

Abstract
Epigenetic modification of cytosine methylation states can be elicited by environmental stresses and may be a key process affecting phenotypic plasticity and adaptation. Parasites are potent stressors with profound physiological and ecological effects on their host, but there is little understanding in how parasites may influence host methylation states. Here, we estimate epigenetic diversity and differentiation among 21 populations of red grouse (Lagopus lagopus scotica) in north-east Scotland and test for association of gastrointestinal parasite load (caecal nematode Trichostrongylus tenuis) with hepatic genome-wide and locus-specific methylation states. Following methylation-sensitive AFLP (MSAP), 129 bands, representing 73 methylation-susceptible and 56 nonmethylated epiloci, were scored across 234 individuals. The populations differed significantly in genome-wide methylation levels and were also significantly epigenetically (FSC = 0.0227; P < 0.001) and genetically (FSC = 0.0058; P < 0.001) differentiated. Parasite load was not associated with either genome-wide methylation levels or epigenetic differentiation. Instead, we found eight disproportionately differentiated epilocus-specific methylation states (FST outliers) using bayescan software and significant positive and negative association of 35 methylation states with parasite load from bespoke generalized estimating equations (GEE), simple logistic regression (sam) and Bayesian environmental analysis (bayenv2). Following Sanger sequencing, genome mapping and geneontology (go) annotation, some of these epiloci were linked to genes involved in regulation of cell cycle, signalling, metabolism, immune system and notably rRNA methylation, histone acetylation and small RNAs. These findings demonstrate an epigenetic signature of parasite load in populations of a wild bird and suggest intriguing physiological effects of parasite-associated cytosine methylation.
Keywords: DNA methylation, epigenetics, genotype-environment association, host-parasite interactions, induced phenotypes, methylation-sensitive AFLP
Introduction
The traditional paradigm that phenotypic variability and evolutionary change are consequences solely of DNA sequence variation is becoming increasingly challenged (Jablonka & Lamb 2007; Bonduriansky & Day 2008; Danchin et al. 2011; Mesoudi et al. 2013). The emerging field of epigenetics is concerned with dynamic, yet mitotically and sometimes meiotically stable, regulatory patterns of gene expression and chromatin remodelling in the absence of nucleotide sequence variation (Jablonka & Raz 2009; Massicotte et al. 2011; Alabert & Groth 2012). From a molecular ecology perspective, these phenomena complement the DNA sequence-based systems hitherto examined and are likely to provide new insights into the underpinning, regulation and evolution of phenotypic traits (Bossdorf et al. 2008; Angers et al. 2010; Richards et al. 2010; Duncan et al. 2014).
The most intensively studied epigenetic mechanism is enzymatically mediated attachment of a methyl group to cytosine or adenine nucleotides (Angers et al. 2010). Such DNA methylation is taxonomically widespread, but its extent and function are highly taxon specific (Suzuki & Bird 2008; Angers et al. 2010; Jones 2012). In plants, for example, cytosine in any trinucleotide (CpNpN) may be methylated, whereas in vertebrates, methylation is almost exclusively limited to cytosine in CpG dinucleotide sites (Angers et al. 2010; Fulneček & Kovařík 2014). Cytosine methylation may display a number of different effects depending on functional sequence context. Increased methylation of CpG islands in gene promoters is often associated with a decrease in the expression of those genes (Angers et al. 2010; Jones 2012; Duncan et al. 2014). In contrast, methylation in gene bodies or noncoding regions may, for example, silence transposable elements or genomic parasitic sequences (Suzuki & Bird 2008; Zemach et al. 2010; Jones 2012), provide mutational hot spots through increased deamination rate of methylated cytosine (Lutsenko & Bhagwat 1999; Poole et al. 2003; Jones 2012), or recruit protein complexes and factors that are involved in chromatin remodelling (Jaenisch & Bird 2003; Bannister & Kouzarides 2011).
Mitotic stability of methylation patterns during ontogenesis is a key mechanism that not only mediates cell differentiation, but in concert with malleability of methylation states also provides a framework for environmental factors to influence phenotype expression during early developmental stages (Bird 2002; Skinner 2011; Feil & Fraga 2012; D’Urso & Brickner 2014). Moreover, compelling evidence is accumulating for environmentally induced changes in methylation patterns long after ontogenesis (Duncan et al. 2014). Not only may such changes underpin phenotypic plasticity during an individual's lifetime (Jaenisch & Bird 2003; Bossdorf et al. 2008; Angers et al. 2010; Stevenson & Prendergast 2013; Duncan et al. 2014), but some of these patterns may also be vertically transmitted, either directly through meiotic stability of methylation patterns or indirectly by transmission of extragenomic molecules in gametes (Jablonka & Raz 2009; Petronis 2010; Skinner 2011; Smith & Ritchie 2013; Duncan et al. 2014). Recent studies highlight a role for methylation in broad eco-evolutionary processes such as biological invasion (Richards et al. 2012), sexual selection (Crews et al. 2007), domestication (Xiang et al. 2013), inbreeding depression (Vergeer et al. 2012), seasonal timing of physiology (Stevenson & Prendergast 2013), transition between maturation stages (Morán & Pérez-Figueroa 2011) and reproductive labour division in social insects (Amarasinghe et al. 2014). On a population epigenetics level, differentiation of methylation states is frequently observed among populations in different environments (Herrera & Bazaga 2010; Lira-Medeiros et al. 2010; Liu et al. 2012; Schulz et al. 2013). Such epigenetic differentiation has also been demonstrated to be meiotically persistent (Salmon et al. 2008; Herrera et al., 2013,2014), implying a potential role for local adaptation and speciation (Bossdorf et al. 2008; Roux et al. 2011; Richards et al. 2012; Smith & Ritchie 2013).
Particularly useful insights on the mechanistic contribution of epigenetics to plasticity and adaptation come from exploring the epigenetic effects of particular environmental stresses (Feil & Fraga 2012). For example, osmotic stress caused by transition from fresh water to sea water induces methylation-mediated acclimation processes in brown trout (Morán et al. 2013). Similarly, methylation-mediated nutritional plasticity as a result of changes in the nutritional environment has been found in a nectar-eating yeast (Herrera et al. 2012) and in horned beetle larvae (Snell-Rood et al. 2013). Numerous studies on plants have identified methylation effects of various abiotic stressors, for example temperature (Paun et al. 2010), nutrient availability (Verhoeven et al. 2010), water availability (Paun et al. 2010) and osmotic stress (Chinnusamy & Zhu 2009; Tan 2010). Compelling evidence for methylation responses to biotic factors, such as pathogens and herbivory, also comes from plant studies (Herrera & Bazaga 2011; Dowen et al. 2012), where even the experimental application of damage-associated plant hormones elicits heritable methylation changes associated with a concomitant stress response (Verhoeven et al. 2010). Parasites are extremely potent stressors with profound eco-evolutionary importance (Bérénos et al. 2011; Gómez-Díaz et al. 2012; Mostowy & Engelstädter 2012), yet little is known about parasite-associated epigenetic effects in animal hosts. Notably, helminth parasites have been linked to carcinogenesis (Fried et al. 2011), most prominently in bladder cancer, where patients with schistosome infection have consistently different tumoral methylation patterns compared with noninfected patients (Gutiérrez et al. 2004). Considering the large gamut of physiological and evolutionary consequences of parasite infection, studying host–parasite systems in an ecological epigenetics context promises to be an exciting, yet challenging, avenue of research (Poulin & Thomas 2008; Gómez-Díaz et al. 2012; Biron & Loxdale 2013; Poulin 2013).
An extremely well-studied natural host–parasite system that is well suited for exploring ecological epigenetics is the red grouse (Lagopus lagopus scotica) and its gastrointestinal nematode parasite Trichostrongylus tenuis. Red grouse are endemic to the heather moorlands of upland Scotland and Northern England, where their environment is intensively managed for sport shooting purposes (Martínez-Padilla et al. 2014). Male grouse are highly philopatric and territorial, particularly in autumn when elevated testosterone enhances aggression among young cocks and their kin during recruitment. Young cocks attempt to establish a territory in the immediate vicinity of their kin, resulting in spatial kin structures within populations (Watson et al. 1994; Piertney et al. 1999, 2000; MacColl et al. 2000) and also some degree of genetic structure among populations (Piertney et al. 1998, 2000). T. tenuis exhibits a direct life cycle and is a major driver of red grouse ecology (Martínez-Padilla et al. 2014). Infectious larvae are ingested with heather shoots and establish in the caecum where adult parasites cause haemorrhaging that results in poor physiological condition and compromised survival and fecundity (Watson et al. 1987; Hudson et al. 1992; Delahay et al. 1995; Delahay & Moss 1996). At least 90% of grouse in a population are infected (Wilson 1983) and, although a specific immune response is mounted (Webster et al. 2011a), grouse typically cannot acquire full immunity and therefore continue to bear specific parasite burdens that vary considerably among individuals (Shaw & Moss 1989).
Chronic parasite infection has marked effects on grouse behaviour and physiology. High parasite load reduces territorial aggression in male grouse (Fox & Hudson 2001; Mougeot et al. 2005a), which has knock-on effects on recruitment and kin structure (Moss & Watson 1991; Mougeot et al. 2005b) and may ultimately contribute to the instability of grouse population dynamics (Hudson et al. 1998; Redpath et al. 2006; Martínez-Padilla et al. 2014). Moreover, parasite infection has a range of physiological consequences that underpin sexual selection processes in grouse populations (e.g. Mougeot et al. 2004; Seivwright et al. 2005; Martínez-Padilla et al. 2007, 2010; Vergara et al. 2012). Both male and female grouse possess carotenoid-based supra-orbital combs that function in males as testosterone-dependent signals of condition. Parasite load is intricately linked to various components of condition, such as immune function (Mougeot et al. 2004; Mougeot & Redpath 2004; Mougeot 2008), oxidative status (Mougeot et al. 2009, 2010a) or physiological stress (Bortolotti et al. 2009; Mougeot et al. 2010b), suggesting a key role in signal modulation. Indeed, parasite infection not only elicits transcriptomic up-regulation of immune system processes and stress responses (Webster et al. 2011a, b), but also interacts with testosterone to depresses immunity and oxidative damage responses consistent with transcriptomically mediated handicap mechanisms (Wenzel et al. 2013). Taken together, these studies highlight a key role of parasites to alter physiological processes in red grouse and suggest changes in gene expression as an important mechanism. Such gene expression changes could potentially be regulated or modulated by epigenetic mechanisms such as cytosine methylation, through, for example, changes in gene promoter methylation or chromatin remodelling (Bird 2002; Jaenisch & Bird 2003; Angers et al. 2010; Jones 2012). More generally, parasite-associated changes in methylation patterns in the absence of genetic variation could act as a regulatory component of physiological stress responses to parasite infection (Poulin & Thomas 2008; Gómez-Díaz et al. 2012).
