

Abstract
Spinocerebellar ataxia type 31 (SCA31) is an autosomal dominant form of pure cerebellar ataxia that is caused by a disease-specific insertion containing penta-nucleotide repeats (TGGAA)n. Neuropathologically, cerebellar Purkinje cells are preferentially affected and reduced in number in SCA31, and they are often surrounded by halo-like amorphous materials. In the present study, we performed neuropathological analyses on two SCA31 brains, and discussed the serial morphological changes of Purkinje cells in SCA31.We found that bent, elongated, often folded nuclei were observed frequently in degenerating Purkinje cells with the halo-like structure. Conversely, Purkinje cells without this structure developed marked atrophy with severely slender and condensed nuclei. On the basis of these pathological findings, we propose two different processes for Purkinje cell degeneration in SCA31, namely, shrinkage of Purkinje cells with or without the halo-like amorphous materials. The former, but not the latter, was considered to be specific to SCA31. Correspondingly, fragmentation of the Golgi apparatus was observed more frequently in Purkinje cells with the halo-like structure than in those without this structure. We consider that the profound nuclear deformity and fragmentation of the Golgi apparatus are closely linked with the formation of the halo-like structure in SCA31.
Keywords: Golgi apparatus, halo-like structure, Purkinje cell, somatic sprouting, spinocerebellar ataxia type 31 (SCA31)
Introduction
Spinocerebellar ataxia type 31 (SCA31) is a subtype of autosomal dominant cerebellar ataxia and is characterized by adult-onset, pure cerebellar ataxia. SCA31 is found commonly in the Japanese population,1 while it is found rarely in other east Asian countries such as Korea and China,2,3 and extremely rarely in Caucasian populations.1 The genetic defect of SCA31 is a specific insertion of complex penta-nucleotide repeats containing (TGGAA)n in the 16q22.1 region.4,5
To date, the neuropathological findings of at least four Japanese SCA31 patients have been reported in the literature.6–10 On the basis of the literature, cerebellar Purkinje cells are predominantly involved in SCA31.6–10 They are often surrounded by halo-like amorphous materials6,7,9 or fuzzy, coat-like, finely fibrillary materials.8 Using immunohistochemistry, this unique structure was shown to be positively stained with anti-calbindin-D-28 K (CaBP28K) and anti-synaptophysin (SYP) antibodies.6–9 Thus, it is considered to consist mainly of the somatic sprouts of Purkinje cells and presynaptic terminals innervated either from basket cells, neurons of the inferior olivary nucleus, or other neurons that connect with Purkinje cells.6,7,9
One of several intriguing questions in SCA31 is how an insertional mutation of complicated penta-nucleotides in an intron of BEAN/TK2 is associated with Purkinje cell degeneration. One of the ways to approach this question may lie in the careful observation of the morphological changes of Purkinje cells in SCA31 because these cells are the major and preferential target of SCA31 pathology. Thus, the aim of this study was to characterize neuropathologically how Purkinje cells degenerate in SCA31, with a particular interest in the morphological changes of the intracellular structures of these cells.
Materials and Methods
Subjects
This study was approved by the Ethics Committees of Shinshu University School of Medicine (approval number #2318). Autopsied brains were taken from two Japanese patients (a 77-year-old man8 and an 81-year-old man) with genetically proven SCA31 and from four age-matched Japanese controls (aged 57–84 years, three male and one female, mean 74.3 years). The clinical and pathological findings of one patient (case 1) were previously reported.8 His age at onset was approximately 58 years and the total duration of cerebellar ataxia was approximately 19 years. The other patient (case 2) was reported to develop cerebellar ataxia at approximately 65 years of age. He had not been followed up regularly and had been institutionalized in a nursing home until he was admitted to hospital because of bronchopneumonia at the age of 81 years. One month after his admission, he died of respiratory failure due to bronchopneumonia. The total duration of cerebellar ataxia in this case was approximately 18 years. We confirmed that the control subjects had no pathological features suggesting neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease or dementia with Lewy bodies. None of the cases or control subjects had been maintained on an artificial respirator during their lifetime.
