

Abstract
A-kinase anchoring proteins (AKAPs) regulate cAMP-dependent protein kinase (PKA) signaling in space and time. Dual-specific AKAP2 (D-AKAP2/AKAP10) binds with high affinity to both RI and RII regulatory subunits of PKA and is anchored to transporters through PDZ domain proteins. Here, we describe a structure of D-AKAP2 in complex with two interacting partners and the exact mechanism by which a segment that on its own is disordered presents an α-helix to PKA and a β-strand to PDZK1. These two motifs nucleate a polyvalent scaffold and show how PKA signaling is linked to the regulation of transporters. Formation of the D-AKAP2: PKA binary complex is an important first step for high affinity interaction with PDZK1, and the structure reveals important clues toward understanding this phenomenon. In contrast to many other AKAPs, D-AKAP2 does not interact directly with the membrane protein. Instead, the interaction is facilitated by the C-terminus of D-AKAP2, which contains two binding motifs—the D-AKAP2AKB and the PDZ motif—that are joined by a short linker and only become ordered upon binding to their respective partner signaling proteins. The D-AKAP2AKB binds to the D/D domain of the R-subunit and the C-terminal PDZ motif binds to a PDZ domain (from PDZK1) that serves as a bridging protein to the transporter. This structure also provides insights into the fundamental question of why D-AKAP2 would exhibit a differential mode of binding to the two PKA isoforms.
Keywords: PKA signaling, A-kinase anchoring proteins, D-AKAP2 specificity, AKAP10, PDZK1 crystal structure
Introduction
Phosphorylation of target proteins by cyclic AMP (cAMP)-dependent protein kinase (PKA) is a crucial step in the regulation of signaling pathways. A diverse family of proteins called the A-kinase anchoring proteins (AKAPs) mediates the spatiotemporal targeting of PKA to various parts of the cell.1,2 The discovery of numerous AKAPs and the emergent knowledge of their involvement in regulating a variety of cellular processes, including PKA-mediated phosphorylation, have underscored the importance of characterizing AKAP-mediated protein complexes. A next challenge is to understand at the molecular level how multiprotein complexes are assembled by AKAPs.
PKA is a homodimer of two regulatory (R) subunits bound to two catalytic (C) subunits, and, based on the R subunits the enzyme, is classified into type I or type II classes (with α and β subclasses).3 While the four R subunits share a similar domain organization, they are not functionally redundant4–6 and are localized differently in the cell.1 Each R subunit contains an N-terminal dimerization/docking (D/D) domain followed by a linker containing an inhibitor site and finally, two tandem cAMP-binding domains.7 The D/D domains dimerize and form an X-type, anti-parallel, four-helix bundle that serves as a docking surface for AKAPs.8–10 AKAPs, in turn, interact with the D/D domains through an amphipathic helix of ∼20 residues.9–13 In recent years, several AKAPs such as D-AKAP2,14 D-AKAP1,15 and AKAP22016 have been identified as dual-specific because of their ability to interact with both types of the R subunits, not just RII subunit.
Dual-specific AKAP2 (D-AKAP2 or AKAP10) is a multi-domain protein that is involved in the late stages of endocytosis.17 It consists of two tandem regulator of G-protein signaling (RGS)-like homology domains, followed by a 27-residue PKA-binding (AKB) domain and a PSD-95/DlgA/ZO-1 (PDZ)-binding motif at the C-terminus.14 The RGS domains were shown more recently to directly interact with two small GTPases, Rab4, and Rab11,17 which belong to a family of proteins that is known to play a key role in the regulation of endocytic membrane trafficking.18–20 The AKB domain (D-AKAP2AKB) at pH 7.0 is predominantly a random coil. Upon binding to the D/D domains of RIα and RIIα with high affinity (KD = 48 and 2 nM, respectively), it forms an amphipathic helix showing that it has all the information necessary and sufficient for binding to PKA.12,13,21 The affinities of the AKB for the full length R subunits is comparable to the published affinities for the isolated D/D domains (unpublished data). The type I PDZ motif (-X-S/T-X-ϕ) of D-AKAP2 (-STKL) interacts with the multi-PDZ domain proteins PDZK1 and NHERF-1.22 PDZK1, in particular, is a physiologically important molecule that has been implicated in regulating the expression and localization of membrane-associated transporters, and maintenance of serum cholesterol levels.23–26
The binding of D-AKAP2AKB (and AKAPs, in general) to the PKA R subunits has been studied extensively using a variety of methods including mutational analyses,9,27 hydrogen/deuterium exchange mass spectrometry,28 peptide arrays and peptide disruption studies,11,29–31 and structural analyses.11–13 These studies were undertaken not just for a thorough molecular understanding of AKAP binding but also to explain the dual-specific nature of D-AKAP2. In particular, the atomic-level information from the structures of D-AKAP2AKB in complex with RIα and RIIα D/D domains revealed an unusual phenomenon: the mode of D-AKAP2AKB binding to RIα is somewhat different than to RIIα.12,13 In particular, the α-helical register of D-AKAP2 is shifted by a single turn thereby presenting a different surface to two proteins through a simple mechanism. This phenomenon was used to elucidate a molecular explanation for the sequence and spatial determinants of D-AKAP2 specificity and was proposed to be true for all RI-, RII-, and dual-specific AKAPs.13 It was also confirmed by the recent structure of an RI-specific AKAP (smAKAP)32 bound to the D/D domain of RIα (manuscript submitted). The shift in the D-AKAP2AKB helical register and its close proximity to the PDZ motif lead to three questions: (1) how does the short C-terminal D-AKAP2AKB that on its own is disordered and has two protein binding motifs embedded in its sequence, nucleate a polyvalent scaffold, (2) what effect does PKA binding to D-AKAP2 have on binding to the PDZ domain, and (3), why do we observe a shift in the helical register?
To answer these questions and to elucidate how a polyvalent scaffold is nucleated by these three proteins, we determined the crystal structure of D-AKAP2AKB in complex with PKA RIIα D/D domain and the 4th PDZ domain of PDZK1 (PDZK1D4). The structure reveals the exact mechanism by which D-AKAP2AKB interacts with both proteins simultaneously; D-AKAP2AKB presents an α-helix for its interaction with PKA RII D/D domains and an antiparallel β-strand for its interaction with PDZK1D4. Binding of the D-AKAP2AKB to the RIIα D/D domain was necessary to achieve a stable ternary complex. Although a short linker region of D-AKAP2 prevents any significant contact between these two interaction surfaces, truncating the linker by 1, 2, or 3 residues does not interfere with the ability to form a high affinity complex with the PKA RIIα D/D and PDZK1D4 based on gel filtration. Modeling the D-AKAP2AKB: RIα D/D complex structure onto the present structure also allows us to hypothesize a reason for the differential binding of D-AKAP2 to the PKA R subunits.
