

Abstract
The oral pathogen Porphyromonas gingivalis is a keystone pathogen in the development of chronic periodontitis. Gingipains, the principle virulence factors of P. gingivalis are multidomain, cell-surface proteins containing a cysteine protease domain. The lysine specific gingipain, Kgp, is a critical virulence factor of P. gingivalis. We have determined the X-ray crystal structure of the lysine-specific protease domain of Kgp to 1.6 Å resolution. The structure provides insights into the mechanism of substrate specificity and catalysis.
Keywords: Porphyromonas gingivalis, keystone pathogen, periodontitis, lysine specific protease, Kgp, gingipains
Introduction
The oral pathogen Porphyromonas gingivalis is a keystone pathogen in the development of chronic periodontitis. The extracellular proteinases (RgpA/B and Kgp, the gingipains) of P. gingivalis have been implicated as major virulence factors that are critical for colonization, penetration into host tissue, and the establishment of dysbiosis and disease.1–5 Kgp has also been implicated as the most important gingipain for pathogenesis as gene inactivation produced an isogenic mutant that was avirulent in inducing alveolar bone resorption in the murine periodontitis model.1 All three gene products contain a signal peptide of ∼22 amino acids in length, an unusually long propeptide of ∼200 amino acids in length, and a catalytic domain of ∼480 amino acids. In addition, rgpA and kgp encode hemagglutinin-adhesin (HA) domains in the polypeptide region C-terminal to the catalytic domains.6–8 The catalytic domains of RgpB and RgpA share a high-degree of sequence homology. However, RgpB lacks the HA domains and is located in a monomeric form on the outer membrane. Two crystal structures of RgpB are available, one with the peptide mimetic FFRCMK,9 and one in complex with the inhibitory propeptide domain.10
To provide a structural basis for understanding the differences in specificity/catalysis between RgpB and Kgp, we have determined the three-dimensional crystal structure of Kgp from the W50 strain of P. gingivalis.
Results and Discussion
Structure description
The structure of the Kgp catalytic domain is similar to that observed in the related Rgp protease with a central ten stranded β-sheet surrounded by ten α-helices forming an α-β sandwich. An immunoglobulin superfamily-like domain comprising of six antiparallel β-strands, is situated on the opposite side from the substrate binding face (Fig. 1).
Figure 1.
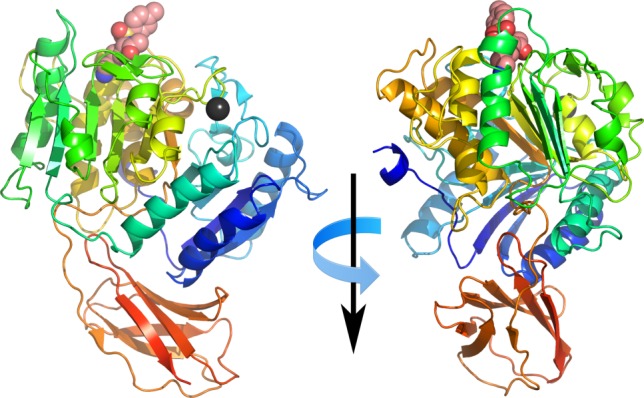
Cartoon representation showing orthogonal views of Kgp. Protein colored from the N-terminus (blue) to C-terminus (red). Covalently bound TCLK inhibitor represented as spheres, carbon atoms colored salmon, oxygen colored red, nitrogen colored blue and sulfur colored yellow. Bound lead atom shown as black sphere.
The lead ion used to derive phase information in the structure determination displays distorted octahedral geometry and is bound to the bifurcated residues Asp313 and Glu491, the main-chain carbonyl of Phe482, and three water molecules. This site is occupied by a calcium ion in the Rgp structure (Asp307 and Glu487) [For ease of comparison, all residue numbering in this discussion will be relative to the gene products, in the case of RgpB that is the numbering used in Ref. (10). The numbering used in the original RgpB structure paper9 can be obtained by subtracting 229 from the RgpB residue indices used in this discussion. Residues referring to RgpB will also be in italics.]