Light microscopic examination and immunohistochemistry
The brains were fixed in 10% formalin, and multiple tissue blocks were embedded in paraffin. The 6-μm-thick sections were stained with HE, KB, Bodian, PAS, Alcian blue and Berlin blue.
Immunohistochemical staining was performed by the avidin-biotin-peroxidase complex (ABC) method (VECTASTAIN ABC Elite kit; Vector, Burlingame, CA, USA). The primary antibodies used in this study are listed in Table 1.11 Non-specific binding of the ABC system reagents was blocked by pretreating the sections with 0.3% hydrogen peroxide in methanol and a normal blocking serum, and then incubating them with the required primary antibody overnight at 4°C. The sections were then incubated for 1 h with the secondary reagent containing a biotinylated anti-rabbit or anti-mouse IgG antibody (diluted 1:200) at 37°C, and finally with the ABC solution for 1 h at room temperature (RT). The sections were subjected to a peroxidase reaction with 30 μL ImmPACT™ DAB Chromogen Concentrate (Vector) (diluted 1:2, with 50 mmol/L Tris HCl (pH 7.6)) in 1 mL ImmPACT™ Diluent (Vector) for 5 min at RT. As antibody controls, the primary antisera were either omitted or replaced with normal rabbit or mouse serum. Several specimens of neural and non-neural tissue from the cases served as positive or negative tissue controls.
Table 1.
Primary antibodies used in this study
| Primary target | Antibody type | Dilution | Increase of antigenicity† | Source |
|---|---|---|---|---|
| Calbindin-D-28 K (CaBP28K) | Rabbit polyclonal | 1:600 | Autoclaving | Sigma-Aldrich, USA |
| Parvalbumin (PV) | Mouse/monoclonal (clone PARV-19) | 1:1000 | Boiling | Sigma-Aldrich, USA |
| Ubiquitin (UB) | Mouse/monoclonal (clone Ubi-1) | 1:20000 | Autoclaving | Millipore, USA |
| Synaptophysin (SYP) | Mouse/monoclonal (clone 171B5) | 1:500 | Autoclaving | Obata K, et al.11 |
| Phosphorylated TDP DNA binding protein 43 (p-TDP43) | Mouse/monoclonal (clone 11-9) | 1:5000 | Autoclaving | Cosmo Bio, Japan |
| Phosphorylated neurofilament | Mouse/monoclonal (clone SMI31) | 1:1000 | Boiling | Covance, USA |
| Non-phosphorylated neurofilament | Mouse/monoclonal (clone SMI32) | 1:500 | Autoclaving | Covance, USA |
| Trans-Golgi network protein 2 (TGOLN2) | Mouse/monoclonal (clone 2F11) | 1:1000 | Autoclaving | Abnova, Taiwan |
Antigenicity was increased by autoclaving (121°C, 20 min) in 0.01 mol/L citrate-buffered solution (pH 6.0) or boiling in a microwave oven (500 W, 7 min) in 0.01 mol/L citrate-buffered solution (pH 7.6).
The fragmentation of the Golgi apparatus was judged by the morphology and signal intensity of positive granular dots detected using an anti-trans-Golgi network protein 2 (TGOLN2) antibody under 1000-fold magnification on a light microscope. We surveyed sequentially approximately 100–150 Purkinje cells at the folia in the mid-portion of the vermis and divided the cells into four groups as follows: Purkinje cells having normal Golgi apparatus with or without the halo-like structure, and Purkinje cells having fragmented Golgi apparatus with or without the halo-like structure.
For the immunofluorescent examination of CaBP28K and PV, after boiling in a microwave oven, sections from the cerebellum were incubated in normal goat serum for 30 min, then the antibodies for CaBP28K (diluted 1:240) and PV (diluted 1:100) were added. The secondary antibodies used were Alexa Fluor 546 goat anti-rabbit IgG (diluted 1:200; Molecular Probes, Eugene, OR, USA) and Alexa Fluor 488 goat anti-mouse IgG (diluted 1:200; Molecular Probes). The sections were then immersed in an autofluorescence eliminator reagent (Millipore, Temecula, CA, USA) for 1 min at RT to remove any of the autofluorescent pigment lipofuscin that had accumulated. The sections were observed under a confocal laser microscope (LSM5; Carl Zeiss, Oberkochen, Germany).