Results and Discussion
D-AKAP2AKB shows specificity for binding to the PDZ domains of NHERF1, NHERF2, and PDZK1
D-AKAP2 has a C-terminal class I PDZ motif that binds to a PDZ domain, which then serves as an adaptor for coupling D-AKAP2 to receptors or transporters (Fig. 1). PDZK1 and NHERF1 are two PDZ domain proteins that bind with high affinity to D-AKAP2.22 Since NHERF1 and PDZK1 both contain more than one PDZ domain, we used an array of PDZ domains (Fig. 1) to determine whether there was specificity for binding of D-AKAP2 to a particular PDZ domain. We showed that D-AKAP2 binds well to the third and fourth PDZ domains of PDZK1 and to specific PDZ domains of NHERF1 and 2. Two D-AKAP2 constructs, GST-AKB that represents the C-terminal AKB and PDZ motifs (residues 623–662) and GST-RGS-AKB-CT that includes the second RGS domain (residues 373–662) gave similar results, as shown in Figure 1(a,c), respectively; both constructs contain the PDZ-binding motif at their C-terminus. D-AKAP2 binds very well to the fourth (spot G1) and third (spot F12) PDZ domains of PDZK1. D-AKAP2 also showed good binding to the first domain of NHERF1 (spot B3) and second domain of NHERF2 (spot B6). Although some other PDZ motifs showed weaker binding, this array demonstrated a clear specificity for of D-AKAP2 binding to individual domains in PDZK1 and NHERF1/2.
Figure 1.
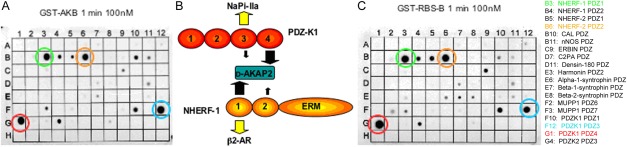
Peptide arrays of PDZ domains reveal specificity of D-AKAP2 for PDZK1 and NHERF. Peptide array of PDZ domains overlaid with the D-AKAP2AKB (a) or with the larger fragment of D-AKAP2 that includes RGS-B (c). Bound AKAP was detected with anti-GST antibody. The corresponding PDZ domains that reacted most strongly with D-AKAP2 are B3: NHERF-1 PDZ1; B6: NHERF-2 PDZ2; F12: PDZK1 PDZ3; and G1: PDZK1 PDZ4. (b) Cartoon model of PDZK1 and NHERF-1 bound to D-AKAP2, which thus serves as an adaptor for coupling D-AKAP2 to receptors or transporters.
Ternary complex purification and crystallization
In an attempt to isolate and crystallize a complex of D-AKAP2, PDZK1, and PKA we purified the individual components from each protein, the D/D domain of RIIα, the AKB from D-AKAP2, and full length PDZ-K1 as well as the 4th PDZ domain (PDZK1D4). Following the purification of these individual components (described in the Material and Methods), purification of the D-AKAP2AKB: PKA RIIα D/D: PDZK1D4 ternary complex required optimization. Using gel filtration as a strict criterion for complex formation, D-AKAP2AKB and PDZK1D4 alone did not form a stable complex. Since the relative affinity of PDZ domains to PDZ motifs is low, it is not surprising that our initial attempts to purify such a complex on a gel filtration column were unsuccessful, and this is true for many other PDZ domain complexes. Previous successful attempts at crystallization and structure determination of PDZ motif: PDZ domain binary complexes have typically involved two different strategies: one, soaking or cocrystallizing peptides containing the PDZ motifs into crystals of PDZ domains and two, designing a construct wherein the PDZ motif is artificially tethered to the C-terminus of a PDZ domain through a peptide linker.
In our case, formation of a high affinity complex containing all three proteins was achieved, but it required two distinct steps. In the first step, the formation of the well-characterized RIIα D/D: D-AKAP2AKB complex was achieved by incubating equimolar quantities for 30 min on ice. The next step involved the addition of equimolar PDZK1D4 (for another 30 min) to this binary complex. Simply incubating equimolar amounts of all three proteins together simultaneously did not yield a high affinity complex. The ternary complex was purified away from the individual proteins on a S75 gel filtration column (Supporting Information Fig. S1) and concentrated to ∼10 mg/mL prior to crystallization. While a similar complex consisting of the full-length PDZK1 (containing four PDZ domains) was also purified, only the ternary complex containing PDZK1D4 yielded crystals. Based on SDS gels and elution from the gel filtration column, the complex seemed to represent a 1:1:1 complex but this was not further characterized.
Structure solution
The crystal structure of the D-AKAP2AKB: RIIα D/D: PDZK1D4 ternary complex was determined to 3.8 Å resolution by molecular replacement, as implemented in Phaser, using the D-AKAP2AKB: RIIα D/D (PDB ID: 2HWN)12 and PDZK1D4 (PDB ID: 2VSP) structures independently as search models. Careful consideration was given to all aspects of structure solution and refinement, taking into account the relatively low resolution of the data (Table I). The Matthews coefficient33 suggested three ternary complexes in the asymmetric unit, and the initial search located three molecules of PDZK1D4 and only two molecules of the D-AKAP2AKB: RIIα D/D complex. Since the search model for D-AKAP2AKB comprised of only a 20-residue peptide, the presence of intact ternary complexes was confirmed by Fo – Fc electron density for the remaining D-AKAP2AKB C-terminal residues that interact with PDZK1D4. Initial rounds implementing non-crystallographic symmetry and rigid-body refinements yielded weak density for the third D-AKAP2AKB molecule. The refinement protocol, as implemented in Phenix,34 included using noncrystallographic symmetry, rigid-body, and secondary structure restraints in addition to positional, B-factor and TLS minimization. The refinement strategy was fortified by using the structures of D-AKAP2AKB: RIIα D/D complex12 and PDZK1D4 as reference models. Such an approach that exploits information from high-resolution structures has been successfully used as a powerful option in macromolecular structure refinement of low-resolution structures.35 To circumvent the problem of model bias, simulated-annealed omit maps for various parts of the model, including the regions unique to the ternary complex compared to the search models, were calculated between rounds of model building. Careful model building using the program, Coot, and refinement carried out to 3.8 Å resolution led to an Rfree of 32.8% (Table I). Model quality was monitored using Molprobity.34
Table I.