Paramount to approaching these intriguing ideas in an ecological context is exploring correlational epigenetic signatures of parasite load in natural populations. Here, we present an ecological epigenetics study on parasite-associated genome-wide cytosine methylation patterns in a landscape system of red grouse populations at high autumnal testosterone levels in north-east Scotland. Our objectives are threefold: First, we epigenotype individuals at genome-wide anonymous CpG sites using methylation-sensitive AFLP (MSAP) to estimate methylation levels and patterns of epigenetic differentiation among populations. Second, we test for associations of the identified genome-wide and locus-specific methylation patterns with parasite load. Finally, we sequence those identified loci to ascertain the potential physiological effects of parasite-associated methylation changes. Our results highlight significant epigenetic differentiation among the sampled grouse populations and significant association of parasite load with locus-specific methylation states, but not genome-wide methylation levels or spatial epigenetic structure.
Materials and methods
Sample collection
A total of 234 shot grouse were sampled in autumn 2012 following driven or walked-up sporting shoots at 21 sites near Deeside, Aberdeenshire (Fig.1, Table1). Age was determined as ‘young’ (<1 year) or ‘old’ (>1 year), and, where possible, only old birds were sampled in order to minimize bias by sampling of kin groups. Weight was measured to the nearest 10 g with a spring balance, and supra-orbital comb size (width and length) was measured to the nearest mm. Caecal content samples were taken for parasite load estimation following faecal parasite egg counts (standard McMaster chamber slide method; Seivwright et al. 2004). Liver samples were taken for DNA extraction because liver is a homoeostatically and immunologically important organ that is well suited to explore systemic parasite-specific effects (Racanelli & Rehermann 2006; Webster et al. 2011a) and typically yields large amounts of contaminant-free high-quality DNA necessary for restriction-based assays (Benjak et al. 2006; Wong et al. 2012). DNA was extracted from 2 to 3 c. 2 mm3 shreds of liver tissue according to Hogan et al. (2008). DNA quality and quantity were assessed with a NanoDrop ND-1000 spectrophotometer, and extracts were diluted to c. 100 ng/μL. PCR-based sex determination using a gonosome-linked locus (Griffiths et al. 1998) was performed as described in Wenzel et al. (2012).
Fig 1.
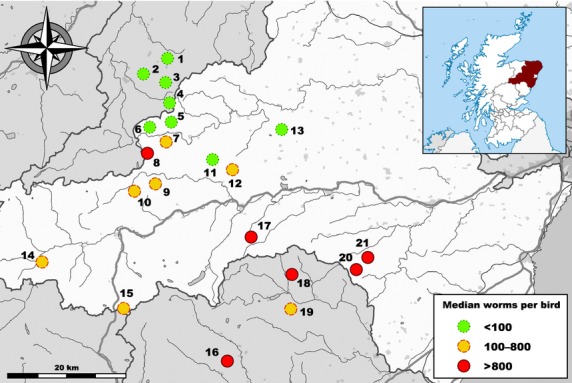
Sites in Aberdeenshire, Angus and Moray that were sampled following grouse shoots in autumn 2012. Detailed locations, sample sizes and parasite loads are given in Table1.
Table 1.
Sampling locations, sample sizes (M = male, F = female, Y = young) and parasite loads (median worms per bird with 25% and 75% quantiles)
| Sampling locations | Sample sizes | Worms per bird | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Site | Estate | Long. | Lat. | Total | M | F | Y | 25% | Median | 75% |
| 1 | Glenlivet | 57.29 | −3.18 | 10 | 4 | 6 | 0 | 4 | 4 | 632 |
| 2 | Glenlivet | 57.25 | −3.28 | 10 | 7 | 3 | 0 | 4 | 4 | 4 |
| 3 | Edinglassie | 57.24 | −3.20 | 10 | 6 | 4 | 0 | 4 | 4 | 4 |
| 4 | Edinglassie | 57.21 | −3.19 | 10 | 7 | 3 | 0 | 4 | 4 | 4 |
| 5 | Allargue | 57.19 | −3.29 | 10 | 4 | 6 | 0 | 4 | 4 | 4 |
| 6 | Allargue | 57.19 | −3.23 | 10 | 6 | 4 | 10 | 4 | 4 | 4 |
| 7 | Delnadamph | 57.16 | −3.26 | 10 | 5 | 5 | 0 | 380 | 582 | 1394 |
| 8 | Delnadamph | 57.14 | −3.30 | 11 | 9 | 2 | 0 | 947 | 1215 | 1715 |
| 9 | Invercauld | 57.10 | −3.29 | 10 | 3 | 7 | 5 | 4 | 513 | 1586 |
| 10 | Invercauld | 57.08 | −3.35 | 10 | 5 | 5 | 5 | 150 | 500 | 2264 |
| 11 | Dinnet | 57.12 | −3.11 | 10 | 8 | 2 | 0 | 4 | 40 | 112 |
| 12 | Dinnet | 57.11 | −3.06 | 10 | 6 | 4 | 0 | 4 | 180 | 556 |
| 13 | Tillypronie | 57.18 | −2.94 | 9 | 3 | 6 | 8 | 4 | 78 | 200 |
| 14 | Mar Lodge | 56.95 | −3.66 | 11 | 6 | 5 | 3 | 315 | 676 | 1400 |
| 15 | Invercauld | 56.87 | −3.40 | 15 | 13 | 2 | 0 | 222 | 602 | 800 |
| 16 | Airlie | 56.81 | −3.08 | 18 | 13 | 5 | 0 | 812 | 2222 | 4069 |
| 17 | Glen Muick | 56.99 | −3.01 | 20 | 11 | 9 | 0 | 674 | 1586 | 2609 |
| 18 | Invermark | 56.94 | −2.89 | 10 | 6 | 4 | 0 | 600 | 1084 | 1380 |
| 19 | Invermark | 56.89 | −2.89 | 10 | 4 | 6 | 0 | 232 | 603 | 694 |
| 20 | Glen Dye | 56.95 | −2.72 | 10 | 6 | 4 | 5 | 372 | 813 | 1141 |
| 21 | Glen Dye | 56.96 | −2.69 | 10 | 6 | 4 | 5 | 358 | 1006 | 1566 |
| 234 | 138 | 96 | 41 | |||||||
Methylation-sensitive AFLP
Methylation-sensitive AFLP (Reyna-López et al. 1997) allows for assaying methylation at anonymous 5’-CCGG restriction sites in a similar fashion to classic AFLP assays of genetic variation (Vos et al. 1995). Genomic DNA is restricted in two parallel digests per sample, each containing EcoRI and one of the two isoschizomers MspI and HpaII that differ in sensitivity to cytosine methylation configurations of the 5’-CCGG restriction site (Herrera & Bazaga 2010; Pérez-Figueroa 2013).
Approximately 300 ng of genomic DNA was digested for 3 h at 37 °C in each of two parallel reactions, containing 5 U of EcoRI-HF and 5 U of either MspI or HpaII (all New England Biolabs) in a total volume of 10 μL. Ligation adaptors were prepared from single-stranded oligonucleotides (EcoRI: 5’–CTCGTAGACTGCGTACC–3’ and 5’–AATTGGTACGCAGTCTAC–3’, Vos et al. 1995; MspI/HpaII: 5’–GACGATGAGTCTAGAA–3’ and 5’–CGTTCTAGACTCATC–3’, Xu et al. 2000) by mixing equal amounts followed by incubation in a G-Storm GS-1 thermocycler at 95 °C for 5 min and slowly cooling down to room temperature within c. 30 min. A ligation mix containing 5 pmol of EcoRI adaptor, 50 pmol of MspI/HpaII adaptor and 1 U of T4 DNA ligase (New England Biolabs) in a volume of 5 μL was added to the digests and incubated at 20 °C overnight. The enzymes were then heat-deactivated at 65 °C for 20 min.
Preselective PCRs were carried out in a total volume of 10 μL containing 1 μL of the digestion/ligation reaction, 0.5 μm each of EcoRI+A (5’–GACTGCGTACCAATTCA–3’; Vos et al. 1995) and MspI/HpaII+T (5’–GATGAGTCTAGAACGGT–3’; Xu et al. 2000) preselective primers, 0.15 U of VELOCITY DNA polymerase (Bioline), 1X VELOCITY HI-FI buffer (containing 2 mm MgCl2), and 0.2 mm of each nucleotide. The PCR profile comprised an initial elongation step at 72 °C for 2 min followed by 30 cycles of denaturation at 95 °C for 20 s, annealing at 56 °C for 30 s and elongation at 72 °C for 1 min, and a final elongation step at 60 °C for 2 min.
Eight primer combinations (Table2) were chosen for selective PCR based on the criteria of fragment number, ease of scoring and levels of polymorphism across four test individuals representing the whole study system. Selective PCRs were carried out in a total volume of 20 μL containing 2 μL of preselective PCR product (diluted 1:10), 0.5 μm of each primer, 0.4 U of Taq DNA polymerase (Qiagen), 1X CoralLoad PCR buffer, 2.5 mm MgCl2 and 0.2 mm of each nucleotide. The PCR profile comprised an initial denaturation step at 95 °C for 2 min, 13 TouchDown (Don et al. 1991) cycles of denaturation at 95 °C for 20 s, annealing decreasing from 65 °C to 56 °C for 30 s and elongation at 72 °C for 1 min, 23 cycles of denaturation at 95 °C for 20 s, annealing at 56 °C for 30 s and elongation at 72 °C for 1 min (increasing by 2 s per cycle), and a final elongation step at 72 °C for 2 min. Two 10 μL aliquots of PCR product were loaded onto two independent 2% agarose–sodium borate gels, electrophoretically separated out for 60 min at 12 V/cm and poststained with Midori Green DNA stain. Band profiles between 125 bp and 600 bp were scored by eye using high-contrast gel photographs.