The peptide surrogate moiety of the TLCK (N-α-tosyl-lysyl chloromethylketone hydrochloride) inhibitor covalently bound to the Sγ of Cys477 and is clearly defined in the electron density map within the S1 binding pocket. The Nζ of the inhibitor lysine makes extensive H-bond interactions with the following atoms acting as H-bond acceptors: the Oγ of Thr442, the backbone carbonyl oxygen of Asn475, and one of the Oδ of Asp516. The conserved acidic residue Asp388, may play a role in the catalytic mechanism since the side-chain carboxyl oxygen atom is located 2.7 Å from the Nε of His444, the catalytic histidine, forming a catalytic triad with Cys477. In addition, there is a chain of three well-defined water molecules that acts as H-bond acceptors in their interactions with the Nζ of the lysine. The other end of this H-bonded chain of water molecules is in turn H-bonded to the backbone carbonyl oxygen of Asp516 (Fig. 2).
Figure 2.
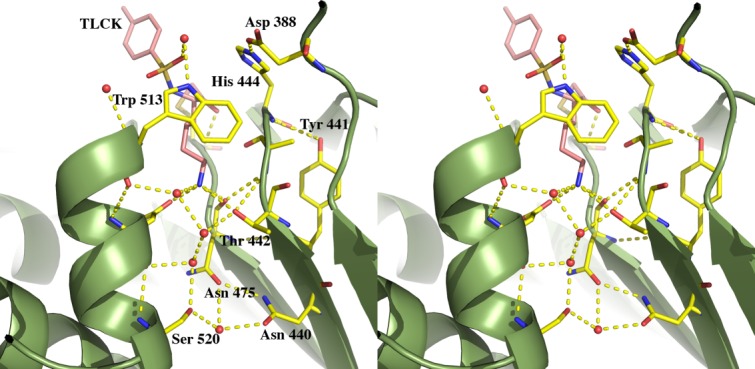
Stereo cartoon showing the Kgp active site residues (yellow) and the bound inhibitor TLCK (pink) as sticks. Water molecules are represented as red spheres. Hydrogen bonds are shown as yellow dashes.
As expected, the overall structure of Kgp is similar to that of RgpB (Supporting Information Fig. S1). Using the program LSQMAN11 to superpose our Kgp structure onto RgpB gave an r.m.s.d. of 1.47 Å relative to 1CRV9 and 4IEF,10 based on 388 and 386 Cα positions, respectively. LSQMAN was also used to generate a structure-based sequence alignment (Supporting Information Fig. S2), illustrating the 22.9% sequence identity and 60% sequence similarity between the two structures. The overlay of the two structures reveals some notable features. The α-helices and loops connecting the β-strand core of the catalytic domain show the greatest structural diversity. Particularly noteworthy is the large, surface-exposed loop extending from Val521 to Ser540 of Kgp that breaks the analogous, continuous α-helix connecting the fifth and sixth β-strands of the core observed in RgpB Ala514-Cys528 (Loop1 in Supporting Information Fig. S1). In RgpB there is a large, surface-exposed loop extending from the fifth β-strand (residues 378–392) while in the Kgp, this loop is relatively short and almost forms a continuous helix from residues 391–406 (Loop2 in Supporting Information Fig. S1). Comparison with the structure of RgpB bound to its cognate propeptide (PDB id: 4IEF) shows that these loops in Kgp may interact with its propeptide particularly at Arg31 and Thr72-Gly74, and may require some changes in the conformation of the propeptide. The propeptides being markedly variable in this region could account for the observed specificity of the recombinant propeptides toward their cognate proteases. The Arg31 equivalent in Kgp (Arg32) is three residues N-terminal to the single cysteine (Cys35) unique to Kgp. Because of this cysteine, the recombinant Kgp propeptide forms homodimers that are unable to inhibit their cognate protease.6
Substrate specificity
The molecular basis for the substrate specificities of Rgp and Kgp can now be determined with the availability of crystal structures of both proteases. The active site pocket of RgpB when bound to the inhibitor FFRCMK (PDB id: 1CRV) is very similar to that seen in the natively inhibited, prodomain bound apo enzyme (PDB id: 4IEF). Briefly, the guanidinium group of Arg126 forms a salt bridge with Asp392. The stacking of Trp513 (above) and Thr438 and Val471 (below) further stabilizes this interaction. The sides of the pocket are lined with Met517 and Gly439. The inhibitor also hydrogen bonds to the main-chain carbonyl of Trp513.