Crystallization, data collection statistics, and refinement statistics
| A. Crystallization | |
| Crystallization method; temperature (°C) | Microbatch; 22 |
| Crystallization conditions | 100 mM HEPES (pH 7), 20% PEG MME |
| Cryoprotectant | None |
| B. Data collection statisticsa | |
| Source; wavelength (Å) | ALS 5.0.1; 0.9774 |
| Detector | ADSC Q315R (3 × 3 CCD array) |
| Number of images | 130 |
| Space group | P3221 |
| Unit cell parameters (Å) | a = 121.5, c = 88.3 |
| Resolution limits (Å) | 100 – 3.80 (3.94 – 3.80) |
| Wilson B (Å2) | 117 |
| Unique reflections | 7687 (758) |
| Multiplicity | 7.7 (7.9) |
| Completeness (%) | 99.9 (100) |
| Average I/σ | 17.4 (2.9) |
| Rmeasb (%); Rmrgd-Fb (%) | 7.1 (45.4); 8.3 (31.1) |
| C. Refinement | |
| Resolution (Å) | 100 – 3.80 (4.18 – 3.80) |
| Reflections (working set/test set) | 7666 (1724)/ 572 (155) |
| Total number of modeled/expected atoms | |
| PDZK1D4 | 1737/1953 |
| D-AKAP2AKB | 606/1068 |
| RIIα D/D | 1202/2343 |
| Number of solvent atoms | 8 |
| Rcryst/Rfree (%) | 26.8 (27.1)/32.8 (37.3) |
| Average atomic displacement factors | |
| PDZK1D4 (Å2) | 113 |
| D-AKAP2AKB (Å2) | 139 |
| RIIα D/D (Å2) | 197 |
| Solvent atoms (Å2) | 72 |
| Root-mean-square deviations | |
| Bond lengths (Å) | 0.02 |
| Bond angles (°) | 1.84 |
| Ramachandran analysisc | |
| Favored (%) | 93.1 |
| Disallowed (%) | 6.7 |
Numbers in parentheses correspond to values in the highest resolution bin.
Rmeas is the multiplicity-weighted merging R factor and Rmrgd-F is an indicator of the quality of the reduced data (Diederichs and Karplus, 1997).
Ramachandran plot quality as defined in MolProbity.
D-AKAP2AKB: PKA RIIα D/D: PDZK1D4 ternary complex structure
Overall structure
The weak diffraction and the low resolution data collected from the crystal led us to believe that one or more components of the asymmetric unit might be disordered. Of the three proteins in the ternary complex, PDZK1D4 is the best ordered. Reasonable density allowed for the modeling of nearly all residues of the three PDZK1D4 molecules in the asymmetric unit; only two N-terminal, His-tag residues (Gly1, Ser0) and three C-terminal residues (Lys457, Lys458 and Ala459) for all three molecules were not modeled. In the case of D-AKAP2AKB, the C-terminal residues that interact with PDZK1D4 were better ordered than the N-terminal residues that interact with the PKA D/D domain. 17 – 22 (out of 45) residues (including residues that were part of the GST tag) were not modeled in each D-AKAP2AKB molecule due to weak density [ Fig. 2(a)]. These residues lie in the region that precedes the helical AKB motif whereas the AKB helix was well ordered. Finally, in the case of the PKA RIIα D/D domains, a complete dimer (out of the three expected dimers) was not modeled due to lack of clear, continuous density. Residues in PDZK1D4, D-AKAP2AKB and RIIα D/D that were modeled as Ala are listed (see Material and Methods section). The relative disorder among the three components of the complex in the asymmetric unit is reflected in the average atomic displacement parameter (or B-factors) values with the PDZK1D4 molecules exhibiting the lowest numbers and the RIIα D/D domains showing the highest (Table I). In summary, nearly 34% of the total expected atoms were not modeled due to weak electron density (Table I). Since there were no significant differences between the two complete ternary complexes in the asymmetric unit, we chose the one with the higher number of modeled residues for structural analyses and figure preparation.
Figure 2.

Crystal structure of the D-AKAP2AKB: RIIα D/D: PDZK1D4 ternary complex. (a) Domain organization of D-AKAP2 and its interacting proteins. The RGS domains (RGS-A and RGS-B) interact with the small GTPases Rab4 and Rab11, the D-AKAP2AKB interacts with the D/D domains of PKA RI and RII subunits and the C-terminal type I PDZ motif, represented as –TKL, is known to interact with PDZK1 and NHERF1. The putative PKA phosphorylation site is shown as KKAS. The domains of the three proteins (D-AKAP2AKB, RIIα D/D, and PDZK1D4) used in this study are circled. The sequence of D-AKAP2AKB used for crystallization is shown and numbered, and every tenth residue indicated by a dot. Residues not modeled and modeled as an Ala in the structure are indicated in black and blue, respectively. Residues involved in an α-helix (interacting with RIIα D/D) and β-strand (interacting with PDZK1D4) are indicated by a solid bar and an arrow respectively. (b) Overall structure of the D-AKAP2AKB: RIIα D/D: PDZK1D4 ternary complex. The monomers of RIIα D/D are depicted in gold and brown, D-AKAP2AKB in red and PDZK1D4 in dark cyan. The N- and C-termini of each protein are indicated. D-AKAP2AKB has been divided into three clusters: the N-terminal region that interacts with RIIα D/D through an α-helix, the C-terminal region that interacts with PDZK1D4 through a β-strand and finally, the linker region that connects the N- and C-termini clusters. All structural figures were prepared using Pymol (http://www.pymol.org). (c) Surface representation of the ternary complex showing the importance of the D-AKAP2 linker in separating the RIIα D/D and PDZK1D4 molecules. For simplicity, the orientation and the coloring scheme match that of Figure 2b.
The ternary complex mediated by D-AKAP2AKB assumes an extended conformation rather than a compact globular structure [ Fig. 2(b,c)]. Based on the structure, the D-AKAP2AKB residues that are well ordered can be divided into three distinct clusters: the N-terminal region that forms an α-helix and anchors PKA, the C-terminal residues containing the PDZ motif that form a β-strand to interact with PDZK1 and finally, a linker region that spatially separates these two regions [ Figs. 2(b,c) and 3(a)]. In the following sections we describe these clusters and compare the present ternary complex structure to the previously solved structures of D-AKAP2AKB: RIIα D/D complex12 and a PDZ domain: PDZ motif (PDZK1D3: SR-BI) complex.36 Since the structures of D-AKAP2AKB: RIIα D/D and the PDZK1D4 domain were used to generate reference model restraints for ternary complex refinement, for the comparisons, only differences that are clearly reflected in the electron density are discussed.
