

Abstract
There are two classifications of hereditary hyperprolinemia: type I (HPI) and type II (HPII). Each type is caused by an autosomal recessive inborn error of the proline metabolic pathway. HPI is caused by an abnormality in the proline-oxidizing enzyme (POX). HPII is caused by a deficiency of Δ-1-pyrroline-5-carboxylate (P5C) dehydrogenase (P5CDh). The clinical features of HPI are unclear. Nephropathy, uncontrolled seizures, mental retardation or schizophrenia have been reported in HPI, but a benign phenotype without neurological problems has also been reported. The clinical features of HPII are also unclear. In addition, the precise incidences of HPI and HPII are unknown. Only two cases of HPI and one case of HPII have been identified in Japan through a questionnaire survey and by a study of previous reports. This suggests that hyperprolinemia is a very rare disease in Japan, consistent with earlier reports in Western countries. The one case of HPII found in Japan was diagnosed in an individual with influenza-associated encephalopathy. This suggests that HPII might reduce the threshold for convulsions, thereby increasing the sensitivity of individuals with influenza-associated encephalopathy. The current study presents diagnostic criteria for HPI and HPII, based on plasma proline level, with or without measurements of urinary P5C. In the future, screening for HPI and HPII in healthy individuals, or patients with relatively common diseases such as developmental disabilities, epilepsy, schizophrenia or behavioral problems will be important.
Keywords: hyperprolinemia type I, hyperprolinemia type II, inborn error of metabolism, P5C, proline
Hyperprolinemia type I (HPI) results from a deficiency of proline oxidase (POX; enzyme commission no. [EC] 1.5.99.8), also called proline dehydrogenase(PRODH). The POX gene (PRODH) is located on chromosome 22 (22q11.21). This region is deleted in a congenital malformation syndrome known as velo-cardio-facial syndrome. This gene locus is also related to susceptibility to schizophrenia. Another form of hyperprolinemia is called hyperprolinemia type II (HPII). HPII is caused by a deficiency of Δ-1-pyrroline-5-carboxylate (P5C) dehydrogenase (P5CDh; EC 1.5.1.12) activity.1,2 The P5CDh gene (ALDH4A1) is located on chromosome1 (1p36.13).
Schafer et al. were the first to document direct involvement of an error in proline metabolism in human disease in 1962.3The affected family members had hyperprolinemia, cerebral dysfunction, renal anomalies, hereditary nephropathy, and deafness. Subsequently, many families with hyperprolinemia were reported in the literature.4–14 Several phenotypes of hyperprolinemia were identified, including those in some asymptomatic family members. Several authors have discussed the potential coincidental association between these clinical features and hyperprolinemia. Recently, our understanding of the associations between proline metabolism, biological function, and disease has increased dramatically. There are now at least six enzymes, three transporters, and seven structural genes known to be directly involved in the interconversions of proline and its immediate metabolites. The functional characteristics, tissue distribution, regulation, and subcellular location of these proteins have been determined, and, for some, structural information is available.1,2 The exact clinical features and the frequencies of HPI or HPII, however, remain unknown. The aim of this study was therefore to assess the incidence of HPI and HPII in Japan, and to evaluate the cases reported previously. In addition, we propose diagnostic criteria for hyperprolinemia and identify a need for future studies on this disorder.
l-Proline metabolism
Tissue and fluid concentrations of l-proline are primarily related to the balance between the enzymatic activities of POX and P5C reductase. In this context, P5C plays a pivotal role in maintaining the concentration of proline in body fluids. Inborn errors of proline metabolism can, therefore, result from a disturbance of P5C-associated enzyme activity. HPI and HPII are differentiated by distinct biochemical and genetic deficiencies in the proline catabolic pathway (Fig. 1). HPI is caused by a deficiency in POX, a mitochondrial inner-membrane enzyme that converts proline to P5C, the first step in the conversion of proline to glutamate. P5CDh, a mitochondrial matrix NAD(+)-dependent dehydrogenase, catalyzes the second step of the proline degradation pathway. Deficiency of this enzyme is associated with HPII, an autosomal recessive disorder characterized by accumulation of P5C and proline (Fig. 1).1,2
Fig 1.