Table 2.
Selective primer combinations used in methylation-sensitive AFLP (MSAP), numbers of scored bands, estimated scoring-error rates and consequential classification into methylation-susceptible (MSL) and nonmethylated (NML) loci
| EcoRI | MspI/HpaII | Bands | Error rate | MSL | NML |
|---|---|---|---|---|---|
| GACTGCGTACCAATTCACA | GATGAGTCTAGAACGGTTGA | 18 | 0.17 | 9 | 9 |
| GACTGCGTACCAATTCACA | GATGAGTCTAGAACGGTTCA | 17 | 0.14 | 12 | 5 |
| GACTGCGTACCAATTCACA | GATGAGTCTAGAACGGTTA | 17 | 0.13 | 9 | 8 |
| GACTGCGTACCAATTCAGA | GATGAGTCTAGAACGGTTGA | 13 | 0.17 | 5 | 8 |
| GACTGCGTACCAATTCAGA | GATGAGTCTAGAACGGTGTT | 17 | 0.12 | 10 | 7 |
| GACTGCGTACCAATTCAGA | GATGAGTCTAGAACGGTAC | 18 | 0.15 | 9 | 9 |
| GACTGCGTACCAATTCATC | GATGAGTCTAGAACGGTAGA | 15 | 0.17 | 9 | 6 |
| GACTGCGTACCAATTCATC | GATGAGTCTAGAACGGTTCA | 14 | 0.21 | 10 | 4 |
| 129 | 73 | 56 |
Statistical analysis
Individual band presence or absence states compared between the EcoRI-MspI and EcoRI-HpaII digests occur in four possible patterns: +/+, –/+, +/– and –/–. In the case of vertebrates, where methylation almost exclusively occurs on the inner cytosine (CpG) of the restriction site (Angers et al. 2010; Fulneček & Kovařík 2014), these patterns are interpreted as absence of methylation, hemimethylation (methylation present on one strand only), methylation of the inner cytosine on both strands (hereafter: full methylation) and absence of restriction site, respectively (Xu et al. 2000; Pérez-Figueroa 2013). Epigenetic variation can then be assessed from bands with polymorphic methylation states, whereas information on genotypic variation can be extracted from polymorphic bands with methylation states below a particular scoring-error threshold, representing band presence (+/+, –/+ or +/–) or absence (–/–) equivalent to classic AFLP loci (Herrera & Bazaga 2010; Pérez-Figueroa 2013). Total primer combination-specific error thresholds eT = eM + eH − 2eMeH (Herrera & Bazaga 2010) were estimated from discordant scores in MspI (eM) and HpaII (eH) profiles of 26 individuals that were processed twice from the same DNA extract. Using the r package msap (Pérez-Figueroa 2013) in r 3.0.3 (R Core Team 2014), bands with methylation frequencies (–/+ or +/– states) above or below these error thresholds (eT) were then classified as methylation-susceptible (MSL) or nonmethylated (NML) loci, respectively. Information content in MSL and NML was estimated using Shannon's diversity index, and differences were tested for with a Wilcoxon rank-sum test. Independence between MSL and NML information was tested for by applying a Mantel test with 105 randomizations on individual-by-individual MSL and NML distance matrices (Pérez-Figueroa 2013).
Genome-wide levels of full methylation, hemimethylation and absence of methylation were estimated for each individual based on methylation-state frequencies in MSL. Differences in median methylation levels among populations were tested for significance using Kruskal–Wallis tests. Associations of methylation levels with individual parasite load were tested for by Spearman rank correlation. Epigenetic (MSL) and genetic (NML) differentiation among populations was visualized by principal coordinates analysis (PCoA). Using two-level amova with 105 randomizations in the r package pegas (Paradis 2010), epigenetic (MSL) and genetic (NML) variances were partitioned into hierarchical components by assigning the populations to three similarly sized groups according to median parasite load (Fig.1). Pairwise epigenetic and genetic differentiation was estimated and tested for using amova-based differentiation statistics (FST). Multiple testing was accounted for by calculating false discovery rate (FDR) adjusted P-values (= q-values) using the r package fdrtool (Strimmer 2008). Associations of pairwise differentiation with geographical distances and differences in median parasite load were examined using Mantel tests with 105 randomizations.
Disproportionately differentiated methylation states were identified using FST outlier approaches (Paun et al. 2010; Chwedorzewska & Bednarek 2012; Schrey et al. 2012). The methylation states of each band were coded as up to three binary variables, each of which represents either the fully methylated, hemimethylated or unmethylated state of the epilocus (‘Mix2’ algorithm in the r script msap_calc; Schulz et al. 2013). Methylation states with a frequency below 0.05 or above 0.95 and states derived from an NML were removed. Linkage disequilibrium was tested for in genepop 4.2.1 (Raymond & Rousset 1995; Rousset 2008) with 10 000 MCMC dememorizations, 100 batches of 5000 MCMC iterations and a significance threshold of α = 0.05. Tests for FST outliers were carried out using bayescan 2.0 (Foll & Gaggiotti 2008; Pérez-Figueroa et al. 2010), running 2·106 iterations (run length 105; thinning interval 20) after 20 pilot runs (104 iterations each) and a burn-in of 5·105, and selecting outliers at q≤0.05. For comparison and corroboration, the analysis was repeated using the dfdist algorithm implemented in mcheza (Antao & Beaumont 2011), running 105 simulations with ‘neutral FST’ and ‘force mean FST’ options, and selecting loci outside the upper tail of the 95% CI.
Associations of binary methylation states with individual parasite load (e.g. Paun et al. 2010; Herrera & Bazaga 2011) were examined by fitting multiple generalized estimating equations (GEE) with exchangeable correlation structure within populations using geepack (Halekoh et al. 2006), thus accounting for within-population correlation of methylation states caused by a shared environment due to kin and population structure (Piertney et al. 1998, 1999, 2000). Potential effects of sex (Boks et al. 2009; Liu et al. 2010), age (Fraga et al. 2005; Boks et al. 2009) and physiological condition (Fitzpatrick & Wilson 2003; Meaney & Szyf 2005; Franco et al. 2008) on methylation were accounted for by including supra-orbital comb area as an additional explanatory variable. Comb area was strongly associated with sex (t = 7.23; P < 0.001), age (t = −2.65; P = 0.009) and weight (t = 3.59; P < 0.001) but not with parasite load (t = 1.39; P = 0.17). These relationships confirm comb area as a sexual signal and an indicator of physiological condition that is not necessarily reflected in parasite load (Mougeot et al. 2004, 2009; Martínez-Padilla et al. 2010), but instead in testosterone-dependent immune function, oxidative status or physiological stress (Mougeot & Redpath 2004; Bortolotti et al. 2009; Mougeot et al. 2010a). Including this single proxy variable rather than a range of interrelated variables reflects the biological relationships of the system and avoids multicollinearity in the model (Graham 2003).
The association analysis was repeated with two other software packages to provide congruence across different model approaches: First, using sam (Joost et al. 2008) which implements a logistic regression with a single explanatory variable and ignores any potential correlation among observations. Second, using bayenv2 (Günther & Coop 2013) which tests for association with a single explanatory variable using both a linear model and Spearman rank correlation while accounting for among-population structure through neutral parameterization by control data. Neutral parameterization was performed twice, using either NML as genetic AFLP loci or a set of 260 neutral SNPs (M. A. Wenzel et al., unpublished) for comparison and corroboration. bayenv2 was run for 106 iterations both for neutral parameterization and locus testing. Methylation states were considered to be meaningfully associated with parasite load if the regression coefficients (β1) of the GEE or sam models were outside the 5% / 95% percentiles of the distribution or if P ≦ 0.05. Analogous criteria for the bayenv models were |ρ| ≧ 0.2 or Bayes factor ≧ 2. FDR correction was made for the GEE and sam models, but significance after FDR correction (q ≦ 0.1) was taken as additional confidence rather than a strict criterion.
Functional characterization of epiloci
Those methylation states that were FST outliers or associated with parasite load according to at least one of the three model approaches were pooled, and the corresponding epiloci (MSAP bands) were identified. These bands were then gel-extracted, cloned and Sanger-sequenced to obtain locus identity and physiological functions as a means to ascertain how differential methylation at these loci may take effect.
Bands were picked from the gel using a 10 μL pipette tip, which was then placed in a 0.2 mL PCR tube containing 12 μL of water and incubated at room temperature for 30 min. Upon removal of the tip, 8 μL of PCR mixture was added; the final volume of 20 μL then contained 0.5 μm of each preselective PCR primer, 0.5 U of DNA polymerase (Sigma-Aldrich), 2.5 mm MgCl2, 10 mm Tris-HCl, 5 mm KCl, and 0.2 mm of each nucleotide. The PCR profile comprised an initial denaturation step at 95 °C for 2 min, 18 TouchDown (Don et al. 1991) cycles of denaturation at 95 °C for 15 s, annealing decreasing from 65 °C to 56 °C for 15 s and elongation at 72 °C for 20 s, 20 cycles of denaturation at 95 °C for 15 s, annealing at 56 °C for 15 s and elongation at 72 °C for 20 s, and a final elongation step at 72 °C for 2 min. PCR products were ligated into Promega pGEM®-T Easy Vector plasmids and transformed into JM109 competent cells according to the manufacturer's instructions. Colonies were screened using standard T7 and SP6 primers in a PCR mixture containing 0.5 μm of each primer, 0.5 U of DNA polymerase (Sigma-Aldrich), 2.5 mm MgCl2, 10 mm Tris-HCl, 5 mm KCl and 0.2 mm of each nucleotide. The PCR profile consisted of an initial denaturation step at 95 °C for 2 min followed by 35 cycles of denaturation at 95 °C for 20 s, annealing at 55 °C for 20 s and elongation at 72 °C for 50 s, and a final elongation step at 72 °C for 2 min. Three to four clones per band were Sanger-sequenced on an ABI 3730XL automatic capillary sequencer (Beckman Coulter Genomics, Takeley, UK).