Figure 3.

Intermolecular interactions and quality of ternary complex structure. (a) Schematic figure detailing the interactions between the main chain and side chains of the D-AKAP2AKB, the RIIα D/D monomer and the PDZK1D4 domain. Solid and dashed arrows indicate the hydrophobic and polar interactions respectively. (b) Quality of the electron density maps to 3.8 Å resolution. Stereoview illustrating the quality of the 2Fo – Fc final electron density map contoured at 1σ for the ternary complex. The figure is centered on the D-AKAP2AKB: RIIα D/D interface, which shows relatively weaker density compared with the PDZK1D4 molecules. The final, refined model is represented as ribbons and labeled.
D-AKAP2AKB interaction with RIIα D/D
Two antiparallel regions from the N-terminus of RIIα dimerize to form the D/D domain, which is an X-type, antiparallel, four helix bundle with the α1 helices from each monomer forming a hydrophobic interface for AKAP binding [ Figs. 3(b) and 4(a)]. The N-terminus of D-AKAP2, specifically residues Ala635 through Tyr652, forms a well-defined, amphipathic helix that binds diagonally across the (∼55°) D/D interface making extensive hydrophobic and polar interactions. In the absence of the D/D domain, the D-AKAP2AKB is disordered.21 The R subunit residues involved in the dimerization and docking have been discussed elsewhere in detail.12 For clarity, the monomer whose N-terminus interacts with the N-terminus of D-AKAP2 is labeled as “A” and the other monomer is labeled “B” [ Fig. 4(a)]. In addition to this primary interaction, the helix of D-AKAP2AKB (all three molecules in the asymmetric unit) is stabilized by packing interactions with other D-AKAP2AKB molecules in the asymmetric unit or the unit cell (data not shown). This added stability possibly accounts for a lower atomic displacement factor compared to the D/D domains (Table I).
Figure 4.
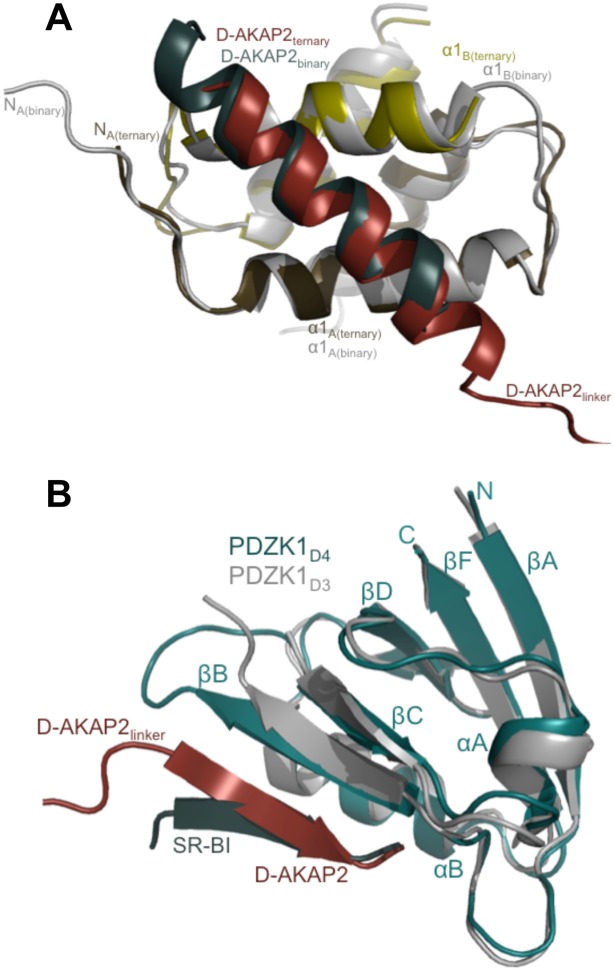
Comparison of the ternary complex with D-AKAP2AKB: RIIα D/D and PDZ domain: PDZ motif complex structures. (a) Structural overlay of D-AKAP2AKB: RIIα D/D from the ternary complex with the structure of the D-AKAP2AKB: RIIα D/D complex crystal structure (PDB ID: 2HWN).12 The ternary complex is colored as Figure 2b. For the D-AKAP2AKB: RIIα D/D structure, the D/D domain is colored gray and the D-AKAP2AKB is colored teal. The helices and termini are labeled with the respective colors. For clarity, the PDZK1D4 domain is not shown. (b) Structural overlay of the PDZK1D4 from the ternary complex with the PDZK1D3 domain from the PDZK1D3: SR-BI complex (PDB ID: 3R69).36 PDZK1D4 from the ternary complex is colored as Figure 2b. The PDZK1D3 is colored gray and the SR-BI PDZ motif is colored teal. All the proteins are labeled with their respective colors. For clarity, the D-AKAP2AKB and the RIIα D/D domains are not shown. The domains are well aligned including the PDZ motifs. Slight variations are seen in the residues leading to the PDZ motif.
As expected, the core of the dimerization and docking interface aligns very well with that of the D-AKAP2AKB: RIIα D/D structure including the angle of the D-AKAP2 binding [ Fig. 4(a)].12 Similar to the previous structure, the N-terminus of chain A is better ordered than chain B though certain differences exist. In the ternary complex, clear main-chain density is observed for residues starting with Ile3 in chain A. Ile3 and Ile5 make critical hydrophobic interactions with D-AKAP2AKB residues—Ile638 and Ala639—and play an important role in defining AKAP specificity and selectivity. In the binary complex, there is clear, additional density for the first two residues of the chain. While these residues do not make critical interactions, their absence in the ternary complex likely causes the first turn of the D-AKAP2AKB helix to be disordered [ Fig. 4(a)]. However, PKA residues that line pockets I and II, and D-AKAP2 residues that occupy them are all well ordered in the structure.13
The N-terminus of chain B is relatively disordered with Gly8 being the first modeled residue compared to Ile5 in the binary complex. Unlike chain A where Ile3 and Ile5 make important interactions with D-AKAP2AKB, in chain B these residues do not make any critical interactions and their absence, therefore, does not impact the binding of D-AKAP2. These findings reinforce the asymmetry of the RIIα D/D: D-AKAP2AKB complex.
D-AKAP2AKB interaction with PDZK1D4
PDZK1D4 is comprised of 85 amino acid residues that assume a canonical, well-conserved PDZ fold.37 As expected, there are 6 β-strands (labeled βA through βF), a short α-helix (labeled αA) and a long α-helix (labeled αB) [ Figs. 2(b) and 4(b)]. The PDZ-motif binding site is a cleft formed between αB and βB with the N- and C-termini of the protein on the opposite side of the peptide-binding site.