In situ terminal dUTP nick-end labeling (TUNEL) was carried out using an in situ apoptosis detection kit (NeuroTACS II™; Trevigen, Gaithersburg, MD, USA) on 6-μm-thick formalin-fixed paraffin-embedded sections of the cerebellum of the control subjects and cases. The sections were digested with proteinase K (Trevigen), and then incubated with terminal deoxynucleotide transferase (TdT; Trevigen). The sections were further incubated at 37°C with TdT dNTP (deoxyribonucleotide) Mix, TdT enzyme, TdT labeling buffer and Mn2+ (all from Trevigen). The sections were incubated for 10 min with streptavidin-conjugated horseradish peroxidase at RT. The sections were subjected to a peroxidase reaction with diaminobenzidine, resulting in a localized brown color within the nuclei of apoptotic cells. As a negative control, DNase-digested sections were used, and distilled water or phosphate-buffered saline was substituted for the TdT solution.
Statistical analysis
A chi-square test was used to compare the frequency of fragmentation of the Golgi apparatus in the Purkinje cells between the SCA31 cases (n = 2) and controls (n = 4). We also compared the frequency of fragmentation of the Golgi apparatus between the Purkinje cells with and without the halo-like structure in the SCA31 cases. All statistical analyses were performed using Excel 2013 (Windows 7 Professional Package; Microsoft Co., Redmond, WA, USA) with the significance level set to P < 0.01.
Results
The Purkinje cells of the control subjects were plump (Fig. 1A), surrounded by thin neurites (Fig. 1B), and did not have the halo-like structure. Case 1 showed an extensive loss of Purkinje cells, while case 2 showed a moderate loss. Some of the remaining Purkinje cells appeared normal, but many were atrophic or surrounded by the halo-like structure (Fig. 1C, Table 2). Only a few of the Purkinje cells with this structure had normal-looking nuclei, and most of them had deformed nuclei (Fig. 1C–F). Ghost-like Purkinje cells were seen in the halo-like structure (Fig. 1G) or surrounded by bundles of lines, which were considered to be axons (Fig. 1H,I). Conversely, simply shrunken Purkinje cells without the halo-like amorphous materials were also observed (Fig. 1C,J, arrowheads) in the SCA31 cases. These cells appeared condensed and the outline of their nuclei became obscure from the early stage of degeneration (Fig 1C). Among the apparently abnormal-looking Purkinje cells, the incidence of Purkinje cells with the halo-like structure was 42.3% and 26.4% in cases 1 and 2, respectively (Table 2).
Figure 1.

Changes of Purkinje cell morphology in cases with spinocerebellar ataxia type 31 (SCA31) (A and B: control subject; C–J: SCA31 case). (A) Purkinje cells of a control subject have round nuclei. (B) Thin neurites (arrows) around a Purkinje cell are seen in a control subject. (C) Two normal-sized Purkinje cells are shown in an SCA31 case. One has a dark cytoplasm and nucleus, suggesting that it has started to shrink (arrowhead). The other apparently has the halo-like amorphous materials with a bent nucleus (thin arrow). (D and E) Bright and elongated nuclei (thin arrows) are occasionally folded up at their end (D, thin arrow) in Purkinje cells with the halo-like amorphous materials. (F) Degenerated Purkinje cell with the typical halo-like amorphous materials. Purkinje cells are shrunken and their nuclei become severely deformed, rod-shaped, and condensed. (G) Ghost-like feature of a Purkinje cell is shown, where the outline of the nucleus is obscure. (H) Ghost-like feature of a Purkinje cell surrounded by bundles of lines (thin arrows). (I) The cell body of Purkinje cell is obscured in the empty basket formed by thick neurites (arrows). (J) A severely thin Purkinje cell with a shrunken and condensed nucleus that was not surrounded by the halo-like amorphous materials. A, C–H, and J: HE stain, B and I: Bodian stain. Bars; 20 μm.
Table 2.