Sequences were aligned and trimmed in geneious 7.1.2 (Drummond et al. 2014) and queried against the ncbi RefSeq protein database using blastx (Altschul et al. 1997). Additionally, as most MSAP loci are expected to fall outside coding regions (Caballero et al. 2013), the clone sequences were mapped to the chicken (galGal4) and turkey (melGal1) genomes using blat (Kent 2002). The genomic locations of the best hits (highest match score) were then used to identify the nearest ensembl annotations (retrieved from the UCSC table browser; Karolchik et al. 2004). Associated gene names, gene descriptions and geneontology terms (The Gene Ontology Consortium 2000) were retrieved from ensembl biomarts (Kinsella et al. 2011). Frequencies of each go annotation among the gene sequences were calculated using blast2go (Conesa et al. 2005; Conesa & Götz 2008) and custom r scripts.
Results
Estimated individual parasite loads among the 234 sampled birds ranged from 4 to 9283 worms per bird and population medians ranged from 4 to 2222 worms per bird (Table1). A total of 129 MSAP bands (13–18 per primer combination) were scored in all individuals. Error rates ranged from 0.12 to 0.21, resulting in 73 bands (c. 125–600 bp) to be classified as methylation-susceptible (MSL) and 56 (c. 125–500 bp) as nonmethylated (NML) loci (Table2). Of these, 62 (85%) and 33 (59%) were polymorphic, with marginally different Shannon diversity indices of I = 0.44 ± 0.22 SD and I = 0.36 ± 0.20 SD, respectively (W = 1248; P = 0.08). Individual-based epigenetic variation was independent of genetic variation (r = 0.02; P = 0.23).
The 21 populations varied significantly in median genome-wide full methylation (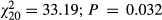 ), hemimethylation (
), hemimethylation (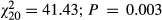 ) and absence of methylation (
) and absence of methylation (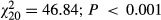 ), but there was no geographical pattern (Fig.2). Parasite load was weakly positively correlated with genome-wide hemimethylation (ρ = 0.15; P = 0.02), but not correlated with full methylation (ρ = −0.10; P = 0.11) or absence of methylation (ρ = −0.07; P = 0.29). Principal coordinates analysis of epigenetic and genetic variation explained only 4–6% of the total variation and showed substantial overlap among populations (Fig. S1, Supporting information). Nevertheless, global epigenetic differentiation among populations was significant (FSC = 0.0227; P < 0.001) and considerably stronger than genetic differentiation (FSC = 0.0058; P < 0.001). Pairwise epigenetic and genetic differentiation (FST) ranged from −0.012 to 0.091 (113 of 210 pairs significant at FDR < 0.05) and from −0.012 to 0.048 (16 of 210 pairs significant at FDR < 0.05), respectively (Fig.3). There was no isolation-by-distance pattern in either epigenetic (r = 0.02; P = 0.38) or genetic (r = −0.13; P = 0.87) differentiation. Seven populations that were not clustered geographically (locations 3, 4, 9, 10, 15, 18 and 19) showed disproportionate epigenetic differentiation compared with all other populations (Fig.3). Of these, populations 9 and 10 were also disproportionately genetically differentiated, causing a weak correlation between pairwise epigenetic and genetic differentiation matrices (r = 0.21; P = 0.06). Assigning populations to groups by median parasite load (Fig.1) did not explain any proportion of the molecular variances (Table3). Similarly, pairwise differences in median parasite load did not explain pairwise epigenetic (r = −0.08; P = 0.75) or genetic (r = −0.11; P = 0.80) differentiation.
), but there was no geographical pattern (Fig.2). Parasite load was weakly positively correlated with genome-wide hemimethylation (ρ = 0.15; P = 0.02), but not correlated with full methylation (ρ = −0.10; P = 0.11) or absence of methylation (ρ = −0.07; P = 0.29). Principal coordinates analysis of epigenetic and genetic variation explained only 4–6% of the total variation and showed substantial overlap among populations (Fig. S1, Supporting information). Nevertheless, global epigenetic differentiation among populations was significant (FSC = 0.0227; P < 0.001) and considerably stronger than genetic differentiation (FSC = 0.0058; P < 0.001). Pairwise epigenetic and genetic differentiation (FST) ranged from −0.012 to 0.091 (113 of 210 pairs significant at FDR < 0.05) and from −0.012 to 0.048 (16 of 210 pairs significant at FDR < 0.05), respectively (Fig.3). There was no isolation-by-distance pattern in either epigenetic (r = 0.02; P = 0.38) or genetic (r = −0.13; P = 0.87) differentiation. Seven populations that were not clustered geographically (locations 3, 4, 9, 10, 15, 18 and 19) showed disproportionate epigenetic differentiation compared with all other populations (Fig.3). Of these, populations 9 and 10 were also disproportionately genetically differentiated, causing a weak correlation between pairwise epigenetic and genetic differentiation matrices (r = 0.21; P = 0.06). Assigning populations to groups by median parasite load (Fig.1) did not explain any proportion of the molecular variances (Table3). Similarly, pairwise differences in median parasite load did not explain pairwise epigenetic (r = −0.08; P = 0.75) or genetic (r = −0.11; P = 0.80) differentiation.
Fig 2.
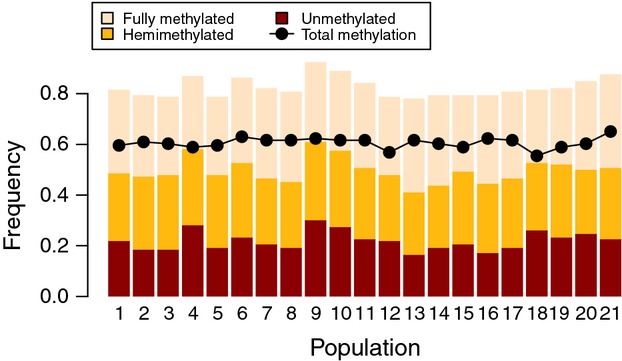
Median genome-wide methylation levels (full methylation, hemimethylation and absence of methylation) per population. Dots represent the summed frequencies of full methylation and hemimethylation.
Fig 3.
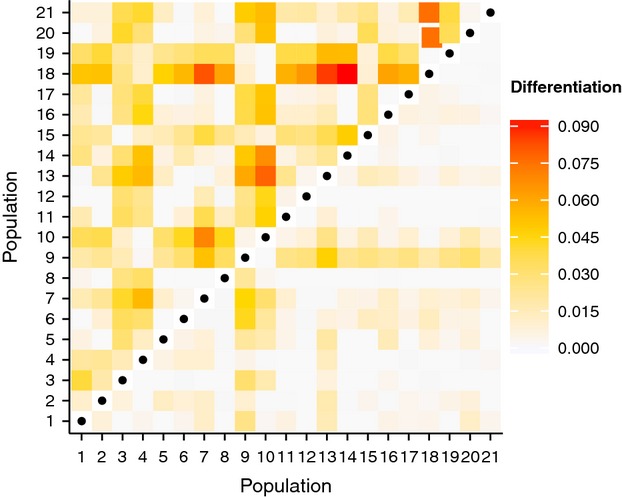
Pairwise epigenetic (above diagonal) and genetic (below diagonal) differentiation (amova-based FST) among populations.
Table 3.
Two-level amova of methylation-susceptible (MSL) or nonmethylated (NML) MSAP bands among populations grouped by median parasite load (Fig. 1)
| DF | SSD | MSD | Variance | Fixation index | |
|---|---|---|---|---|---|
| MSL | |||||
| Among groups | 2 | 0.7597 | 0.3799 | −0.0001 (−0.13%) | FCT = −0.0001; P = 0.36 |
| Among populations within groups | 18 | 6.7840 | 0.3769 | 0.0070 (2.27%) | FSC = 0.0227; P < 0.001 |
| Within populations | 213 | 63.9168 | 0.3001 | 0.3001 (97.74%) | |
| Total | 233 | 71.4605 | 0.3067 | ||
| NML | |||||
| Among groups | 2 | 0.5473 | 0.2737 | −0.0004 (−0.13%) | FCT = −0.0013; P = 0.99 |
| Among populations within groups | 18 | 5.4284 | 0.3016 | 0.0016 (0.58%) | FSC = 0.0058; P < 0.001 |
| Within populations | 213 | 60.3803 | 0.2835 | 0.2835 (99.56%) | |
| Total | 233 | 66.3560 | 0.2848 | ||
In contrast, epilocus-by-epilocus analyses revealed associations of locus-specific methylation states with parasite load. The 62 polymorphic MSL were transformed into 132 binary methylation states with no evidence of linkage disequilibrium among them. bayescan highlighted eight FST outlier methylation states, all of which were corroborated in mcheza. Overall, 35 individual methylation states (9 fully methylated, 12 hemimethylated and 14 unmethylated states), representing 25 epiloci, were significantly associated with parasite load using at least one model approach (GEE, sam, bayenv2). Of these, 19 methylation states were positively associated with parasite load and 16 states negatively. In the cases of ten epiloci, two methylation states were associated with parasite load and the unmethylated state always had the opposite directionality of association of the methylated state. Two FST outliers were not associated with parasite load. Regression results of each analysis method are summarized as volcano plots (Fig.4). FST outliers and significantly associated methylation states are organized by corresponding epiloci and statistical support based on congruence across analysis approaches (Table4).
Fig 4.
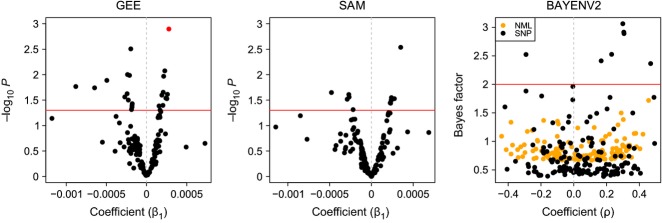
Coefficients and statistical significance of regression tests between epilocus-specific methylation states and parasite load using generalized estimating equations (GEE), logistic regression (sam) and Bayesian environmental analysis (bayenv2 with neutral parameterization by either NML or SNP genetic data). Each dot represents one methylation state. The red lines indicate significance thresholds (P ≦ 0.05 or Bayes factor ≧ 2), and red dots represent models with FDR-corrected q ≦ 0.1.
Table 4.