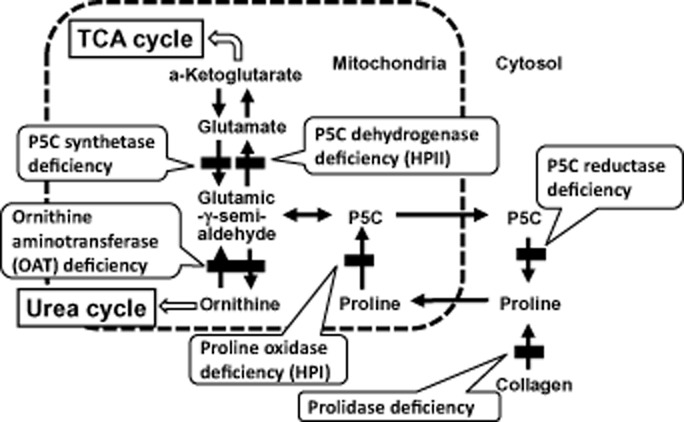
l-Proline metabolism and enzyme deficiencies. The metabolism of proline in mammals involves two other amino acids, glutamate and ornithine, and six enzymes. Conversion of P5C to glutamic-γ-semi-aldehyde is a non-enzymatic step. HPI, hyperprolinemia type I; HPII, hyperprolinemia type II; P5C, Δ-1-pyrroline-5-carboxylate; TCA, tricarboxylic acid.
Human disease in proline metabolism
In human inborn errors of metabolism of l-proline, HPI, HPII, P5C synthetase deficiency, P5C reductase deficiency, ornithine aminotransferase deficiency and prolidase deficiency are known as single-gene disorders. The characteristics and molecular biology of these enzymes in humans are summarized in Table 1. Of these diseases, hyperprolinemia is seen only in HPI and HPII.2
Table 1.
Human enzymes involved in proline metabolism
| Proline oxidase | P5C dehydrogenase | P5C synthetase | P5C reductase | OAT | Prolidase | |
|---|---|---|---|---|---|---|
| EC | EC 1.5.99.8 | EC 1.5.1.12 | None | EC 1.5.1.2 | EC 2.6.1.13 | EC 3.4.13.9 |
| Subcellular location | Mitochondrial inner membrane | Mitochondrial matrix | Mitochondrial inner membrane | Cytoplasm | Mitochondrial matrix | Cytoplasm |
| Subunit size (kDa) | 63 | 62 | 81 | 32 | 49 | 54.3 |
| Structure | Unknown | Homodimer | Hexamer | Homopolymer | Homohexamer | Homodimer |
| Cofactor | Unknown | NAD+ | ATP, NAD(P)H | NAD(P)H | Vitamin B6 | Mn2+,Ca2+, Mg2+,Fe2+ |
| Activity in tissues | Liver, kidney, brain | Ubiquitous | Small intestine mucosa, colon, pancreas, brain, thymus | Ubiquitous | Ubiquitous | Ubiquitous |
| Disease association | HPI | HPII | Hypoprolinemia | Cutis laxa | Gyrate atrophy | Prolidase deficiency |
| Map location | 22q11.21 | 1p36.13 | 10q24.1 | 17q25.3 | 10q26.13 | 19q13.11 |
| Gene/locus MIM | PRODH 606810 | ALDH4A1 606811 | ALDH18A1 138250 | PYCR1 179035 | OAT 613349 | PEPD 613230 |
ALDH, aldehyde dehydrogenase; EC, enzyme commission number; HPI, hyperprolinemia type I; HPII, hyperprolinemia type II; MIM, mendelian inheritance in man; OAT, ornithine aminotransferase; P5C, Δ-1-pyrroline-5-carboxylate; PEPD, peptidase; PRODH, proline dehydrogenase; PYCR1, pyrroline-5-carboxylate reductase 1.