Frequency of the appearance of the halo-like structure and fragmentation of the Golgi apparatus in Purkinje cells
| No. of Purkinje cells examined | No. of Purkinje cells with the halo-like structure (%) | Fragmentation of the Golgi apparatus (%) | No. of Purkinje cells without the halo-like structure (%) | Fragmentation of the Golgi apparatus (%) | |
|---|---|---|---|---|---|
| Case 1 | 137 | 58 (42.3) | (+) 46 (79.3) | 79 (57.7) | (+) 17 (21.5) |
| (−) 12 (20.7) | (−) 62 (78.5) | ||||
| Case 2 | 106 | 28 (26.4) | (+) 28 (100.0) | 78 (73.6) | (+) 10 (12.8) |
| (−) 0 (0) | (−) 68 (87.2) | ||||
| Total | 243 | 86 | (+) 74 | 157 | (+) 27 |
| (−) 12 | (−) 130 | ||||
| Control 1 | 119 | 0 (0) | 119 (100.0) | (+) 8 (6.7) | |
| (−) 111 (93.3) | |||||
| Control 2 | 124 | 0 (0) | 124 (100.0) | (+) 17 (13.7) | |
| (−) 107 (86.3) | |||||
| Control 3 | 123 | 0 (0) | 123 (100.0) | (+) 15 (12.2) | |
| (−) 108 (87.8) | |||||
| Control 4 | 142 | 0 (0) | 142 (100.0) | (+) 19 (13.4) | |
| (−) 123 (86.6) | |||||
| Total | 508 | 0 | 508 | (+) 59 | |
| (−) 449 |
Immunohistochemically, thick somatic sprouts from the cytoplasm of the Purkinje cells, positive for CaBP28K and PV, formed the halo-like amorphous materials in the SCA31 cases (Fig. 2A–C). Cactus-like changes of Purkinje cells were observed frequently in the molecular layer (Fig. 2A, arrowhead), but torpedoes in the granular cell layer were seen rarely in the SCA31 cases. Some Purkinje cells appeared to have two nuclei (Fig. 2B). The basket or satellite cells in the molecular layer were positive for PV immunohistochemistry (Fig. 2C, arrows). The halo-like amorphous materials were immunopositive for SYP (Fig. 2D) and ubiquitin (data not shown). Ubiquitin- or phosphorylated TAR DNA binding protein 43 (p-TDP43)-positive inclusions were not observed in the Purkinje cells. The axons surrounding the degenerated Purkinje cells were immunopositive for phosphorylated neurofilament (SMI31) and non-phosphorylated neurofilament (SMI32) (data not shown).
Figure 2.
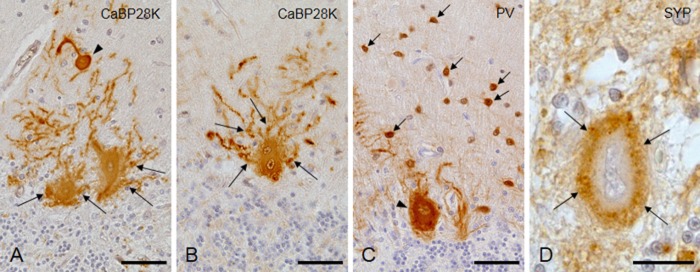
Immunohistochemical study of the cerebellum in case 1. (A and B) The halo-like structure and cactus-like change (arrowhead) are immunopositive for calcium-binding protein-D-28 K (CaBP28K). Thick dendrites are mixed up in the halo-like amorphous materials (arrows). The Purkinje cell shown in Figure 2B appears to have two nuclei. (C) Basket/satellite cells (arrows) and a Purkinje cell (arrowhead) are immunopositive for parvalbumin (PV). (D) The halo-like structure is immunopositive for synaptophysin (SYP). Bars; 50 μm.
Under confocal laser microscopy, double immunohistochemistry for CaBP28K and PV clearly showed that the cell surface of the Purkinje cells was smooth without somatic sprouts in the control subjects (Fig. 3A–C), but that the cells of the SCA31 cases were severely atrophic with many somatic sprouts (Fig. 3D–F). The diameter of the fibrillary materials ranged from 0.2 to 0.9 μm. CaBP28K and PV colocalized in the halo-like amorphous materials (Fig. 3D–F).
Figure 3.