In contrast, with Kgp the bidentate interaction to Asp392 is no longer possible since the substrate is a lysine moiety and Asp392 is substituted for a glycine at position 396. Similarly, Val471 which lines the floor of the pocket is substituted for Asn475 and swings away from the substrate site, facilitated by an extension of the binding cleft with the substitution of Gln520 to Ser520. The lids of both pockets have tryptophan residues; however, the rotamers are different, again due to the extra room afforded by the substitution of Asp392 to Gly396. In the Kgp binding cavity, three water molecules line the side of the cavity where Met517 (located on loop 1) is seen in the RgpB structure (Fig. 2).
In summary, the structure of Kgp and the previously determined structures of RgpB (PDB codes: 1CVR and 4IEF) provide an insight into the structural modifications that accompany the change in substrate specificity of the two enzymes.
Materials and Methods
Bacterial strains and growth conditions
P. gingivalis recombinant strain ECR368 produces rKgp with a Gly681–Ala710 deletion and releases the Kgp catalytic domain in soluble form into the culture supernatant. The recombinant strain was grown under the conditions previously described.6
Purification of Kgp
The Kgp was purified as described previously with the modification of the addition of 1 mM TLCK (Sigma, St. Louis, USA) during the steps following acetone precipitation.6
Crystallization and X-ray diffraction experiments
Initial crystallization trials were carried out in-house using JSCG++ (BioScientific, Gymea, Australia) and Hampton Research HTS screens (Hampton Research, Aliso Viejo, USA) in Rigaku UV+ 96 well sitting drop vapor diffusion plates (AXT, Sydney, Australia). Typically, screens were made by adding 30 μL of crystallant to the well, and then adding 200 nL of crystallant to 200 nL of protein to form the sitting drops, using a Gryphon robot (Art Robbins Instruments, Sunnyvale, USA). Crystals grew in four conditions in each screen at 21°C. Crystal optimizations were carried out by the vapor diffusion hanging drop technique using 24 well VDX plates (Hampton Research). Equal volumes (1 μL) of Kgp protein was added to reservoir solution containing 27% polyethylene glycol 4000, 100 mM sodium acetate pH 5.0 and 0.2 M ammonium sulfate, and then suspended over 500 μL of reservoir solution. Crystals grew after one week as 100 μm × 25 μm × 25 μm hexagonal shaped rods. Crystals formed in the tetragonal space group P43212, with unit cell dimensions a = 116.79 Å, b = 116.79 Å, c = 86.87 Å. One molecule in the asymmetric unit gave a predicted Matthews coefficient of 2.98 Å3 D−1 and a corresponding solvent content of 58.77%. Native crystals were flash-cooled prior to data collection by passing them briefly through Paratone-N (Hampton Research) before being plunged directly into liquid nitrogen. All diffraction data were collected at the Australian Synchrotron, beamline MX2 at 100 K using the program Blue-Ice.12 Data were integrated using XDS,13 and scaled using SCALA.14
Heavy atom derivatization
Crystals were soaked in mother liquor supplemented with either 5 mM trimethylead acetate or 5 mM potassium dicyanoaurate (I) for 36 h. Derivative crystals were briefly “back-soaked” in mother liquor containing 20% ethylene glycol before being cryo-cooled.