Hyperprolinemia type I is diagnosed biochemically on high plasma proline without urinary excretion of P5C. In contrast, the presence of P5C in the urine is indicative of HPII. The clinical phenotype of HPI is not well characterized. Prior to the identification of PRODH, more than 15 families were reported in the literature with proline levels ranging from twofold to 10-fold above normal.3–16 Several phenotypes were found in these families. Whereas some patients were asymptomatic, others had neurological, renal and/or auditory defects. The potential for a coincidental association between these clinical features and HPI has been discussed.10–13
Hyperprolinemia has been reported in patients with a microdeletion in the 22q11 region.15–17 Since the identification of PRODH, various mutations of this enzyme (heterozygous and homozygous) have been identified in hyperprolinemic patients with different phenotypes (i.e. patients with schizophrenia, a 22q11 microdeletion and/or early neurological impairment).17–20 Jacquet et al. identified a complete homozygous deletion of PRODH located on chromosome 22q11 in a child with a severe form of HPI.17,18 That child had severe psychomotor delay and status epilepticus associated with a very high level of plasma proline (2246 μmol/L). These studies show, unambiguously, that the severe form of HPI, characterized by neurological manifestations, results from homozygous inactivating alterations of PRODH.
The PRODH mutations were divided into three groups: those leading to mild (<30%); moderate (30–70%); and severe (>70%) reductions of POX activity. Serum proline level seems to correlate with the severity of POX deficiency.19,20 Higher serum proline was associated with lower IQ in patients with the 22q11 deletion (including the region of PRODH) syndrome.19,21 Recently, Guilmatre et al. also reported genotype/phenotype correlations in HPI.22
Hyperprolinemia type II is due to the absence of P5CDh activity. Valle et al. measured P5CDh activity in normal fibroblasts and fibroblasts from patients with HPII.23 The HPII fibroblast cells had no detectable P5CDh activity over a range of reaction conditions, whereas normal cells had easily measurable activity. This enzymatic defect accounts for the biochemical abnormalities in HPII, which is usually characterized by mental retardation and convulsions.
Geraghty et al. surveyed ALDH4A1 from four affected members of a family with HPII.24 Four mutant alleles were found. A 1 bp deletion (G) at nucleotide 21 (A7fs(-1)) and a 1 bp insertion of a T following nucleotide 1563 (G521fs(+1)) cause a frameshift. The other two are missense mutations (S352L and P16L). To test the functional consequences of three of these mutations, they were expressed in a P5CDh-deficient strain of Saccharomyces cerevisiae. In contrast to yeast expressing wild-type human P5CDh, yeast expressing S352L and G521fs(+1) failed to grow on proline and had no detectable P5CDh activity. Interestingly, the G521fs(+1) allele, segregated in the large Irish traveler pedigree reported by Flynn et al.,25 was used to define the HPII phenotype. To our knowledge, this is the first description of the molecular basis for this inborn error.
Hyperprolinemia type II was identified in a pedigree of Irish travelers (nomads), among whom consanguineous marriage and high fertility are common. Flynn et al. studied 312 subjects in 71 families closely related to a proband with HPII. Thirteen additional cases of HPII were discovered. A further 50 subjects were found to have mild hyperprolinemia. Flynn et al. found a strong association between HPII and seizures during childhood but no significant association with cognitive function. Most adults in this group with HPII enjoyed normal health.25
Some neuropsychiatric disorders appear to be related to hyperprolinemia. The prevalence of schizophrenia among patients with the 22q11 interstitial deletion associated with DiGeorge syndrome was high. This suggests the existence of a susceptibility gene for schizophrenia within the DiGeorge syndrome chromosomal region. This deletion was associated with hyperprolinemia in schizophrenia patients. In addition, two heterozygous mutations (L441P and L289M) in PRODH, identified in three of 63 patients with schizophrenia, but not in the 68 controls, were also associated with increased plasma proline.17,18 Bender et al. identified at least 16 missense mutations in PRODH in studies of HPI and schizophrenia, 10 of which were present at polymorphic frequencies.26 Although there is limited information on plasma proline level in the individuals with known PRODH genotype, extant data suggest that severe hyperprolinemia (>800 mol/L) and the most severe effect on function occurs in individuals with large deletions and/or missense mutations (L441P and R453C) in PRODH; modest hyperprolinemia (300–500 mol/L) is associated with PRODH alleles exhibiting a moderate reduction in POX activity. Interestingly, three of the four alleles associated with, or found in, schizophrenia (V427M, L441P, and R453C) resulted in a severe reduction of POX activity and hyperprolinemia.