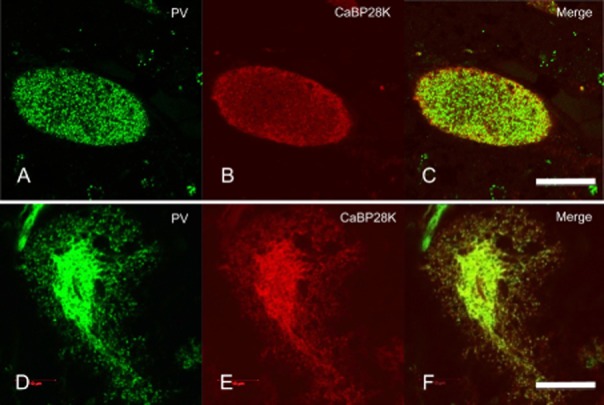
Confocal laser microscopic observation of parvalbumin (PV) and calcium-binding protein-D-28 K (CaBP28K) fluorescence in Purkinje cells of a control subject (A–C), and case 1 with spinocerebellar ataxia type 31 (SCA31) (D–F). The atrophic Purkinje cells of the SCA31 cases have many dendritic processes (somatic sprouting), which consist of a surrounding halo-like amorphous materials (D–F). Bars; 20 μm.
Immunohistochemistry for TGOLN2 revealed the large granular appearance of the Golgi apparatus in the cytoplasm of the Purkinje cells of the control subjects (Fig. 4A, arrowheads) and in the normal-looking Purkinje cells of the SCA31 cases (Fig. 4B, arrowheads). Fragmentation of the Golgi apparatus was seen in a small number of Purkinje cells in the controls (6.7–13.7 %, 59/508 in total), but it was observed significantly more frequently in the SCA31 cases (101/243 in total) (P = 7.18 x 10−21, Table 2). In SCA31 cases, the Golgi apparatus of the Purkinje cells with the halo-like structure (Fig. 4C,D, arrows) was frequently fragmented and reduced in number (Fig. 4C,D, arrowheads, Table 2). Conversely, the Golgi apparatus of the shrunken Purkinje cells without the halo-like structure was reduced in number, but fragmentation was observed much less frequently than those with the structure (Fig. 4E,F, arrowheads, Table 2). The difference in the frequency of Golgi fragmentation between the Purkinje cells with and without the halo-like structure (74/86 vs. 27/157 in total) was statistically significant (P = 1.88 x 10−25, Table 2). TUNEL for detecting apoptosis was negative in the cerebellum of the control subjects and SCA31 cases. No significant PAS-, Alcian blue- or Berlin blue-positive structures were identified.
Figure 4.
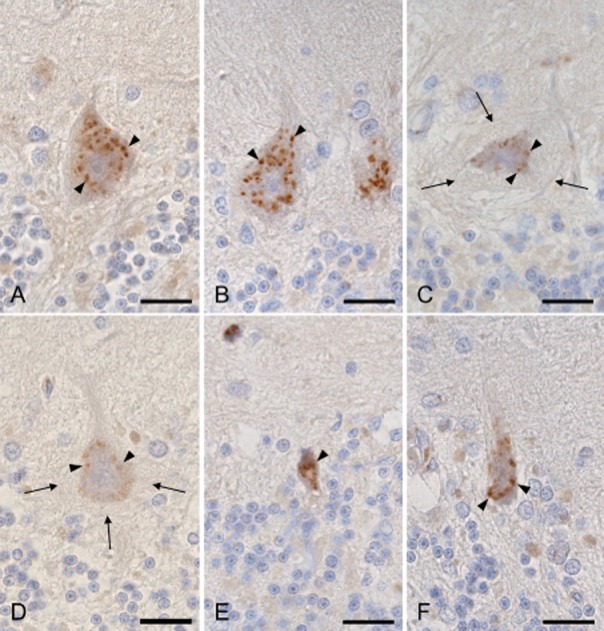
Immunohistochemistry for trans-Golgi network protein 2 (TGOLN2). (A) Granular appearance of a Purkinje cell from a control subject (arrowheads). (B) Normal-sized Purkinje cells without the halo-like amorphous materials from case 1 with spinocerebellar ataxia type 31 (SCA31) show the relative preservation of the Golgi apparatus (arrowheads). (C and D) Purkinje cells with the halo-like amorphous materials (arrows) from case 1 (C) and 2 (D) with SCA31 show severely fragmented and a reduced amount of the Golgi apparatus (arrowheads). (E and F) Shrunken Purkinje cells without the halo-like amorphous materials from cases 1 (E) and 2 (F) with SCA31 show reduced, but not fragmented Golgi apparatus (arrowheads). Bars; 20 μm.