Structure determination
Electron density maps produced using a native dataset to 2.7 Å resolution (data not shown) and phase information via molecular replacement using the RgpB structure (PDB id: 1CVR) were relatively poor. Instead, experimental phase information was generated by the SIRAS (single isomorphous replacement with anomalous scattering) method using a second, high resolution native dataset, together with a lead derivative (Table I). The heavy atoms were located and a solvent flattened electron density map calculated using SHELXC/D/E.13 Experimentally phased electron density maps from either derivative were extremely clear and allowed the manual building of 301 residues (out of 452) using COOT15 in the first build. The crystal to detector distance was optimized prior to collecting the lead data and was thus chosen for refinement.
Table I.
Summary of X-Ray Data and Refinement Statistics
| Native | Pb derivative | |
|---|---|---|
| Space group | P43212 | P43212 |
| Cell dimensions Å (a, b, c) | 116.79, 116.79, 86.87 | 116.94, 116.94, 86.85 |
| Resolution (Å)a | 48.46–1.93 (2.03–1.93) | 48.50–1.60 (1.69–1.60) |
| Number of reflectionsb | 1317580 [45868] | 923558 [78616] |
| Completeness (%)a | 99.6 (97.5) | 98.3 (90.0) |
| Anomalous completeness (%)a | 96.3 (78.0) | |
| I/σIa | 20.3 (6.3) | 20.2 (1.5) |
| Rmerge (%)a | 15.3 (70.5) | 7.4 (63.3) |
| Rp.i.m. (%)a | 2.9 (13.3) | 2.20 (33.5) |
| Redundancya | 28.7 (28.0) | 11.7 (4.7) |
| Anomalous redundancya | 6.1 (2.5) | |
| Wilson B factor (Å2) | 16.8 | 15.4 |
| Refinement | ||
| Resolution Å | 48.5–1.6 | |
| No. of reflections | 78336 | |
| Rwork (%) /Rfree (%)c | 12.2/14.8 | |
| Fo, Fc correlation | 0.97 | |
| Non-hydrogen atomsd | ||
| Protein | 3597 (15.01) | |
| Water | 479 (29.13) | |
| Ligands | 20 (22.11) | |
| Lead ions | 1 (11.82) | |
| Acetate ions | 4 (30.81) | |
| Sulfate ions | 20 (36.59) | |
| Ethylene glycol | 40 (36.62) | |
| Sodium ions | 2 (13.10) | |
| Potassium ions | 1 (25.21) | |
| r.m.s.d. from ideal geometry | ||
| Bond lengths (Å) | 0.008 | |
| Bond angles (°) | 1.257 | |
| Molprobity analysis | ||
| Ramachandran favoured % | 96.3 | |
| Ramachandran outliers % | 0.4 | |
| Rotamer outliers | 0.8 | |
Values in parentheses represent the highest resolution shell.
Values in square brackets represent the number of merged reflections.
Rfree is based on 5.04% (3924 reflections) of the total number reflections excluded from refinement. All data collected was used for refinement.
Value in parentheses represent the average B factor in Å2.
Anisotropic ADP refinement was carried out using the program PHENIX.16 Successive rounds of model building using COOT and further refinement were carried out using data between 48–1.6 Å resolution resulted in the final model having one molecule in the asymmetric unit with an Rfactor of 12.2% and an Rfree of 14.8%. Ramachandran outliers (Ala 443 and Ile 478) are clearly located in the electron density map and lie close to the covalently bound TCLK inhibitor. Secondary structural elements were calculated using DSSP.17 A summary of the crystallographic data collection and refinement statistics are given in Table I.
Atomic coordinates
Atomic coordinates and structure factor amplitudes of the lead derivative used for refinement have been deposited into the PDB with the accession code 4TKX.
Acknowledgments
This research was partly undertaken on the MX2 beamline at the Australian Synchrotron, Victoria, Australia. We thank the beamline staff for their assistance. Funding from the Victorian Government Operational Infrastructure Support Scheme to St Vincent's Institute is gratefully acknowledged.