Proline oxidase functions are also related to tumor growth, hypoalimentation stress reaction, and fetal growth. Recent studies found that POX can regulate tumor growth both by tumor suppression and increasing tumor survival.27,28
Hyperprolinemia in Japan
The present study investigated hyperprolinemia patients in Japan in 2009. The study was approved by the Ethics Committee of the Faculty of Life Science, Kumamoto University. A questionnaire was sent to the Departments of Pediatrics, Neonatology, Genetics, and Transplant surgery in 928 institutions in Japan. Institutions were asked to confirm that they had diagnosed and/or treated HPII. A secondary detailed questionnaire concerning HPII was then sent to those confirming institutions. We also examined previous patient reports on hyperprolinemia in Japan. Two reports of HPI and one report of HPII were identified and analyzed. According to the secondary questionnaire, the HPII patient was a 4-year-old boy. His parents were not consanguineous, and his brother died at 11 months of age due to sudden infant death syndrome. The HPII patient had no seizures and had normal development until 4 years of age, when he suddenly developed high fever progressing to generalized convulsions and unconsciousness. He was admitted to hospital and diagnosed with influenza encephalopathy. His blood proline was elevated (2767 μmol/L), while his blood ornithine was normal. Urinary P5C was detected. After treatment for encephalopathy, the patient recovered, although a high level of blood proline and urinary P5C excretion continued. He was then diagnosed with HPII. He currently is not undergoing any treatment, has no convulsions, his IQ score is 96, and his growth is within the normal range.29,30
Two cases of HPI have been reported in Japan; one in 1987 and one in 1991. The 1987 case was accompanied by chromosomal abnormality. This patient had below-normal activity of POX and hyperprolinemia, although no relation between blood proline level and clinical features was established.31The 1991 case involved a 9-year-old girl who had photosensitive epilepsy. She had developmental delay and developed epilepsy at 7 years of age. Her blood proline level was three–fourfold higher than normal. P5C was not detected in the urine. POX activity in the liver was 23.5% of normal. A proline-restricted diet was not effective in preventing convulsions.32Overall, details of these two cases are limited because the reports are more than 20 years old.
Amino acids were analyzed in plasma samples from 200 severely handicapped patients living in a special institution in Kumamoto, as well as in plasma samples from 500 healthy individuals. None of the samples indicated hyperprolinemia, although some of the samples from normal individuals had plasma proline 2.5 SD higher than normal. Analysis of plasma proline in samples from elderly patients in a nursing home identified one case of suspected hyperprolinemia in a 90-year-old patient but, despite additional analysis, the patient was too old to confirm causality (data not shown).
Most newborn screening tests are done by measuring amino acids in whole blood samples collected on specialized filter paper. Almost all amino acids can be analyzed by this method. In Korea, blood proline is analyzed as part of newborn screening, and four neonates were identified as having HPI.33 Similar newborn screening for proline is needed to determine the incidence of hyperprolinemia in Japan.