Epilocus-specific methylation states significantly associated with parasite load or identified as FST outliers. Each epilocus is listed with size, type of methylation state (U = unmethylated, H = hemimethylated, M = fully methylated), sign of regression coefficient (β1 for GEE/sam and ρ for bayenv2) and strength of statistical support. Results for bayenv2 are given for separate analyses using either nonmethylated MSAP loci (NML) or a set of 260 neutral SNPs for neutral parameterization
| Epilocus (MSAP band) | bayenv2b | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Name | Size | State ID | Type | Coefficient | GEEa | sama | NML | SNP | bayescanc | mchezad |
| 1 | MSAP_4.10_01 | 500 | ML201 | U | – | ** | ** | 2 | *** | *** | |
| ML234 | H | + | ** | *** | *** | ||||||
| 2 | MSAP_3.12_05 | 400 | ML189 | H | + | 2** | *** | *** | |||
| ML245 | U | – | *** | *** | |||||||
| 3 | MSAP_3.12_04 | 450 | ML192 | M | + | 2 | *** | *** | |||
| ML244 | U | – | 2 | *** | *** | ||||||
| 4 | MSAP_2.17_02 | 475 | ML238 | M | – | *** | *** | ||||
| ML117 | U | + | ** | 3 | 4 | ||||||
| 5 | MSAP_2.13_10 | 275 | ML159 | M | + | 3 | 2 | *** | |||
| 6 | MSAP_2.13_03 | 500 | ML224 | H | – | 3 | 2** | *** | |||
| ML220 | U | + | 3 | 3*** | |||||||
| 7 | MSAP_2.6_09 | 300 | ML202 | H | + | *** | ** | 3 | 2** | *** | ** |
| ML218 | U | – | ** | ** | 3 | 2 | ** | ||||
| 8 | MSAP_3.16_01 | 600 | ML209 | H | + | ** | ** | 3 | 2 | ** | |
| ML128 | U | – | ** | * | |||||||
| 9 | MSAP_4.13_04 | 450 | ML207 | H | + | ** | 3 | * | |||
| 10 | MSAP_4.10_15 | 125 | ML98 | U | – | ** | * | ||||
| ML194 | M | + | ** | ** | 2 | ||||||
| 11 | MSAP_3.16_17 | 130 | ML172 | U | – | ** | ** | ||||
| 12 | MSAP_3.12_01 | 600 | ML204 | H | + | ** | ** | 4 | 3** | ||
| ML210 | U | – | 3 | 2 | |||||||
| 13 | MSAP_4.13_08 | 250 | ML213 | H | + | ** | ** | 2 | 2 | ||
| 14 | MSAP_2.17_14 | 160 | ML228 | U | – | ** | ** | ||||
| 15 | MSAP_3.16_07 | 320 | ML272 | M | – | ** | ** | 2 | 2 | ||
| 16 | MSAP_4.10_05 | 390 | ML143 | U | + | ** | 2 | 3 | |||
| 17 | MSAP_3.6_09 | 225 | ML100 | M | – | * | * | ||||
| 18 | MSAP_4.13_06 | 390 | ML284 | H | + | * | * | 4 | 3 | ||
| 19 | MSAP_3.12_09 | 320 | ML288 | M | + | * | * | ||||
| 20 | MSAP_4.13_01 | 600 | ML99 | U | – | * | * | ||||
| 21 | MSAP_4.13_11 | 150 | ML275 | H | + | * | |||||
| 22 | MSAP_3.12_02 | 550 | ML105 | M | + | 4 | 4** | ||||
| 23 | MSAP_2.6_01 | 600 | ML107 | H | + | 4 | 4* | ||||
| 24 | MSAP_3.16_03 | 500 | ML153 | U | – | 2 | |||||
| ML262 | H | + | 2 | 2** | |||||||
| 25 | MSAP_3.16_13 | 200 | ML281 | M | – | 3 | 3 | ||||
*: absolute value of coefficient outside 5% / 95% percentiles; **: P ≦ 0.05; ***: q ≦ 0.1.
2: |ρ| ≧ 0.2; 3: |ρ| ≧ 0.3; 4: |ρ| ≧ 0.4; **: Bayes factor ≧ 2; ***: Bayes factor ≧ 3.
***: q ≦ 0.05.
*: P ≧ 0.90; **: P ≧ 0.95; ***: P ≧ 0.99.
Sanger sequencing of cloned epiloci yielded one to four sequences per epilocus (77 unique sequences, available from genbank Accession nos KJ655444–KJ655520), suggesting some incidence of clone band size homoplasy. Of these, only six (representing five MSAP bands) provided characterized RefSeq protein hits with an expected value below 1. At least one sequence per band could be mapped to the chicken or turkey genomes and most sequences were mapped to noncoding regions. The distance between the midpoints of the mapped sequence and the nearest ensembl gene annotation ranged from 96 bp to 630 Kbp (c. 0.003 mM to 1.764 cM; Andreescu et al. 2007). The geneontology annotations of these protein hits or nearest annotated genes included numerous biological process categories (Fig.5), most notably immune system, epigenetic mechanisms, cell cycle/proliferation and energy metabolism (Table5). Immune system genes included B-cell (PRDM1/BLIMP1, IKZF3) and T-cell (EOMES) proliferation regulators, MHC binding proteins (MARCH1) and enzymes involved in somatic hypermutation (DNTT). Intriguing additional findings included an rRNA methyltransferase (TFB2M), a histone acetyltransferase (KAT2B), two microRNAs (MIR1575, MIRLET7G), one small nucleolar RNA (SNORD111) and a transposable element (POGK). Complete characterizations for every sequence including full geneontology terms are presented in Table S1 (Supporting information).
Fig 5.
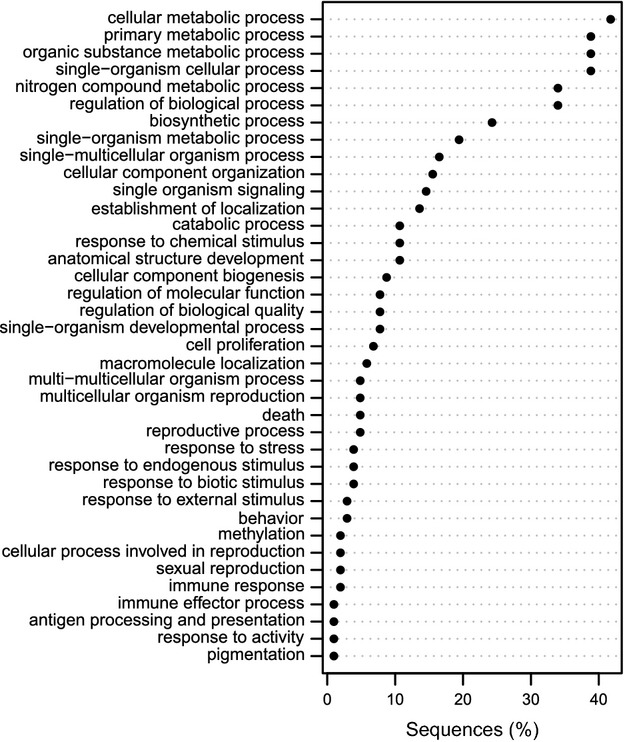
Frequencies of level-3 ‘biological process’ geneontology (go) annotations of genes near MSAP band clone sequences mapped to chicken and turkey genomes.
Table 5.
Sequence characterization of select MSAP bands detailing gene names, descriptions, key geneontology terms and accessions of either blastx top hit (with expected E-value) or nearest ensembl annotation (with distance in base pairs) in chicken (ENSGAL) or turkey (ENSMGA) genomes
| Band ID | Gene name | Description | Accession | E-value | Distance | Key go terms |
|---|---|---|---|---|---|---|
| Immune system | ||||||
| 12 | PRDM1/BLIMP1 | PR domain containing 1, with ZNF domain | ENSGALT00000024824 | 6.28E+03 | positive regulation of B-cell differentiation (GO:0045579); negative regulation of B-cell proliferation (GO:0030889) | |
| 17 | EOMES | eomesodermin | ENSGALT00000042658 | 7.56E+04 | CD8-positive, alpha-beta T-cell differentiation involved in immune response (GO:0002302) | |
| 5 | IKZF3 | IKAROS family zinc finger 3 (Aiolos) | ENSMGAT00000003138 | 6.74E+03 | regulation of B-cell differentiation (GO:0045577) | |
| 10, 11, 14 | MARCH1 | membrane-associated ring finger (C3HC4) 1, E3 ubiquitin protein ligase | ENSMGAT00000002357 | 7.50E+04 | MHC protein binding (GO:0042287); immune response (GO:0006955) | |
| 22 | DNTT | DNA nucleotidylexotransferase | ENSGALT00000033285 | 6.63E+04 | DNA binding (GO:0003677) | |
| Epigenetic mechanisms | ||||||
| 17 | TFB2M | mitochondrial 12S rRNA dimethylase 2 | ENSGALT00000017328 | 1.55E+04 | rRNA methylation (GO:0031167) | |
| 8 | KAT2B | histone acetyltransferase KAT2B isoform X2 | XP_426001.4 | 4.06E-08 | lysine N-acetyltransferase activity (GO:0004468) | |
| 15 | MIR1575 | gga-mir-1575 | ENSGALT00000042383 | 4.85E+04 | N/A (microRNA) | |
| 10 | MIRLET7G | gga-let-7g | ENSGALT00000028969 | 9.60E+01 | N/A (microRNA) | |
| 23 | SNORD111 | small nucleolar RNA SNORD111 | ENSGALT00000042232 | 2.48E+02 | N/A | |
| Cell cycle and proliferation | ||||||
| 22, 3 | PHACTR3 | phosphatase and actin regulator 3 isoform X3 | XP_004947133.1 | 3.90E-37 | actin binding (GO:0003779) | |
| ENSGALT00000007780 | 1.43E+04 | |||||
| 25 | DIAPH3 | diaphanous-related formin 3 | ENSMGAT00000016825 | 3.90E+04 | cellular component organization (GO:0016043) | |
| 8 | TUC338 | transcribed ultra-conserved region 338 | ENSGALT00000044640 | 4.95E+05 | N/A (hepatocyte proliferation) | |
| 8 | WDR26 | WD repeat domain 26 | ENSGALT00000015154 | 9.62E+02 | protein binding (GO:0005515) | |
| 23 | SF3B3 | splicing factor 3B subunit 3, partial | XP_005009141.1 | 1.31E-33 | mRNA processing (GO:0006397) | |
| 24 | POGK | pogo transposable element with KRAB domain | ENSMGAT00000013461 | 8.00E+04 | regulation of transcription, DNA-templated (GO:0006355) | |
| Energy metabolism | ||||||
| 20 | SDHA | succinate dehydrogenase [ubiquinone] flavoprotein subunit A | XP_005419422.1 | 1.99E-04 | mitochondrial respiratory chain complex II (GO:0005749) | |
| 19 | LARGE | like-glycosyltransferase | ENSMGAT00000014370 | 2.58E+05 | glycoprotein biosynthetic process (GO:0009101) | |
| 18 | PPARGC1A | peroxisome proliferator-activated receptor gamma, coactivator 1 alpha | ENSGALT00000023263 | 1.32E+05 | positive regulation of cellular respiration (GO:1901857) | |
| 22 | ACOX2 | acyl-CoA oxidase 2, branched chain | ENSGALT00000011555 | 4.72E+03 | fatty acid metabolic process (GO:0006631) |
Discussion
We highlight significant fine-scale epigenetic structure among red grouse populations in north-east Scotland and associations of parasite load with methylation patterns on a locus-specific, but not genome-wide level. Some parasite-associated epiloci were mapped to genomic regions close to immune genes or genes for epigenetic factors such as histone acetyltransferases and microRNAs, providing intriguing correlational evidence for epigenetically regulated host–parasite interactions in a wild bird species.
