32:255–274. doi: 10.1023/a:1016507810083. [DOI] [PubMed] [Google Scholar]
- 13.Longo L, Lee J, Blaber M. Simplified protein design biased for pre-biotic amino acids yields a foldable, halophilic protein. Proc Natl Acad Sci USA. 2013;110:2135–2139. doi: 10.1073/pnas.1219530110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oren A, Larimer F, Richardson P, Lapidus A, Csonka LN. How to be moderately halophilic with broad salt tolerance: clues from the genome of Chromohalobacter salexigens. Extremophiles. 2005;9:275–279. doi: 10.1007/s00792-005-0442-7. [DOI] [PubMed] [Google Scholar]
- 15.Paul S, Bag SK, Das S, Harvill ET, Dutta C. Molecular signature of hypersaline adaptation: insights from genome and proteome composition of halophilic prokaryotes. Genome Biol. 2008;9:R70.71–19. doi: 10.1186/gb-2008-9-4-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kennedy SP, Ng WV, Salzberg SL, Hood L, DasSarma S. Understanding the adaptation of Halobacterium species NRC-1 to its extreme environment through computational analysis of its genome sequence. Genome Res. 2001;11:1641–1650. doi: 10.1101/gr.190201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rode BM. Peptides and the origin of life. Peptides. 1999;20:773–786. doi: 10.1016/s0196-9781(99)00062-5. [DOI] [PubMed] [Google Scholar]
- 18.Dundas I. Was the environment for primordial life hypersaline? Extremophiles. 1998;2:375–377. doi: 10.1007/s007920050081. [DOI] [PubMed] [Google Scholar]
- 19.Cleaves HJI. The origin of the biologically coded amino acids. J Theor Biol. 2010;263:490–498. doi: 10.1016/j.jtbi.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 20.Wong JT-F. Coevolution theory of the genetic code at age thirty. Bioessays. 2005;27:406–425. doi: 10.1002/bies.20208. [DOI] [PubMed] [Google Scholar]
- 21.Trifonov EN. Consensus temporal order of amino acids and evolution of the triplet code. Gene. 2000;261:139–151. doi: 10.1016/s0378-1119(00)00476-5. [DOI] [PubMed] [Google Scholar]
- 22.Merino E, Jensen RA, Yanofsky C. Evolution of bacterial trp operons and their regulation. Curr Opin Microbiol. 2008;11:78–86. doi: 10.1016/j.mib.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernandez-Montes G, Diaz-Mejia JJ, Perez-Rueda E, Segovia L. The hidden universal distribution of amino acid biosynthetic networks: a genomic perspective on their origins and evolution. Genome Biol. 2008;9:R95. doi: 10.1186/gb-2008-9-6-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ringvold A, Anderssen E, Kjonniksen I. UV absorption by uric acid in diurnal bird aqueous humor. Invest Ophthalmol Vis Sci. 2000;41:2067–2069. [PubMed] [Google Scholar]
- 25.Beadle BM, Shoichet BK. Structural basis of stability—function tradeoffs in enzymes. J Mol Biol. 2002;321:285–296. doi: 10.1016/s0022-2836(02)00599-5. [DOI] [PubMed] [Google Scholar]
- 26.Longo L, Lee J, Blaber M. Experimental support for the foldability-function tradeoff hypothesis: segregation of the folding nucleus and functional regions in FGF-1. Protein Sci. 2012;21:1911–1920. doi: 10.1002/pro.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J, Blaber SI, Dubey VK, Blaber M. A polypeptide “building block” for the ß-trefoil fold identified by “top-down symmetric deconstruction”. J Mol Biol. 2011;407:744–763. doi: 10.1016/j.jmb.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Longo LM, Kumru OS, Middaugh CR, Blaber M. Evolution and design of protein structure by folding nucleus symmetric expansion. Structure. doi: 10.1016/j.str.2014.08.008. (in press). doi: 10.1016/j.str.2014.08.008. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 29.Broom A, Doxey AC, Lobsanov YD, Berthin LG, Rose DR, Howell PL, McConkey BJ, Meiering EM. Modular evolution and the origins of symmetry: reconstruction of a three-fold symmetric globular protein. Structure. 2012;20:1–11. doi: 10.1016/j.str.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 30.Dyer KF. The quiet revolution: A new synthesis of biological knowledge. J Biol Educ. 1971;5:15–24. [Google Scholar]
- 31.King JL, Jukes TH. Non-Darwinian evolution. Science. 1969;164:788–798. doi: 10.1126/science.164.3881.788. [DOI] [PubMed] [Google Scholar]
- 32.Doi N, Kakukawa K, Oishi Y, Yanagawa H. High solubility of random-sequence proteins consisting of five kinds of primitive amino acids. Prot Eng Des Sel. 2005;18:279–284. doi: 10.1093/protein/gzi034. [DOI] [PubMed] [Google Scholar]