Diagnostic criteria for HPI and HPII
The exact clinical features of HPI are unclear. Nephropathy, uncontrolled seizures, mental retardation, or schizophrenia have been associated with HPI, but HPI without neurological manifestations is also known. The clinical features of HPII are similarly variable. Therefore, clinical signs and symptoms cannot be used to diagnose HPI or HPII. Patients who have uncontrolled seizures or developmental delay or encephalopathy of unknown origin should undergo blood amino acid analysis. When hyperprolinemia is identified, the patient may be diagnosed with HPI or HPII. A flow chart for the diagnosis of HPI and HPII is given in Figure 2. In HPI, blood proline is usually fivefold–10-fold higher than the normal range of 51–271 μmol/L. In HPII, blood proline is even higher than in HPI, usually exceeding 1500 μmol/L. Lactic acidosis can lead to secondary hyperprolinemia. Thus, any diagnosis of HPI or HPII must rule out lactic acidosis. If either HPI or HPII is suspected, analysis of urinary organic acids on gas chromatography–mass spectroscopy is useful. If P5C is detected in urinary samples, a diagnosis of HPII is confirmed. If P5C is not detected, the diagnosis is HPI. The activities of the different enzymes affected in HPI and HPII can be determined using cultured lymphocytes, peripheral leukocytes, or cultured fibroblast. Each affected gene has been cloned previously, and, therefore, gene analysis is also possible. Many of the mutations on these genes are described in the previous sections.
Fig 2.
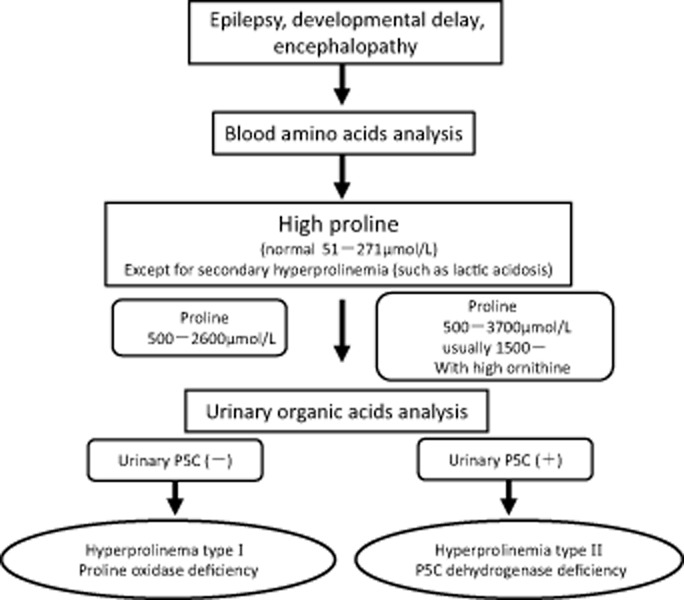
Diagnostic criteria for hyperprolinemia types I and II. P5C, Δ-1-pyrroline-5-carboxylate.
Treatment and prognosis
Treatment of the hyperprolinemia seen in HPI has included dietary therapy such as the restriction of proline. Unfortunately, this results in only modest control of plasma proline level and has no impact on clinical phenotype.34 As a result, dietary therapy may not be necessary.1
The seizures associated with the rare inherited disorder HPII are usually precipitated by an infection during childhood. The control of seizures in HPII is clearly important. It is necessary to protect against febrile convulsions and status epilepticus. As in HPI, dietary therapy is unable to significantly reduce plasma proline level in HPII. P5C is a unique endogenous vitamin antagonist. The inactivation of vitamin B6 by P5C may contribute to seizures in HPII, which are so far unexplained. The evidence for this largely rests on the diagnosis of HPII and vitamin B6 deficiency in a well-nourished child with seizures. Pyridoxine deficiency was implicated in her seizures and may have been the result of inactivation of vitamin B6 by the proline metabolite, P5C. Long-term vitamin B6 supplementation may therefore prevent these seizures.35,36
Recently, oxidative stress was detected in the brain tissue from rats with hyperprolinemia. Antioxidants such as vitamin E, vitamin C, and glutathione may be effective therapeutic agents in this disorder. van de Ven et al. suggested that these agents should be used for hyperprolinemia in patients as young as possible.37
If acute manifestations of hyperprolinemia such as convulsions are controlled safely, the prognosis for these disorders is good. Normal development can be expected, although the long-term outcome in terms of neurological disturbances is unknown. In general, individuals with hyperprolinemia should be monitored closely due to their frequent behavioral problems.38
Conclusions
Hereditary hyperprolinemia is a very rare disease in Japan. The patients that have been identified have a variety of neurological abnormalities. Screening and investigation of healthy individuals as well as patients with relatively common disorders such as developmental disabilities, epilepsy, schizophrenia, and behavioral problems will be important in Japan. Analysis of plasma amino acids can be used to identify HPI and HPII. The current study has presented diagnostic criteria for these diseases. In addition, a strong relationship between proline metabolism and tumor growth, as well as hypoalimentation stress reaction and fetal growth has been reported. Further studies will provide additional insights into the problems associated with hyperprolinemia, and may lead to new therapeutic strategies.