The sampled grouse populations differed significantly in genome-wide methylation levels and were significantly epigenetically differentiated, providing first evidence of significant epigenetic differentiation among wild bird populations on a small geographical scale. This is in surprising contrast with introduced house sparrow populations in Kenya and Florida, which are not epigenetically differentiated within Kenya or even across continents, possibly due to high epigenetic diversity compensating for low genetic diversity and inbreeding (Schrey et al. 2012; Liebl et al. 2013). The magnitude of epigenetic differentiation observed among grouse was considerably stronger than genetic differentiation derived from nonmethylated MSAP loci. Similarly, epigenetic diversity was also marginally greater than genetic diversity. There was no evidence for an association of epigenetic with genetic variation, suggesting autonomy of the captured epigenetic variation (Richards 2006). In concert, these findings suggest that DNA methylation may be an important source of ecologically relevant variation in these grouse populations (Lira-Medeiros et al. 2010; Richards et al. 2012).
Epigenetic differentiation among populations could be caused by neutral epimutations subject to random drift as a consequence of limited dispersal (Massicotte et al. 2011). Significant epigenetic differentiation (FST = 0.3) consistent with such short-distance dispersal has been reported among great roundleaf bat populations in China (Liu et al. 2012). However, this scenario is unlikely to be the case in grouse, because epigenetic differentiation was not attributable to isolation by distance, despite genetic evidence for population structure from microsatellite data (Piertney et al. 1998, 1999). A more likely explanation for the observed epigenetic differentiation may be environmental heterogeneity across the landscape, such as differential parasite load. Considerable epigenetic differentiation (FST = 0.1–0.3) is commonly found among plant populations in contrasting environments on similar spatial scales as our study (Herrera & Bazaga 2010; Lira-Medeiros et al. 2010; Herrera et al. 2013; Schulz et al. 2013). Notably, Chwedorzewska & Bednarek (2012) found marked epigenetic differentiation (FST = 0.5) among Polish and Antarctic annual bluegrass populations, and Richards et al. (2012) report strong epigenetic differentiation (FST = 0.5–0.8) of Japanese knotweed populations during invasion into different environments. However, the observed genome-wide epigenetic patterns among grouse populations were not attributable to parasite load. Instead, these patterns were predominantly driven by the disproportionate differentiation of seven populations that were neither geographically clustered nor similar in parasite load. Intriguingly, two of these populations were both epigenetically and genetically disproportionately differentiated. These patterns may be caused by demographic or adaptive processes due to environmental factors that were not considered in this study, but warrant further investigation.
Our main objective was to ascertain whether parasite load is linked to epigenetic variation in wild grouse populations. Neither population-based genome-wide epigenetic differentiation nor individual-based genomewide methylation levels were associated with parasite load, apart from a weak positive association with genome-wide hemimethylation. However, epilocus-by-epilocus analyses revealed associations of methylation states at particular epiloci with parasite load and also disproportionate differentiation (FST outliers). This is no contradiction because epilocus-specific associations with parasite load can be either positive or negative, which precludes the detection of association when methylation levels are averaged across loci to provide genome-wide estimates (Paun et al. 2010; Schrey et al. 2012). Indeed, environmental factors may well impact a finite number of individual epiloci rather than genome-wide methylation, which becomes manifested in differentiation or association at specific epiloci (Paun et al. 2010; Chwedorzewska & Bednarek 2012) even in the absence of genome-wide differentiation (Schrey et al. 2012). The observed FST outliers suggest such an impact by unknown environmental factors, because not all outliers were also associated with parasite load. Nevertheless, our findings vividly demonstrate a locus-specific relationship between epigenetic variation and a biotic environmental stressor.
Given that controlled transcriptomic experiments have previously demonstrated that parasite infection alters gene expression in liver, spleen and caecum tissues in red grouse (Webster et al. 2011a, b), one possible interpretation of epilocus-specific association with parasite load is that parasites cause epilocus-specific methylation changes that impact gene expression (Angers et al. 2010; Paun et al. 2010; Jones 2012). Similarly to transcriptomic changes, such parasite-driven methylation changes would then present a transient response to an environmental factor during the bird's lifetime without assuming inheritance of methylation states (Skinner 2011). Among our association results, methylation was predominantly positively associated with parasite load (76%) and absence of methylation negatively (79%), often consistently in complement at the same epilocus. This suggests a predominant pattern of methylation-mediated positive association of parasite load with down-regulation of gene expression (Angers et al. 2010; Jones 2012). The rarer inverted observation that methylation was negatively associated with parasite load and absence of methylation positively suggests that parasite infection may also cause demethylation at some loci and concomitant up-regulation of gene expression (Angers et al. 2010; Jones 2012). The physiological processes highlighted by the geneontology terms of the sequenced epiloci were manifold, corroborating the view that parasite infection impacts physiological condition through a wide range of vital cellular processes rather than single categories such as the immune system (Hill 2011; Webster et al. 2011a, b). Immune system processes were only a small subset, yet methylation at all but one of those immune genes was positively associated with parasite load, consistent with immunosuppressive effects of helminth infection (Maizels & Yazdanbakhsh 2003; Biron & Loxdale 2013). Most intriguingly, some epiloci were linked to genes that are themselves involved in epigenetic mechanisms, including rRNA methylation, histone acetylation and RNA interference by small RNAs. These mechanisms are primarily involved in regulating ribosomal translation and chromatin remodelling (He & Hannon 2004; Shahbazian & Grunstein 2007; Metodiev et al. 2009; Bannister & Kouzarides 2011). In consequence, parasite-linked changes in methylation patterns at these loci may regulate the expression of epigenetic factors that regulate gene expression or chromatin remodelling elsewhere in the genome, providing an enticing, yet speculative perspective on the consequences of environmentally induced epigenetic states (Feil & Fraga 2012).
An alternative functional interpretation of methylation changes is facilitation of nucleotide sequence mutations rather than regulation of gene expression. Methylated cytosine is substantially more liable to deamination than unmethylated cytosine (Lutsenko & Bhagwat 1999; Poole et al. 2003), suggesting that increased locus-specific methylation following parasite infection may create mutational hot spots in gene bodies that could provide genetic variation during, for example, somatic hypermutation in immune genes of proliferating hepatic cells (Racanelli & Rehermann 2006; Jones 2012). Another important function of methylation is the silencing of transposable elements that become released following demethylation (Suzuki & Bird 2008; Zemach et al. 2010; Jones 2012). The few observed cases of association of absence of methylation with parasite load could therefore be explained as a release of transposable elements that could create somatic genetic variation to facilitate systemic responses to parasite infection. This interpretation would be consistent with the frequently observed phenomenon that demethylation increases phenotypic variance (Bossdorf et al. 2010; Vergeer et al. 2012). One epilocus was indeed mapped to the vicinity of a transposable element, but its association with parasite-linked hemimethylation would impede transposition at high parasite load rather than induce it, suggesting this may be coincidental.
These functional interpretations of parasite-associated methylation have to remain speculative because no independent genomic data are available for red grouse. However, our finding that most epiloci were mapped to noncoding sequence regions is consistent with gene regulation either directly through methylation changes in the CpG islands of gene promoters (Angers et al. 2010; Jones 2012; Duncan et al. 2014) or indirectly through methylation-associated recruitment of complexes that remodel chromatin (Jaenisch & Bird 2003; Bannister & Kouzarides 2011; Feil & Fraga 2012). Given that many epiloci were mapped to the vicinity of a gene, these genes may be directly affected by these epiloci, but this becomes increasingly difficult to reconcile with increasing genomic distances. Although long-range transcriptional regulation exists (Kleinjan & van Heyningen 2005), it is likely that many of those epiloci in noncoding regions are not specifically involved in regulating the genes in their vicinity, but may instead be involved in remodelling chromating with potentially far-reaching regulatory consequences (Jaenisch & Bird 2003; Bannister & Kouzarides 2011; Feil & Fraga 2012). Clearly, functional genomics analyses in the context of a controlled infection experiment would be required to establish causal links between methylation changes and their genomic and physiological consequences (Duncan et al. 2014). Nevertheless, despite their speculative nature, our interpretations describe a number of hypothesis-generating mechanisms that may direct further exciting research.