For its interaction with PDZK1D4, the six C-terminal residues of D-AKAP2 (Glu657, Lys658, Ser659, Thr660, Lys661, and Leu662) adopt a β-strand conformation and form antiparallel hydrogen bonds with βB. The type I PDZ motif is further stabilized in the binding cleft by residues from αB [ Figs. 2(b) and 4(b)]. The residues lining the cleft, especially those belonging to the carboxylate-binding loop (between βA and βB) are well-conserved 37; the sequence of this loop on PDZK1D4 (Lys383-Gly-Glu-Asn-Gly-Tyr-Gly-Phe390) matches the expected motif (Arg/Lys-X-X-X-Gly-ϕ-Gly-ϕ). The exact details of a prototypical PDZ domain: PDZ motif interactions have been described in detail in previous studies36–39 and are therefore, not discussed here further. A structural overlay of the present structure with that of the PDZK1D3: SR-BI structure (PDB ID: 3R69)36 shows that the D-AKAP2AKB: PDZK1D4 interaction follows a typical PDZ domain: PDZ motif interaction [ Fig. 4(b)].
D-AKAP2AKB linker region
A four-residue linker region (Asp653-Gln654-Pro655-Leu656) of the D-AKAP2AKB connects the N-terminal α-helix and the C-terminal β-strand (Figs. 2 and 3). In the crystal, PDZK1D4 molecules in the asymmetric unit and unit cell stabilize these residues. In the absence of these and the N-terminal α-helical RIIα D/D interactions, it is likely that the linker residues and the C-terminal PDZ-motif would be much more flexible. To form a stable ternary complex with PDZK1D4, it was necessary to present D-AKAP2AKB that had been stabilized in a complex with RIIα D/D. Based on our previous structure of the D-AKAP2AKB bound to the D/D domain of RIIα, the entire C-terminal region is disordered in the absence of a PDZ domain.12 Given the high affinity binding between the D-AKAP2AKB and the RIIα D/D domain, it is highly likely that this is the physiological complex that is presented to the transporters.
To test whether flexibility of the linker was required for binding to the PDZK1D4 domain we engineered single, double, and triple deletion mutants in the linker. We found that the length of the D-AKAP2 linker region possesses an unexpected degree of mutability without preventing ternary complex formation. D-AKAP2AKB Δ653, Δ653–654, and Δ653–655 all eluted as stable ternary complexes on a gel filtration column. While the D-AKAP2AKB deletion mutants may exhibit different affinities to either PKA RII or PDZK1D4 than wild-type, the mutations do not prevent stable association of the PDZK1D4 with the D-AKAP2AKB: RIIα D/D complex. Deletion mutants could facilitate the formation of a more compact complex. Crystallization attempts with the D-AKAP2AKB deletion mutants are currently underway, and these structures could clarify the effect that a shortened linker has on complex formation. Our initial hypothesis that four linker residues may be important to effectively prevent steric clashes between PKA and PDZK1 (Figs. 2 and 3) needs to be reconsidered; it is clearly not essential for the simultaneous binding of RIIα and PDZK1. The N-terminal segment of PDZK1D4 could potentially adopt an extended conformation to compensate for the reduced flexibility of D-AKAP2AKB deletion mutants. The significance of this phenomenon in the context of the RIα isoform and the shift in the D-AKAP2 binding helix register is discussed in detail below.
Basis for D-AKAP2AKB helical register shift
Comparison with the D-AKAP2AKB: PKA RIα D/D complex crystal structure
In the case of the RIα D/D, in addition to the two core helices, there is an additional N-terminal α-helix (α0) that interacts with D-AKAP2.13 Positional stabilization of this helix by a disulfide bond (between Cys16 and Cys37) results in the formation of a symmetrical and more significantly, a larger binding surface for AKAP interactions.13,27 As a result of this stabilization, the D-AKAP2AKB helix is longer both at the N- and C-termini [ Fig. 5(a)]. While residues Gln651 and Tyr652 are the last ordered residues and show helical propensity, these residues do not interact with RIα D/D [ Fig. 5(b)]. Instead, the last residue stabilized is Ala650 that forms hydrophobic interactions with Leu13 and Cys37 in the D/D domain.13 On the other hand, in RIIα D/D the binding of an AKAP results in the ordering of the N-terminus of only one of the two monomers. This results in an asymmetrical binding surface for the asymmetrical sequence of D-AKAP2.11,12 Thus, compared to the RIIα D/D structure, the D-AKAP2AKB has one additional α-helical turn and two additional α-helical turns at the N- and C-terminus respectively when bound to RIα D/D [ Fig. 5(a)].
Figure 5.