Acknowledgments
The work was supported by a Grant-in-Aid from the Ministry of Health, Labour and Welfare of Japan. We thank Dr Torayuki Okuyama from National Center for Child Health and Development, Dr Kenji Ihara from Kyusyu University, Dr Tomiko Kuhara at Kanazawa Medical University, Dr Yasutsugu Chinen from Ryukyu University and Dr Toshihiko Ando from Ajinomoto Co. Ltd for useful discussions and information.
References
- 1.Phang JM, Hu C-A, Valle D. Disorders of proline and hydroxyproline metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 1821–1838. 8th edn. [Google Scholar]
- 2.Mitsubuchi H, Nakamura K, Matsumoto S, Endo F. Inborn errors of proline metabolism. J. Nutr. 2008;138:2016S–2020. doi: 10.1093/jn/138.10.2016S. [DOI] [PubMed] [Google Scholar]
- 3.Schafer IA, Scriver CR, Efron ML. Familial hyperprolinemia, cerebral dysfunction and renal anomalies occurring in a family with hereditary nephropathy and deafness. N. Engl. J. Med. 1962;267:51–60. doi: 10.1056/NEJM196207122670201. [DOI] [PubMed] [Google Scholar]
- 4.Kopelman H, Asatoor AM, Milne MD. Hyperprolinaemia and hereditary nephritis. Lancet. 1964;13:1075–1079. doi: 10.1016/s0140-6736(64)92605-4. [DOI] [PubMed] [Google Scholar]
- 5.Efron ML. Familial hyperprolinemia. Report of a second case, associated with congenital renal malformations, hereditary hematuria and mild mental retardation, with demonstration of an enzyme defect. N. Engl. J. Med. 1965;272:1243–1254. doi: 10.1056/NEJM196506172722401. [DOI] [PubMed] [Google Scholar]
- 6.Rokkones T, Loken AC. Congenital renal dysplasia, retinal dysplasia and mental retardation associated with hyperprolinuria and hyper-OH-prolinuria. Acta Paediatr. Scand. 1968;57:225–229. doi: 10.1111/j.1651-2227.1968.tb04682.x. [DOI] [PubMed] [Google Scholar]
- 7.Perry TL, Hardwick DF, Lowry RB, Hansen S. Hyperprolinaemia in two successive generations of a North American Indian family. Ann. Hum. Genet. 1968;31:401–407. doi: 10.1111/j.1469-1809.1968.tb00573.x. [DOI] [PubMed] [Google Scholar]
- 8.Woody NC, Snyder CH, Harris JA. Hyperprolinemia: Clinical and biochemical family study. Pediatrics. 1969;44:554–563. [PubMed] [Google Scholar]
- 9.Fontaine G, Farriaux JP, Dautrevaux M. Type I hyperprolinemia. Study of a familial case. Helv. Paediatr. Acta. 1970;25:165–175. [PubMed] [Google Scholar]
- 10.Hainaut H, Hariga J, Willems C, Heusden A, Chapelle P. Familial essential hyperprolinemia. Presse Med. 1971;79:945–948. [PubMed] [Google Scholar]
- 11.Mollica F, Pavone L, Antener I. Familial hyperprolinemia without mental retardation and hereditary nephropathy. Monogr. Hum. Genet. 1972;6:144–145. doi: 10.1159/000392676. [DOI] [PubMed] [Google Scholar]
- 12.Potter JL, Waickman FJ. Hyperprolinemia. I. A study of a large family. J. Pediatr. 1973;83:635–638. doi: 10.1016/s0022-3476(73)80230-6. [DOI] [PubMed] [Google Scholar]