The rationale of our study was to detect a correlational epigenetic signature of parasite load in red grouse populations that could be intepreted as a transient epigenetic response to parasites, similarly to a transcriptomic response (Webster et al. 2011b). This was prompted by a large body of red grouse research that has identified parasite infection as an important effect on physiology and behaviour (e.g. Fox & Hudson 2001; Mougeot et al. 2005a, 2010a, Mougeot et al. 2010a,b; Martínez-Padilla et al. 2007; Vergara et al. 2012). From this point of view, our results provide evidence for a broad epigenetically mediated physiological response to parasites and suggest that helminths may effect manipulations of host physiology and behaviour at least partially through transient epigenetic mechanisms (Maizels & Yazdanbakhsh 2003; Biron & Loxdale 2013; Poulin 2013). The evolutionary relevance of these mechanisms is difficult to ascertain (Richards et al. 2010; Duncan et al. 2014). Vertical transmission of methylation patterns in an analogous way to genetic polymorphisms exists (Jablonka & Raz 2009; Petronis 2010; Skinner 2011; Smith & Ritchie 2013; D’Urso & Brickner 2014), particularly in plants (Salmon et al. 2008; Verhoeven et al. 2010; Herrera et al., 2013, 2014), but the dearth of transgenerational ecological epigenetics studies on animals leaves a large scope for exciting future research. If inheritance of methylation patterns could be demonstrated in red grouse, parasite load might potentially be a consequence of methylation changes rather than a cause. Inherited methylation patterns may then contribute to an innate resistance to parasites (‘condition’) without necessarily undergoing alterations as a consequence of infection themselves (Poulin & Thomas 2008).
In conclusion, our study highlights the potential for ecological epigenetics to illuminate mechanisms of plasticity and adaptation in the context of host–parasite interactions in natural systems. We also highlight the necessity of independent transcriptomics and genomics data to overcome conceptual difficulties in interpreting epigenetics patterns. In spite of these challenges, DNA methylation may be key to understanding the generation of phenotypic variation and the evolution of complex phenotypes in the absence of genetic variation, indicating that the study of epigenetic causes and consequences of environmentally induced phenotypes may be paramount to understanding how plasticity-conferred functional variation may contribute to eco-evolutionary processes (Bossdorf et al. 2008, 2010; Petronis 2010; Roux et al. 2011).
Acknowledgments
This study was funded by a BBSRC studentship (MA Wenzel) and NERC grants NE/H00775X/1 and NE/D000602/1 (SB Piertney). The authors are grateful to Mario Röder and Keliya Bai for fieldwork assistance; Alex Douglas for statistical advice; Tyler Stevenson, Heather Ritchie and three anonymous reviewers for helpful comments on manuscript drafts; and all estate owners, factors and keepers for access to field sites, most particularly MJ Taylor and Mike Nisbet (Airlie), Neil Brown (Allargue), RR Gledson and David Scrimgeour (Delnadamph), Andrew Salvesen and John Hay (Dinnet), Stuart Young and Derek Calder (Edinglassie), Kirsty Donald and David Busfield (Glen Dye), Neil Hogbin and Ab Taylor (Glen Muick), Alistair Mitchell (Glenlivet), Simon Blackett, Jim Davidson and Liam Donald (Invercauld) Richard Cooke and Fred Taylor (Invermark), Shaila Rao and Christopher Murphy (Mar Lodge), and Ralph Peters and Philip Astor (Tillypronie).
Biography
M.A.W. and S.B.P. conceived and designed the study. M.A.W. performed field and laboratory work and analysed the data. M.A.W. and S.B.P. wrote the manuscript.
Data accessibility
DNA clone sequences: genbank Accession nos KJ655444–KJ655520.
Clone sequence characterization including geneontology terms uploaded as supplementary information (Table S1).
Custom R scripts with input files: Dryad 10.5061/dryad.f727j
Supporting Information
Additional supporting information may be found in the online version of this article.
Principal coordinate analysis plots of epigenetic and genetic variation in the study populations.
Clone sequence characterization following genome mapping and geneontology annotation. Contains chicken/turkey chromosome locations of mapped clone sequences, nearest ensemble gene identifiers and associated geneontology terms.
References
- Alabert C, Groth A. Chromatin replication and epigenome maintenance. Nature Reviews Molecular Cell Biology. 2012;13:153–167. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- Altschul S, Madden T, Schäffer A, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarasinghe HE, Clayton CI, Mallon EB. Methylation and worker reproduction in the bumblebee (Bombus terrestris. Proceedings of the Royal Society of London Series B: Biological Sciences. 2014;281:20132502. doi: 10.1098/rspb.2013.2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreescu C, Avendano S, Brown SR, Hassen A, Lamont SJ, Dekkers JCM. Linkage disequilibrium in related breeding lines of chickens. Genetics. 2007;177:2161–2169. doi: 10.1534/genetics.107.082206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers B, Castonguay E, Massicotte R. Environmentally induced phenotypes and DNA methylation: how to deal with unpredictable conditions until the next generation and after. Molecular Ecology. 2010;19:1283–1295. doi: 10.1111/j.1365-294X.2010.04580.x. [DOI] [PubMed] [Google Scholar]
- Antao T, Beaumont MA. mcheza: a workbench to detect selection using dominant markers. Bioinformatics. 2011;27:1717–1718. doi: 10.1093/bioinformatics/btr253. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Research. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjak A, Konradi J, Blaich R, Forneck A. Different DNA extraction methods can cause different AFLP profiles in grapevine (Vitis vinifera L.) Vitis. 2006;45:15–21. [Google Scholar]
- Bérénos C, Wegner KM, Schmid-Hempel P. Antagonistic coevolution with parasites maintains host genetic diversity: an experimental test. Proceedings of the Royal Society B: Biological Sciences. 2011;278:218–224. doi: 10.1098/rspb.2010.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes & Development. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Biron DG, Loxdale HD. Host–parasite molecular cross-talk during the manipulative process of a host by its parasite. The Journal of Experimental Biology. 2013;216:148–160. doi: 10.1242/jeb.073825. [DOI] [PubMed] [Google Scholar]
- Boks MP, Derks EM, Weisenberger DJ, et al. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS One. 2009;4:e6767. doi: 10.1371/journal.pone.0006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonduriansky R, Day T. Nongenetic inheritance and its evolutionary implications. Annual Review of Ecology, Evolution, and Systematics. 2008;40:103–125. [Google Scholar]
- Bortolotti GR, Mougeot F, Martínez-Padilla J, Webster LMI, Piertney SB. Physiological stress mediates the honesty of social signals. PLoS ONE. 2009;4:e4983. doi: 10.1371/journal.pone.0004983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossdorf O, Richards CL, Pigliucci M. Epigenetics for ecologists. Ecology Letters. 2008;11:106–115. doi: 10.1111/j.1461-0248.2007.01130.x. [DOI] [PubMed] [Google Scholar]
- Bossdorf O, Arcuri D, Richards CL, Pigliucci M. Experimental alteration of DNA methylation affects the phenotypic plasticity of ecologically relevant traits in Arabidopsis thaliana. Evolutionary Ecology. 2010;24:541–553. [Google Scholar]
- Caballero A, Garc´a-Pereira MJ, Quesada H, et al. Genomic distribution of AFLP markers relative to gene locations for different eukaryotic species. BMC Genomics. 2013;14:1–11. doi: 10.1186/1471-2164-14-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnusamy V, Zhu JK. Epigenetic regulation of stress responses in plants. Current Opinion in Plant Biology. 2009;12:133–139. doi: 10.1016/j.pbi.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwedorzewska K, Bednarek P. Genetic and epigenetic variation in a cosmopolitan grass Poa annua from Antarctic and Polish populations. Polish Polar Research. 2012;33:63–80. [Google Scholar]
- Conesa A, Götz S. blast2go: a comprehensive suite for functional analysis in plant genomics. International Journal of Plant Genomics. 2008;2008:619832. doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Götz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. blast2go: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Crews D, Gore AC, Hsu TS, et al. Transgenerational epigenetic imprints on mate preference. Proceedings of the National Academy of Sciences. 2007;104:5942–5946. doi: 10.1073/pnas.0610410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Urso A, Brickner JH. Mechanisms of epigenetic memory. Trends in Genetics. 2014;30:230–236. doi: 10.1016/j.tig.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin É, Charmantier A, Champagne FA, Mesoudi A, Pujol B, Blanchet S. Beyond DNA: integrating inclusive inheritance into an extended theory of evolution. Nature Reviews Genetics. 2011;12:475–486. doi: 10.1038/nrg3028. [DOI] [PubMed] [Google Scholar]
- Delahay RJ, Moss R. Food intake, weight changes and egg production in captive red grouse before and during laying: effects of the parasitic nematode Trichostrongylus tenuis. The Condor. 1996;98:501–511. [Google Scholar]
- Delahay RJ, Speakman JR, Moss R. The energetic consequences of parasitism: effects of a developing infection of Trichostrongylus tenuis (Nematoda) on red grouse (Lagopus lagopus scoticus) energy balance, body weight and condition. Parasitology. 1995;110:473–482. [Google Scholar]
- Don R, Cox P, Wainwright B, Baker K, Mattick J. Touchdown’ PCR to circumvent spurious priming during gene amplification. Nucleic Acids Research. 1991;19:4008. doi: 10.1093/nar/19.14.4008. ) ‘. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen RH, Pelizzola M, Schmitz RJ, et al. Widespread dynamic DNA methylation in response to biotic stress. Proceedings of the National Academy of Sciences. 2012;109:E2183–E2191. doi: 10.1073/pnas.1209329109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A, Ashton B, Buxton S, et al. Geneious v7.1.2. 2014. Available from http://www.geneious.com.