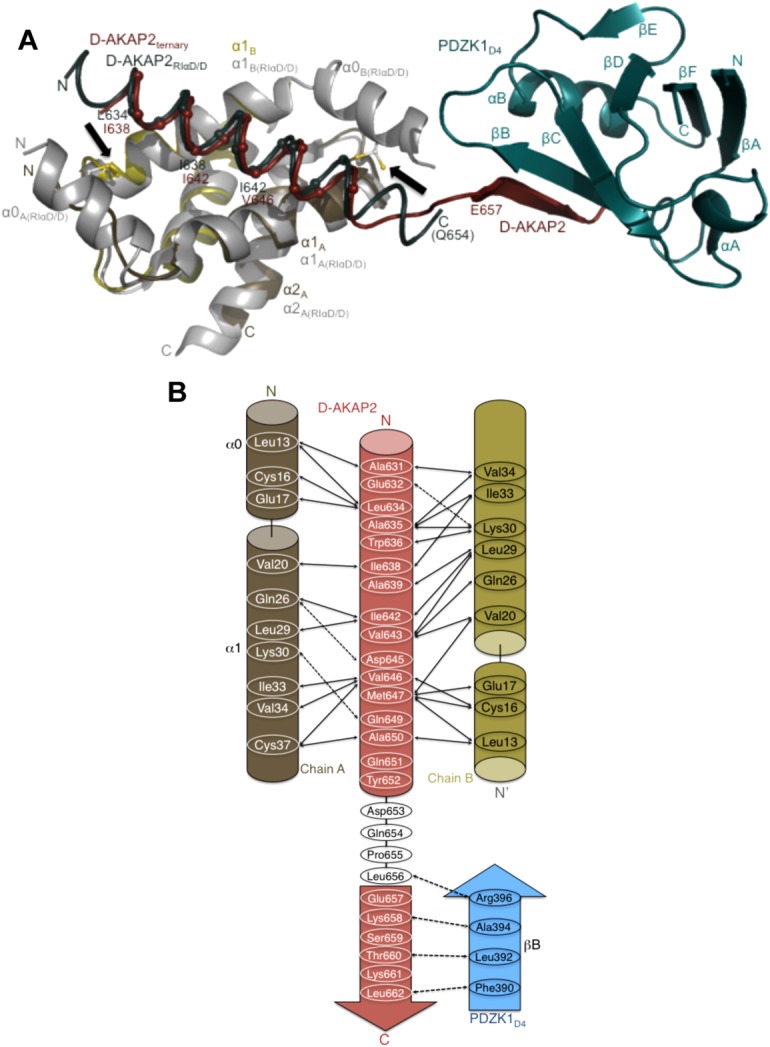
Structural overlay of the ternary complex with the D-AKAP2AKB: RIα D/D and model revealing clues to the reasons for the AKAP helix shift. (a) Structural overlay of the D-AKAP2AKB: RIIα D/D from the ternary complex with the structure of the D-AKAP2AKB: RIα D/D complex crystal structure (PDB ID: 3IM4).13 The ternary complex is colored as Figure 2b. For the D-AKAP2AKB: RIα D/D structure, the D/D domains are colored grey and the D-AKAP2AKB is colored teal. Black arrows indicate the location of the disulfide bonds in the N-terminal helices (α0) of RIα D/D. The helices and termini are labeled with their respective colors. Cα atoms of the D-AKAP2AKB helices are shown and labeled to denote the shift in its helical register when bound to the PKA isoforms. For the ternary complex, the first residue of the β-strand interacting with PDZK1D4, Glu657 is shown. Similarly, the last modeled D-AKAP2 residue in the D-AKAP2AKB: RIα D/D complex, Gln654 is shown and labeled. (b) Schematic figure detailing the potential binding mechanism of PDZK1D4 and RIα D/D to the D-AKAP2AKB. Only with the helical shift would D-AKAP2 be able to bind to PKA and PDZK1 simultaneously. Solid and dashed arrows indicate the hydrophobic and polar interactions respectively in the D-AKAP2AKB: RIα D/D crystal structure and the potential interactions between D-AKAP2AKB and PDZK1D4.13.
The most significant difference is the shift in the helical register of D-AKAP2AKB upon binding to PKA isoforms; the helix is shifted down by an entire turn when bound to RIα D/D [ Fig. 4(a)].13 Thus, for example, with the D/D domains aligned, the space occupied by D-AKAP2AKB residues—Ile642 and Val643—in RIIα D/D is occupied by residues Ile638 and Ala639 in D-AKAP2AKB: RIα D/D [ Fig. 5(a)].
A structural comparison between the D-AKAP2AKB: RIα D/D and the D-AKAP2AKB: RIIα D/D structures (from the ternary complex) reveals a surprising fact. While expected, the downward helical shift does not result in the availability of additional C-terminal residues in RIα D/D for PDZ interactions; the D-AKAP2AKB α-helix extends to Tyr652 in both the structures with Asp653 forming the first linker residue. In RIα D/D, the shift is accompanied by the presence of the N-terminal helix (α0) in the D/D domains that results in the stabilization of the D-AKAP2AKB helix thereby preventing additional residues from being part of the linker region.
Basis for differential binding of D-AKAP2 to PKA isoforms
We have modeled the D-AKAP2AKB: RIα D/D structure13 onto the present ternary complex structure to answer the question of why D-AKAP2 binds differentially to the RIα and RIIα PKA isoforms. By comparing the effects of two different scenarios—the nonoccurrence and occurrence of the D-AKAP2AKB helical shift—we arrive at a potential reason for the shift.
In the absence of a helical shift in RIα, the terminal D-AKAP2AKB residue forming the α-helix would be Leu656 instead of Tyr 652 [ Fig. 5(b)]. Accordingly, the last residue making interactions with the D/D domain would be Gln654 instead of Als650. At the same time, interaction with PDZK1D4 involves six C-terminal residues (Asp 657 through Leu662). Thus, in the absence of the helical shift, there would only be two residues, Pro655 and Leu656, in the linker region.
In the presence of the helical shift, the last residue part of the α-helix would be Tyr652 with Asp653 forming the first residue of the loop. Such an organization would be similar to the one seen in the present ternary complex. Thus, in the presence of the helical shift the six C-terminal D-AKAP2AKB residues would be available for interaction with PDZ proteins.
The differential binding of D-AKAP2 to the PKA RIα and RIIα isoforms can be traced to the occurrence of a single phenomenon: the presence of an intermolecular disulfide bond between Cys16 and Cys37 of the two D/D domains of RIα. The N-terminal α0 helices are spatially constrained as a result and form an extended, symmetrical binding surface for D-AKAP2. The consequence of this phenomenon on D-AKAP2 is the extra α-helical turns at both the N- and C-termini. In the RIIα D/D domains, the N-termini are not constrained by disulfide bonds and, as a result, an asymmetrical binding surface is presented for binding to D-AKAP2.
As a final note, there are intriguing possibilities and unanswered questions raised by the present structure. Under reducing conditions, the N-terminal helices in RIα D/D would no longer be spatially constrained due to the lack of the disulfide bonds. Under such conditions, the D-AKAP2AKB binding to RIα would resemble the binding to RIIα with fewer turns of the α-helix at the termini. Thus, in the absence of the primary driving reason for the helical shift, D-AKAP2 could possibly bind to RIα D/D in a fashion similar to RIIα D/D. Further, it was proposed by Sarma et al.13 that this helical register shift is likely true for all RI-, RII- and dual-specific AKAPs. While our analyses reveals the reason for the shift in D-AKAP2, it does not answer the question of why this shift would occur in all AKAPs. Further structural, biochemical, and biophysical studies of AKAPs binding to PKA and its interacting proteins would not only test the AKAP helical shift model but will also provide reasons for the shift.
Materials and Methods
Protein expression and purification
Rat RIIα D/D (residues 1 – 44) was cloned as a His-tagged protein in pET15b vector (Novagen) with an internal thrombin site. The protein was expressed as described earlier.12 Prior to purification, the cells were suspended in 20 mM Tris (pH 8.0), 100 mM NaCl buffer containing protease inhibitors and 2 mM DTT. Initial purification of His-RIIα D/D was carried out using the Profinia protein purification system (Bio-Rad). A preprogrammed method was used to bind the protein to a Ni-affinity column, followed by imidazole elution and a desalting step. Thrombin was used to cleave the His-tag and the protein was separated from the His-tag and thrombin by S75 gel filtration. The protein was concentrated to ∼15 mg/mL (ε280 nm = 2680 M−1 cm−1).