- 13.Steinlin M, Boltshauser E, Steinmann B, Wichmann W, Niemeyer G. Hyperprolinaemia type I and white matter disease: Coincidence or causal relationship? Eur. J. Pediatr. 1989;149:40–42. doi: 10.1007/BF02024332. [DOI] [PubMed] [Google Scholar]
- 14.Humbertclaude V, Rivier F, Roubertie A, et al. Is hyperprolinemia type I actually a benign trait? Report of a case with severe neurologic involvement and vigabatrin intolerance. J. Child Neurol. 2001;16:622–623. doi: 10.1177/088307380101600820. [DOI] [PubMed] [Google Scholar]
- 15.Campbell HD, Webb GC, Young IG. A human homologue of the Drosophila melanogaster sluggish-A (proline oxidase) gene maps to 22q11.2, and is a candidate gene for type-I hyperprolinaemia. Hum. Genet. 1997;101:69–74. doi: 10.1007/s004390050589. [DOI] [PubMed] [Google Scholar]
- 16.Jaeken J, Goemans N, Fryns JP, Farncois I, Zegher F. Association of hyperprolinaemia type I and heparin cofactor II deficiency with CATCH 22 syndrome: Evidence for a contiguous gene syndrome locating the proline oxidase gene. J. Inherit. Metab. Dis. 1996;19:275–277. doi: 10.1007/BF01799254. [DOI] [PubMed] [Google Scholar]
- 17.Jacquet H, Raux G, Thibaut F, et al. PRODH mutations and hyperprolinemia in a subset of schizophrenic patients. Hum. Mol. Genet. 2002;11:2243–2249. doi: 10.1093/hmg/11.19.2243. [DOI] [PubMed] [Google Scholar]
- 18.Jacquet H, Berthelot J, Bonnemains C, et al. The severe form of type I hyperprolinaemia results from homozygous inactivation of the PRODH gene. Med. Genet. 2003;40:e7. doi: 10.1136/jmg.40.1.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raux G, Bumsel E, Hecketsweiler B, et al. Involvement of hyperprolinemia in cognitive and psychiatric features of the 22q11 deletion syndrome. Hum. Mol. Genet. 2007;16:83–91. doi: 10.1093/hmg/ddl443. [DOI] [PubMed] [Google Scholar]
- 20.Afenjar A, Moutard ML, Doummar D, et al. Early neurological phenotype in 4 children with biallelic PRODH mutations. Brain Dev. 2007;29:547–552. doi: 10.1016/j.braindev.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 21.Shprintzen RJ, Goldberg RB, Young D, Wolford L. The velo-cardio-facial syndrome: A clinical and genetic analysis. Pediatrics. 1981;67:167–172. [PubMed] [Google Scholar]
- 22.Guilmatre A, Legallic S, Steel G, et al. Type I hyperprolinemia: Genotype/phenotype correlations. Hum. Mutat. 2010;31:961–965. doi: 10.1002/humu.21296. [DOI] [PubMed] [Google Scholar]
- 23.Valle DL, Phang JM, Goodman SI. Type 2 hyperprolinemia: Absence of delta1-pyrroline-5-carboxylic acid dehydrogenase activity. Science. 1974;185:1053–1054. doi: 10.1126/science.185.4156.1053. [DOI] [PubMed] [Google Scholar]
- 24.Geraghty MT, Vaughn D, Nicholson AJ, et al. Mutations in the delta1-pyrroline 5-carboxylate dehydrogenase gene cause type II hyperprolinemia. Hum. Mol. Genet. 1998;7:1411–1415. doi: 10.1093/hmg/7.9.1411. [DOI] [PubMed] [Google Scholar]