Glossary
- FFRCMK
D-Phe-Phe-Arg-chloromethylketone
- Kgp
lysine specific gingipain
- Rgp
arginine specific gingipain
- r.m.s.d.
root mean square deviation
- TLCK
N-α-tosyl-lysyl chloromethylketone hydrochloride.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information Figure 1.
Supplementary Information Figure 2.
References
- 1.Pathirana RD, O'Brien-Simpson NM, Brammar GC, Slakeski N, Reynolds EC. Kgp and RgpB, but not RgpA, are important for Porphyromonas gingivalis virulence in the murine periodontitis model. Infect Immun. 2007;75:1436–1442. doi: 10.1128/IAI.01627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pathirana RD, O'Brien-Simpson NM, Visvanathan K, Hamilton JA, Reynolds EC. Flow cytometric analysis of adherence of Porphyromonas gingivalis to oral epithelial cells. Infect Immun. 2007;75:2484–2492. doi: 10.1128/IAI.02004-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pathirana RD, O'Brien-Simpson NM, Visvanathan K, Hamilton JA, Reynolds EC. The role of the RgpA-Kgp proteinase-adhesin complexes in the adherence of Porphyromonas gingivalis to fibroblasts. Microbiol SGM. 2008;154:2904–2911. doi: 10.1099/mic.0.2008/019943-0. [DOI] [PubMed] [Google Scholar]
- 4.O'Brien-Simpson NM, Pathirana RD, Walker GD, Reynolds EC. Porphyromonas gingivalis RgpA-Kgp proteinase–adhesin complexes penetrate gingival tissue and induce proinflammatory cytokines or apoptosis in a concentration-dependent manner. Infect Immun. 2009;77:1246–1261. doi: 10.1128/IAI.01038-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10:717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huq NL, Seers CA, Toh EC, Dashper SG, Slakeski N, Zhang L, Ward BR, Meuric V, Chen D, Cross KJ, Reynolds EC. Propeptide-mediated inhibition of cognate gingipain proteinases. PLoS One. 2013;8:e65447. doi: 10.1371/journal.pone.0065447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curtis MA, Kuramitsu HK, Lantz M, Macrina FL, Nakayama K, Potempa J, Reynolds EC, Aduse-Opoku J. Molecular genetics and nomenclature of proteases of Porphyromonas gingivalis. J Periodontal Res. 1999;34:464–472. doi: 10.1111/j.1600-0765.1999.tb02282.x. [DOI] [PubMed] [Google Scholar]
- 8.Potempa J, Nguyen KA. Purification and characterization of gingipains. Curr Protoc Protein Sci. 2007;49:21.20.1–21.20.27. doi: 10.1002/0471140864.ps2120s49. [DOI] [PubMed] [Google Scholar]
- 9.Eichinger A, Beisel H-G, Jacob U, Huber R, Medrano F-J, Banbula A, Potempa J, Travis J, Bode W. Crystal structure of gingipain R: an Arg-specific bacterial cysteine proteinase with a caspase-like fold. EMBO J. 1999;18:5453–5462. doi: 10.1093/emboj/18.20.5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Diego I, Veillard FT, Guevara T, Potempa B, Sztukowska M, Potempa J, Gomis-Ruth FX. Porphyromonas gingivalis virulence factor gingipain RgpB shows a unique zymogenic mechanism for cysteine peptidases. J Biol Chem. 2013;288:14287–14296. doi: 10.1074/jbc.M112.444927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleywegt GJ. Experimental assessment of differences between related protein crystal structures. Acta Crystallogr D Biol Crystallogr. 1999;55:1878–1884. doi: 10.1107/s0907444999010495. [DOI] [PubMed] [Google Scholar]
- 12.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 13.Kabsch W. Integration, scaling, space–group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 15.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 16.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 17.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information Figure 1.
Supplementary Information Figure 2.