- 33.McDonald GD, Storrie-Lombardi MC. Biochemical constraints in a protobiotic Earth devoid of basic amino acids: the “BAA(-) World”. Astrobiology. 2010;10:989–1000. doi: 10.1089/ast.2010.0484. [DOI] [PubMed] [Google Scholar]
- 34.Riddle DS, Santiago JV, Bray-Hall ST, Doshi N, Grantcharova VP, Yi Q, Baker D. Functional rapidly folding proteins from simplified amino acid sequences. Nat Struct Biol. 1997;4:805–809. doi: 10.1038/nsb1097-805. [DOI] [PubMed] [Google Scholar]
- 35.Walter KU, Vamvaca K, Hilvert D. An active enzyme constructed from a 9-amino acid alphabet. J Biol Chem. 2005;280:37742–37746. doi: 10.1074/jbc.M507210200. [DOI] [PubMed] [Google Scholar]
- 36.Lee J, Blaber M. Experimental support for the evolution of symmetric protein architecture from a simple peptide motif. Proc Natl Acad Sci USA. 2011;108:126–130. doi: 10.1073/pnas.1015032108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alsenaidy MA, Wang T, Kim JH, Joshi SB, Lee J, Blaber M, Volkin DB, Middaugh CR. An empirical phase diagram approach to investigate conformational stability of “second-generation” functional mutants of acidic fibroblast growth factor (FGF-1) Protein Sci. 2012;21:418–432. doi: 10.1002/pro.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brych SR, Blaber SI, Logan TM, Blaber M. Structure and stability effects of mutations designed to increase the primary sequence symmetry within the core region of a β-trefoil. Protein Sci. 2001;10:2587–2599. doi: 10.1110/ps.ps.34701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murzin AG, Lesk AM, Chothia C. β-Trefoil fold. Patterns of structure and sequence in the kunitz inhibitors interleukins-1β and 1α and fibroblast growth factors. J Mol Biol. 1992;223:531–543. doi: 10.1016/0022-2836(92)90668-a. [DOI] [PubMed] [Google Scholar]
- 40.Hecht MH, Sturtevant JM, Sauer RT. Effect of single amino acid replacements on the thermal stability of the NH2-terminal domain of phage λ repressor. Proc Natl Acad Sci USA. 1984;81:5685–5689. doi: 10.1073/pnas.81.18.5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shortle D, Stites WE, Meeker AK. Contributions of the large hydrophobic amino acids to the stability of Staphylococcal nuclease. Biochemistry. 1990;29:8033–8041. doi: 10.1021/bi00487a007. [DOI] [PubMed] [Google Scholar]
- 42.Eriksson AE, Baase WA, Zhang X-J, Heinz DW, Blaber M, Baldwin EP, Matthews BW. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 1992;255:178–183. doi: 10.1126/science.1553543. [DOI] [PubMed] [Google Scholar]
- 43.Lim WA, Farruggio DC, Sauer RT. Structural and energetic consequences of disruptive mutations in a protein core. Biochemistry. 1992;31:4324–4333. doi: 10.1021/bi00132a025. [DOI] [PubMed] [Google Scholar]
- 44.Wolfenden R, Andersson L, Cullis PM, Southgate CCB. Affinities of amino acid side chains for solvent water. Biochemistry. 1981;20:849–855. doi: 10.1021/bi00507a030. [DOI] [PubMed] [Google Scholar]
- 45.Blaber M, Lindstrom JD, Gassner N, Xu J, Heinz DW, Matthews BW. Energetic cost and structural consequences of burying a hydroxyl group within the core of a protein determined from ala->ser and val->thr substitutions in T4 lysozyme. Biochemistry. 1993;32:11363–11373. doi: 10.1021/bi00093a013. [DOI] [PubMed] [Google Scholar]
- 46.Schwendinger MG, Rode BM. Possible role of copper and sodium chloride in prebiotic evolution of peptides. Analyt Sci. 1989;5:411–414. [Google Scholar]
- 47.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 48.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Loerger TR, McCoy AJ, McKee E, Moriarity NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD. Automated structure solution with the PHENIX suite. Methods Mol Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 49.Grek SB, Davis JK, Blaber M. An efficient, flexible-model program for the analyis of differential scanning calorimetry protein denaturation data. Prot Pept Lett. 2001;8:429–436. [Google Scholar]
- 50.Kim JJ, Iyer V, Joshi SB, Volkin DB, Middaugh CR. Improved data visualization techniques for analyzing macromolecule structural changes. Protein Sci. 2012;21:1540–1553. doi: 10.1002/pro.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information