- 25.Flynn MP, Martin MC, Moore PT, Stafford JA, Fleming GA, Phang JM. Type II hyperprolinaemia in a pedigree of Irish travellers (nomads) Arch. Dis. Child. 1989;64:1699–1707. doi: 10.1136/adc.64.12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bender HU, Almashanu S, Steel G, et al. Functional consequences of PRODH missense mutations. Am. J. Hum. Genet. 2005;76:409–420. doi: 10.1086/428142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu W, Le A, Hancock C, et al. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl Acad. Sci. USA. 2012;109:8983–8988. doi: 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu W, Phang JM. Proline dehydrogenase (oxidase) in cancer. Biofactors. 2012;38:398–406. doi: 10.1002/biof.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kato Y, Ihara K, Miyako K, Kuhara T, Inoue Y, Hara T. Acute encephalopathy associated with influenza virus infection in a patient with hyperprolinaemia type II. J. Inherit. Metab. Dis. 2005;28:789–790. doi: 10.1007/s10545-005-0048-3. [DOI] [PubMed] [Google Scholar]
- 30.Kuhara T. Noninvasive human metabolome analysis for differential diagnosis of inborn errors of metabolism. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2007;855:42–50. doi: 10.1016/j.jchromb.2007.03.031. [DOI] [PubMed] [Google Scholar]
- 31.Oyanagi K, Tsuchiyama A, Itakura Y, et al. Clinical, biochemical and enzymatic studies in type I hyperprolinemia associated with chromosomal abnormality. Tohoku J. Exp. Med. 1987;151:465–475. doi: 10.1620/tjem.151.465. [DOI] [PubMed] [Google Scholar]
- 32.Ishikawa Y, Kameda K, Okabe M, Ima T, Nagaoka M, Minami R. A case of type I hyperprolinemia associated with photogenic epilepsy. No To Hattatsu. 1991;23:81–86. [PubMed] [Google Scholar]
- 33.Jang MA, Kim BC, Ki CS, et al. Identification of PRODH mutations in Korean neonates with type I hyperprolinemia. Ann. Clin. Lab. Sci. 2013;43:31–36. [PubMed] [Google Scholar]
- 34.Harries JT, Piesowicz AT, Seakins JW, Francis DE, Wolff OH. Low proline diet in type I hyperprolinaemia. Arch. Dis. Child. 1971;46:72–81. doi: 10.1136/adc.46.245.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farrant RD, Walker V, Mills GA, Mellor JM, Langley GJ. Pyridoxal phosphate de-activation by pyrroline-5-carboxylic acid. Increased risk of vitamin B6 deficiency and seizures in hyperprolinemia type II. J. Biol. Chem. 2001;276:15107–15116. doi: 10.1074/jbc.M010860200. [DOI] [PubMed] [Google Scholar]
- 36.Walker V, Mills GA, Peters SA, Merton WL. Fits, pyridoxine, and hyperprolinaemia type II. Arch. Dis. Child. 2000;82:236–237. doi: 10.1136/adc.82.3.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van de Ven S, Gardeitchik T, Kouwenberg D, Kluijtmans L, Wevers R, Morava E. Long-term clinical outcome, therapy and mild mitochondrial dysfunction in hyperprolinemia. J. Inherit. Metab. Dis. 2014;37:383–390. doi: 10.1007/s10545-013-9660-9. [DOI] [PubMed] [Google Scholar]
- 38.Wyse AT, Netto CA. Behavioral and neurochemical effects of proline. Metab. Brain Dis. 2011;26:159–172. doi: 10.1007/s11011-011-9246-x. [DOI] [PubMed] [Google Scholar]