Discussion
One of the main findings of this study is that profoundly deformed nuclei were evident in the Purkinje cells of the SCA31 cases. Dented, bent, elongated or folded nuclei were observed frequently in the Purkinje cells surrounded by the halo-like amorphous materials. The observed nuclear deformity may be due to the fragility of the nuclear membrane in Purkinje cells. The high incidence of the coexistence of nuclear deformity and the halo-like structure indicates that the pathogenic mechanisms for these features are closely linked. As a remarkable contrast to this finding, some Purkinje cells showed marked atrophy without having the halo-like amorphous materials. In these cells, their slender nuclei appeared dark and condensed. Based on these observations, we speculate that there are two pathways by which the Purkinje cells of SCA31 patients degenerate, that is, shrinkage and death of Purkinje cells with (Fig. 5A) or without (Fig. 5B) the halo-like amorphous materials. The latter pathway is considered not to be specific to SCA31, but is occasionally seen in other cerebellar ataxias. At present, we are not sure whether these pathways intersect with each other, but we suggest that the intersection may possibly take place in the early phase of both processes.
Figure 5.
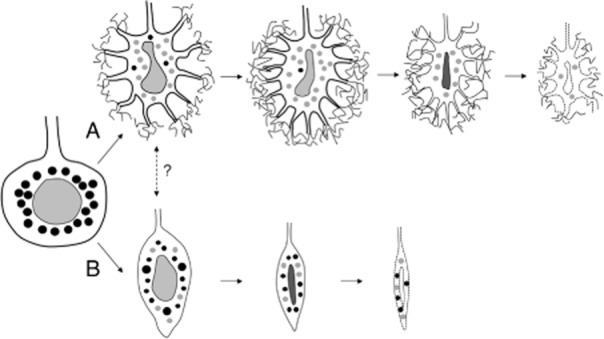
Schematic drawing of the chronological changes of Purkinje cell morphology in spinocerebellar ataxia type 31 (SCA31). Two different processes of Purkinje cell degeneration are shown. One is the shrinkage of Purkinje cells with the halo-like amorphous materials (A). These cells very often have bent and elongated nuclei. The other is the shrinkage of Purkinje cells without the halo-like amorphous materials (B). These cells develop marked atrophy with slender and condensed nuclei. The black circles in the cytoplasm indicate Golgi apparatus with a normal appearance, while the gray circles indicate Golgi apparatus undergoing fragmentation. Fragmentation of the Golgi is observed more frequently in degenerated Purkinje cells with the halo-like structure than in those without this structure. Synaptophysin-positive vesicles are not indicated in this schema.
Furthermore, TGOLN2 immunohistochemistry showed that the fragmentation of the Golgi apparatus occurred more frequently in Purkinje cells with the halo-like structure than in those without this structure. This may support the presence of different degeneration processes between the cells with or without the halo-like structure. The Golgi apparatus is considered to play important roles in not only post-translational processing, but also sorting of newly synthesized proteins.12–14 Fragmentation of the Golgi apparatus has been reported in several neurodegenerative diseases, including amyotrophic lateral sclerosis, corticobasal degeneration, and spinocerebellar ataxia type 2 (SCA2).12–14 This fragmentation may be a consequence of apoptosis or could trigger apoptosis; however, we could not find any evidence of apoptosis by TUNEL or protein aggregation in the affected Purkinje cells. Thus, the significance of the fragmentation of Golgi in SCA31 is unclear at present, but it is of interest that this fragmentation was conspicuous in the Purkinje cells with the halo-like structure. In these cells, the improper sorting of proteins in Purkinje cell dendrites may cause the aberrant outgrowth of dendritic processes, resulting in somatic and dendritic sprouting (cactus formation).