The AKB region of human D-AKAP2 (residues 623 – 662) was cloned, expressed and purified in a manner described earlier.13 Following S75 gel filtration purification, the protein was concentrated to ∼4 mg/mL (ε280 nm = 6970 M−1 cm−1). The fourth domain of human (residues 375 – 449) PDZK1 was cloned into pQTEV vector with an internal TEV protease cleavage site. The His-tagged protein was purified on the Profinia purification system in a fashion similar to RIIα D/D. The His-tag on the PDZ protein was cleaved by incubating the protein with His-tagged TEV protease overnight at 4°C. The cleaved His-tag and the protease were purified away from the protein using a modified protocol on the Profinia system. As a final step, the PDZK1D4 protein was purified on a S75 gel filtration column and concentrated to ∼6 mg/mL. Ternary complex formation is discussed in the main text.
D-AKAP2 truncation mutants were created using site-directed mutagenesis. Three constructs, D-AKAP2 Δ653, Δ653–654, and Δ653–655, were created. Each truncation mutant was transformed, expressed, and purified by the same method as wild-type D-AKAP2AKB. Following purification, ternary complex formation was performed with S75 size-exclusion chromatography.
Peptide array screening
The peptide arrays were preincubated with T-TBS blocking buffer (TBS, pH 8.0/0.05% Tween 20 in the presence of blocking reagent). Various PDZ domains are spotted on the membrane.40 Subsequently, the peptide arrays were incubated with solutions of GST-AKB-CT and GST-RGS-AKB-CT at a final concentration of 1.0 μg/mL for 2 h in T-TBS blocking buffer. After washing three times for 10 min with T-TBS the anti-GST antibody was added to a final concentration of 1 μg/mL in T-TBS blocking buffer for 1 h followed by washing three times for 10 min with T-TBS. Analysis and quantification were done by using a chemiluminescence substrate (Roche Diagnostics chemiluminescence detection kit 1500694) and the LumiImager (Roche Diagnostics). All steps were carried out at room temperature. Binding of the detection antibodies to the peptides was excluded by control incubations with anti-GST antibody alone (data not shown).
Crystallization, structure determination, and refinement
Crystals of the RIIα D/D: D-AKAP2AKB: PDZK1D4 ternary complex appeared in a week at room temperature using microbatch crystallization. The crystals grew from a 1:1 drop containing 20% PEG MME and 0.1M HEPES (pH 7.0) and directly flash-frozen in liquid nitrogen. For structure determination and refinement, a single data set was collected at the advanced light source (ALS) synchrotron (beamline 5.0.1). Data were processed using HKL200041 and model refinement carried out using Phenix,34 with 7.5% of the reflections set aside for cross validation. The low-resolution data demanded careful attention to the refinement details and are discussed in the main text.
The structure was refined to a final R and Rfree of 26.8 and 32.8%, respectively. The following residues were modeled as Ala due to lack of density: For PDZK1D4: Lys375, Arg380, Glu385, Arg396, Lys448 (chains A, E, and I), Lys408 (chain A), Asn386 (chain E), and Lys377, Leu381, Lys383, Lys404, Asn430, Lys438, Arg442 (chain I); for D-AKAP2AKB: Lys637 (chains D and L), and Tyr652 (chains D, H, and L); for RIIα D/D: Arg43 (chain F).
Acknowledgments
Valuable discussions with the members of the Taylor laboratory are appreciated. The authors would like to thank Dr. Jian Wu for help with manuscript preparation. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information Figure 1.
References
- 1.Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 2.Scott JD, Pawson T. Cell signaling in space and time: where proteins come together and when they're apart. Science. 2009;326:1220–1224. doi: 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skålhegg BS, Taskén K. Specificity in the cAMP/PKA signaling pathway. differential expression, regulation, and subcellular localization of subunits of PKA. Front Biosci. 1997;2:d331–d342. doi: 10.2741/a195. [DOI] [PubMed] [Google Scholar]
- 4.Amieux PS, Cummings DE, Motamed K, Brandon EP, Wailes LA, Le K, Idzerda RL, McKnight GS. Compensatory regulation of RIalpha protein levels in protein kinase A mutant mice. J Biol Chem. 1997;272:3993–3998. doi: 10.1074/jbc.272.7.3993. [DOI] [PubMed] [Google Scholar]
- 5.Casey M, Vaughan CJ, He J, Hatcher CJ, Winter JM, Weremowicz S, Montgomery K, Kucherlapati R, Morton CC, Basson CT. Mutations in the protein kinase A R1alpha regulatory subunit cause familial cardiac myxomas and Carney complex. J Clin Invest. 2000;106:R31–R38. doi: 10.1172/JCI10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 7.Taylor SS, Kim C, Vigil D, Haste NM, Yang J, Wu J, Anand GS. Dynamics of signaling by PKA. Biochim Biophys Acta. 2005;1754:25–37. doi: 10.1016/j.bbapap.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 8.Harris NL, Presnell SR, Cohen FE. Four helix bundle diversity in globular proteins. J Mol Biol. 1994;236:1356–1368. doi: 10.1016/0022-2836(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 9.Banky P, Huang LJ, Taylor SS. Dimerization/docking domain of the type Ialpha regulatory subunit of cAMP-dependent protein kinase. Requirements for dimerization and docking are distinct but overlapping. J Biol Chem. 1998;273:35048–35055. doi: 10.1074/jbc.273.52.35048. [DOI] [PubMed] [Google Scholar]
- 10.Newlon MG, Roy M, Morikis D, Carr DW, Westphal R, Scott JD, Jennings PA. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J. 2001;20:1651–1662. doi: 10.1093/emboj/20.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gold MG, Lygren B, Dokurno P, Hoshi N, McConnachie G, Tasken K, Carlson CR, Scott JD, Barford D. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol Cell. 2006;24:383–395. doi: 10.1016/j.molcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, Xuong NH, Jennings PA, Taylor SS. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol Cell. 2006;24:397–408. doi: 10.1016/j.molcel.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarma GN, Kinderman FS, Kim C, von Daake S, Chen L, Wang BC, Taylor SS. Structure of D-AKAP2:PKA RI complex: insights into AKAP specificity and selectivity. Structure. 2010;18:155–166. doi: 10.1016/j.str.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. D-AKAP2, a novel protein kinase A anchoring protein with a putative RGS domain. Proc Natl Acad Sci USA. 1997;94:11184–11189. doi: 10.1073/pnas.94.21.11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J Biol Chem. 1997;272:8057–8064. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- 16.Reinton N, Orstavik S, Haugen TB, Jahnsen T, Taskén K, Skålhegg BS. A novel isoform of human cyclic 3′,5′-adenosine monophosphate-dependent protein kinase, c alpha-s, localizes to sperm midpiece. Biol Reprod. 2000;63:607–611. doi: 10.1095/biolreprod63.2.607. [DOI] [PubMed] [Google Scholar]
- 17.Eggers CT, Schafer JC, Goldenring JR, Taylor SS. D-AKAP2 interacts with Rab4 and Rab11 through its RGS domains and regulates transferrin receptor recycling. J Biol Chem. 2009;284:32869–32880. doi: 10.1074/jbc.M109.022582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stenmark H, Olkkonen VM. The Rab GTPase family. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-5-reviews3007. REVIEWS3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 20.Seachrist JL, Ferguson SSG. Regulation of G protein-coupled receptor endocytosis and trafficking by Rab GTPases. Life Sci. 2003;74:225–235. doi: 10.1016/j.lfs.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 21.Burns LL, Canaves JM, Pennypacker JK, Blumenthal DK, Taylor SS. Isoform specific differences in binding of a dual-specificity A-kinase anchoring protein to type I and type II regulatory subunits of PKA. Biochemistry. 2003;42:5754–5763. doi: 10.1021/bi0265729. [DOI] [PubMed] [Google Scholar]
- 22.Gisler SM, Madjdpour C, Bacic D, Pribanic S, Taylor SS, Biber J, Murer H. PDZK1: II. an anchoring site for the PKA-binding protein D-AKAP2 in renal proximal tubular cells. Kidney Int. 2003;64:1746–1754. doi: 10.1046/j.1523-1755.2003.00267.x. [DOI] [PubMed] [Google Scholar]
- 23.Kocher O, Pal R, Roberts M, Cirovic C, Gilchrist A. Targeted disruption of the PDZK1 gene by homologous recombination. Mol Cell Biol. 2003;23:1175–1180. doi: 10.1128/MCB.23.4.1175-1180.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silver DL, Wang N, Vogel S. Identification of small PDZK1-associated protein, DD96/MAP17, as a regulator of PDZK1 and plasma high density lipoprotein levels. J Biol Chem. 2003;278:28528–28532. doi: 10.1074/jbc.M304109200. [DOI] [PubMed] [Google Scholar]
- 25.Yesilaltay A, Kocher O, Rigotti A, Krieger M. Regulation of SR-BI-mediated high-density lipoprotein metabolism by the tissue-specific adaptor protein PDZK1. Curr Opin Lipidol. 2005;16:147–152. doi: 10.1097/01.mol.0000162319.54795.e5. [DOI] [PubMed] [Google Scholar]
- 26.Kocher O, Birrane G, Tsukamoto K, Fenske S, Yesilaltay A, Pal R, Daniels K, Ladias Jaa, Krieger M. In vitro and in vivo analysis of the binding of the C terminus of the HDL receptor scavenger receptor class B, type I (SR-BI), to the PDZ1 domain of its adaptor protein PDZK1. J Biol Chem. 2010;285:34999–5010. doi: 10.1074/jbc.M110.164418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banky P, Roy M, Newlon MG, Morikis D, Haste NM, Taylor SS, Jennings PA. Related protein - Protein interaction modules present drastically different surface topographies despite A conserved helical platform. J Mol Biol. 2003;330:1117–1129. doi: 10.1016/s0022-2836(03)00552-7. [DOI] [PubMed] [Google Scholar]
- 28.Burns-Hamuro LL, Hamuro Y, Kim JS, Sigala P, Fayos R, Stranz DD, Jennings PA, Taylor SS, Woods VL. Distinct interaction modes of an AKAP bound to two regulatory subunit isoforms of protein kinase A revealed by amide hydrogen/deuterium exchange. Protein Sci. 2005;14:2982–2992. doi: 10.1110/ps.051687305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alto NM, Soderling SH, Hoshi N, Langeberg LK, Fayos R, Jennings PA, Scott JD. Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring. Proc Natl Acad Sci USA. 2003;100:4445–4450. doi: 10.1073/pnas.0330734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burns-Hamuro LL, Ma Y, Kammerer S, Reineke U, Self C, Cook C, Olson GL, Cantor CR, Braun A, Taylor SS. Designing isoform-specific peptide disruptors of protein kinase A localization. Proc Natl Acad Sci USA. 2003;100:4072–4077. doi: 10.1073/pnas.2628038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlson CR, Lygren B, Berge T, Hoshi N, Wong W, Taskén K, Scott JD. Delineation of type I protein kinase A-selective signaling events using an RI anchoring disruptor. J Biol Chem. 2006;281:21535–21545. doi: 10.1074/jbc.M603223200. [DOI] [PubMed] [Google Scholar]
- 32.Burgers PP, Ma Y, Margarucci L, Mackey M, van der Heyden MAG, Ellisman M, Scholten A, Taylor SS, Heck AJR. A small novel A-kinase anchoring protein (AKAP) that localizes specifically protein kinase A-regulatory subunit I (PKA-RI) to the plasma membrane. J Biol Chem. 2012;287:43789–43797. doi: 10.1074/jbc.M112.395970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 34.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schröder GF, Levitt M, Brunger AT. Super-resolution biomolecular crystallography with low-resolution data. Nature. 2010;464:1218–1222. doi: 10.1038/nature08892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kocher O, Birrane G, Yesilaltay A, Shechter S, Pal R, Daniels K, Krieger M. Identification of the PDZ3 domain of the adaptor protein PDZK1 as a second, physiologically functional binding site for the C terminus of the high density lipoprotein receptor scavenger receptor class B type I. J Biol Chem. 2011;286:25171–25186. doi: 10.1074/jbc.M111.242362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee H-J, Zheng JJ. PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun Signal. 2010;8:8. doi: 10.1186/1478-811X-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeleń F, Oleksy A, Smietana K, Otlewski J. PDZ domains - common players in the cell signaling. Acta Biochim Pol. 2003;50:985–1017. [PubMed] [Google Scholar]
- 39.Van Ham M, Hendriks W. PDZ domains-glue and guide. Mol Biol Rep. 2003;30:69–82. doi: 10.1023/a:1023941703493. [DOI] [PubMed] [Google Scholar]
- 40.He J, Bellini M, Inuzuka H, Xu J, Xiong Y, Wang X, Castleberry AM, Hall RA. Proteomic analysis of beta-1-adrenergic receptor interactions with PDZ scaffold proteins. J Biol Chem. 2006;281:2820–2827. doi: 10.1074/jbc.M509503200. [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information Figure 1.