In conclusion, this study suggests that there are two different pathways of degeneration in Purkinje cells with or without the halo-like amorphous materials in SCA31. We speculate that the former pathway with the halo-like structure is closely linked to the nuclear deformity and fragmentation of the Golgi apparatus in the Purkinje cells. Further study is necessary to elucidate the pathogenic mechanism underlying the disruption of the stability of the nuclear membrane, and the correlation between nuclear deformity, dysfunction of cytoplasmic organelles and the formation of the halo-like amorphous materials.
Acknowledgments
The authors are indebted to Ms Yayoi Uehara (Department of Brain Disease Research, Shinshu University), for her excellent technical assistance. This work was supported in part by a Grant-in-Aid for Science Research from the Ministry of Education, Science, and Culture, Japan and a grant from the Research Committee for Ataxic Diseases, the Ministry of Health, Labour, and Welfare, Japan. The authors report no conflicts of interest.
References
- Ishikawa K, Durr A, Klopstock T. Pentanucleotide repeats at the spinocerebellar ataxia type 31 (SCA31) locus in Caucasians. Neurology. 2011;77:1853–1855. doi: 10.1212/WNL.0b013e3182377e3a. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Jeon BS, Lee WY. SCA in Korea and its regional distribution: a multicenter analysis. Parkinsonism Relat Disord. 2011;17:72–75. doi: 10.1016/j.parkreldis.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Ouyang Y, He Z, Li L, Qin X, Zhao Y, Yuan L. Spinocerebellar ataxia type 31 exists in Northeast China. J Neurol Sci. 2012;316:164–167. doi: 10.1016/j.jns.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Sato N, Amino T, Kobayashi K. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85:1–14. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai H, Yoshida K, Shimizu Y, Morita H, Ikeda S, Matsumoto N. Analysis of an insertion mutation in a cohort of 94 patients with spinocerebellar ataxia type 31 from Nagano, Japan. Neurogenetics. 2010;11:409–415. doi: 10.1007/s10048-010-0245-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owada K, Ishikawa K, Toru S. A clinical, genetic, and neuropathologic study in a family with 16q-linked ADCA type III. Neurology. 2005;65:629–632. doi: 10.1212/01.wnl.0000173065.75680.e2. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Mizusawa H. On autosomal dominant cerebellar ataxia (ADCA) other than polyglutamine diseases, with special reference to chromosome 16q22.1-linked ADCA. Neuropathology. 2006;26:352–360. doi: 10.1111/j.1440-1789.2006.00719.x. [DOI] [PubMed] [Google Scholar]
- Shintaku M, Kaneda D. Chromosome 16q-22.1-linked autosomal dominant cerebellar ataxia: an autopsy case report with some new observations on cerebellar pathology. Neuropathology. 2009;29:285–292. doi: 10.1111/j.1440-1789.2008.00947.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Mizusawa H. The chromosome 16q-linked autosomal dominant cerebellar ataxia (16q-ADCA*): a newly identified degenerative ataxia in Japan showing peculiar morphological changes of the Purkinje cell. Neuropathology. 2010;30:490–494. doi: 10.1111/j.1440-1789.2010.01142.x. [DOI] [PubMed] [Google Scholar]
- Niimi Y, Takahashi M, Sugawara E. Abnormal RNA structures (RNA foci) containing a penta-nucleotide repeat (UGGAA)n in the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis. Neuropathology. 2013;33:600–611. doi: 10.1111/neup.12032. [DOI] [PubMed] [Google Scholar]
- Obata K, Nishiye H, Fujita S. Identification of a synaptic vesicle-specific 38,000-dalton protein by monoclonal antibodies. Brain Res. 1986;375:37–48. doi: 10.1016/0006-8993(86)90956-x. [DOI] [PubMed] [Google Scholar]
- Gonatas NK, Stieber A, Mourelatos Z. Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis. Am J Pathol. 1992;140:731–737. [PMC free article] [PubMed] [Google Scholar]
- Gonatas NK, Stieber A, Gonatas JO. Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J Neurol Sci. 2006;246:21–30. doi: 10.1016/j.jns.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Fan J, Hu Z, Zeng L. Golgi apparatus and neurodegenerative diseases. Int J Dev Neurosci. 2008;26:523–534. doi: 10.1016/j.ijdevneu.2008.05.006. [DOI] [PubMed] [Google Scholar]