- Duncan EJ, Gluckman PD, Dearden PK. Epigenetics, plasticity, and evolution: how do we link epigenetic change to phenotype? Journal of Experimental Zoolagy Part B: Molecular and Developmental Evolution. 2014;322:208–220. doi: 10.1002/jez.b.22571. [DOI] [PubMed] [Google Scholar]
- Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nature Reviews Genetics. 2012;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DR, Wilson CB. Methylation and demethylation in the regulation of genes, cells, and responses in the immune system. Clinical Immunology. 2003;109:37–45. doi: 10.1016/s1521-6616(03)00205-5. [DOI] [PubMed] [Google Scholar]
- Foll M, Gaggiotti O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics. 2008;180:977–993. doi: 10.1534/genetics.108.092221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox A, Hudson PJ. Parasites reduce territorial behaviour in red grouse (Lagopus lagopus scoticus. Ecology Letters. 2001;4:139–143. [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences, USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Schoneveld O, Georgakilas AG, Panayiotidis MI. Oxidative stress, DNA methylation and carcinogenesis. Cancer Letters. 2008;266:6–11. doi: 10.1016/j.canlet.2008.02.026. [DOI] [PubMed] [Google Scholar]
- Fried B, Reddy A, Mayer D. Helminths in human carcinogenesis. Cancer Letters. 2011;305:239–249. doi: 10.1016/j.canlet.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Fulneček J, Kovařík A. How to interpret Methylation Sensitive Amplified Polymorphism (MSAP) profiles? BMC Genetics, 2014;15,:2. doi: 10.1186/1471-2156-15-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Díaz E, Jordà M, Peinado MA, Rivero A. Epigenetics of host–pathogen interactions: the road ahead and the road behind. PLoS Pathogens. 2012;8:e1003007. doi: 10.1371/journal.ppat.1003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham MH. Confronting multicollinearity in ecological multiple regression. Ecology. 2003;84:2809–2815. [Google Scholar]
- Griffiths R, Double MC, Orr K, Dawson RJ. A DNA test to sex most birds. Molecular Ecology. 1998;7:1071–1075. doi: 10.1046/j.1365-294x.1998.00389.x. [DOI] [PubMed] [Google Scholar]
- Günther T, Coop G. Robust identification of local adaptation from allele frequencies. Genetics. 2013;195:205–220. doi: 10.1534/genetics.113.152462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez MI, Siraj AK, Khaled H, Koon N, Bhatia K, et al. CpG island methylation in Schistosoma-and non-Schistosoma-associated bladder cancer. Modern Pathology. 2004;17:1268–1274. doi: 10.1038/modpathol.3800177. [DOI] [PubMed] [Google Scholar]
- Halekoh U, Højsgaard S, Yan J. The R package geepack for generalized estimating equations. Journal of Statistical Software. 2006;15:1–11. [Google Scholar]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nature Reviews Genetics. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- Herrera CM, Bazaga P. Epigenetic differentiation and relationship to adaptive genetic divergence in discrete populations of the violet Viola cazorlensis. New Phytologist. 2010;187:867–876. doi: 10.1111/j.1469-8137.2010.03298.x. [DOI] [PubMed] [Google Scholar]
- Herrera CM, Bazaga P. Untangling individual variation in natural populations: ecological, genetic and epigenetic correlates of long-term inequality in herbivory. Molecular Ecology. 2011;20:1675–1688. doi: 10.1111/j.1365-294X.2011.05026.x. [DOI] [PubMed] [Google Scholar]
- Herrera CM, Pozo MI, Bazaga P. Jack of all nectars, master of most: DNA methylation and the epigenetic basis of niche width in a flower-living yeast. Molecular Ecology. 2012;21:2602–2616. doi: 10.1111/j.1365-294X.2011.05402.x. [DOI] [PubMed] [Google Scholar]
- Herrera CM, Medrano M, Bazaga P. Epigenetic differentiation persists after male gametogenesis in natural populations of the perennial herb Helleborus foetidus (Ranunculaceae) PLoS One. 2013;8:e70730. doi: 10.1371/journal.pone.0070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera CM, Medrano M, Bazaga P. Variation in DNA methylation transmissibility, genetic heterogeneity and fecundity-related traits in natural populations of the perennial herb Helleborus foetidus. Molecular Ecology. 2014;23:1085–1095. doi: 10.1111/mec.12679. [DOI] [PubMed] [Google Scholar]
- Hill GE. Condition-dependent traits as signals of the functionality of vital cellular processes. Ecology Letters. 2011;14:625–634. doi: 10.1111/j.1461-0248.2011.01622.x. [DOI] [PubMed] [Google Scholar]
- Hogan FE, Cooke R, Burridge CP, Norman JA. Optimizing the use of shed feathers for genetic analysis. Molecular Ecology Resources. 2008;8:561–567. doi: 10.1111/j.1471-8286.2007.02044.x. [DOI] [PubMed] [Google Scholar]
- Hudson PJ, Dobson AP, Newborn D. Do parasites make prey vulnerable to predation? Red grouse and parasites. Journal of Animal Ecology. 1992;61:681–692. [Google Scholar]
- Hudson PJ, Dobson AP, Newborn D. Prevention of population cycles by parasite removal. Science. 1998;282:2256–2258. doi: 10.1126/science.282.5397.2256. [DOI] [PubMed] [Google Scholar]
- Jablonka E, Lamb MJ. Precis of evolution in four dimensions. Behavioral and Brain Sciences. 2007;30:353–365. doi: 10.1017/S0140525X07002221. [DOI] [PubMed] [Google Scholar]
- Jablonka E, Raz G. Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. The Quarterly Review of Biology. 2009;84:131–176. doi: 10.1086/598822. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Reviews Genetics. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Joost S, Kalbermatten M, Bonin A. Spatial analysis method (sam): a software tool combining molecular and environmental data to identify candidate loci for selection. Molecular Ecology Resources. 2008;8:957–960. doi: 10.1111/j.1755-0998.2008.02162.x. [DOI] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Research. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT–The BLAST-Like alignment tool. Genome Research. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella RJ, Kähäri A, Haider S, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database. 2011;2011:bar030. doi: 10.1093/database/bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan DA, van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. The American Journal of Human Genetics. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebl AL, Schrey AW, Richards CL, Martin LB. Patterns of DNA methylation throughout a range expansion of an introduced songbird. Integrative and Comparative Biology. 2013;53:351–358. doi: 10.1093/icb/ict007. [DOI] [PubMed] [Google Scholar]
- Lira-Medeiros CF, Parisod C, Fernandes RA, Mata CS, Cardoso MA, Ferreira PCG. Epigenetic variation in mangrove plants occurring in contrasting natural environment. PLoS One. 2010;5:e10326. doi: 10.1371/journal.pone.0010326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Morgan M, Hutchison K, Calhoun VD. A study of the influence of sex on genome wide methylation. PLoS One. 2010;5:e10028. doi: 10.1371/journal.pone.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Sun K, Jiang T, Ho JP, Liu B, Feng J. Natural epigenetic variation in the female great roundleaf bat (Hipposideros armiger) populations. Molecular Genetics and Genomics. 2012;287:643–650. doi: 10.1007/s00438-012-0704-x. [DOI] [PubMed] [Google Scholar]
- Lutsenko E, Bhagwat AS. Principal causes of hot spots for cytosine to thymine mutations at sites of cytosine methylation in growing cells: a model, its experimental support and implications. Mutation Research/Reviews in Mutation Research. 1999;437:11–20. doi: 10.1016/s1383-5742(99)00065-4. [DOI] [PubMed] [Google Scholar]
- MacColl ADC, Piertney SB, Moss R, Lambin X. Spatial arrangement of kin affects recruitment success in young male red grouse. Oikos. 2000;90:261–270. [Google Scholar]
- Maizels RM, Yazdanbakhsh M. Immune regulation by helminth parasites: cellular and molecular mechanisms. Nature Reviews Immunology. 2003;3:733–744. doi: 10.1038/nri1183. [DOI] [PubMed] [Google Scholar]
- Martínez-Padilla J, Mougeot F, Pérez-Rodr´guez L, Bortolotti GR. Nematode parasites reduce carotenoid-based signalling in male red grouse. Biology Letters. 2007;3:161–164. doi: 10.1098/rsbl.2006.0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Padilla J, Mougeot F, Webster LMI, Pérez-Rodr´guez L, Piertney SB. Testing the interactive effects of testosterone and parasites on carotenoid-based ornamentation in a wild bird. Journal of Evolutionary Biology. 2010;23:902–913. doi: 10.1111/j.1420-9101.2010.01956.x. [DOI] [PubMed] [Google Scholar]
- Martínez-Padilla J, Redpath SM, Zeineddine M, Mougeot F. Insights into population ecology from long-term studies of red grouse Lagopus lagopus scoticus. Journal of Animal Ecology. 2014;83:85–98. doi: 10.1111/1365-2656.12098. [DOI] [PubMed] [Google Scholar]
- Massicotte R, Whitelaw E, Angers B. DNA methylation: a source of random variation in natural populations. Epigenetics: Official Journal of the DNA Methylation Society. 2011;6:421–427. doi: 10.4161/epi.6.4.14532. [DOI] [PubMed] [Google Scholar]
- Meaney M, Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues in Clinical Neuroscience. 2005;7:103. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesoudi A, Blanchet S, Charmantier A, et al. Is non-genetic inheritance just a proximate mechanism? A corroboration of the extended evolutionary synthesis. Biological Theory. 2013;7:189–195. [Google Scholar]
- Metodiev MD, Lesko N, Park CB, et al. Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metabolism. 2009;9:386–397. doi: 10.1016/j.cmet.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Morán P, Pérez-Figueroa A. Methylation changes associated with early maturation stages in the Atlantic salmon. BMC Genetics. 2011;12:86. doi: 10.1186/1471-2156-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morán P, Marco-Rius F, Meg´as M, Covelo-Soto L, Pérez-Figueroa A. Environmental induced methylation changes associated with seawater adaptation in brown trout. Aquaculture. 2013;392:77–83. [Google Scholar]
- Moss R, Watson A. Population cycles and kin selection in red grouse Lagopus lagopus scoticus. Ibis. 1991;133:113–120. [Google Scholar]
- Mostowy R, Engelstädter J. Host–parasite coevolution induces selection for condition-dependent sex. Journal of Evolutionary Biology. 2012;25:2033–2046. doi: 10.1111/j.1420-9101.2012.02584.x. [DOI] [PubMed] [Google Scholar]
- Mougeot F. Ornamental comb colour predicts T-cell-mediated immunity in male red grouse Lagopus lagopus scoticus. Naturwissenschaften. 2008;95:125–132. doi: 10.1007/s00114-007-0303-6. [DOI] [PubMed] [Google Scholar]
- Mougeot F, Redpath SM. Sexual ornamentation relates to immune function in male red grouse Lagopus lagopus scoticus. Journal of Avian Biology. 2004;35:425–433. [Google Scholar]
- Mougeot F, Irvine JR, Seivwright L, Redpath SM, Piertney S. Testosterone, immunocompetence, and honest sexual signaling in male red grouse. Behavioral Ecology. 2004;15:930–937. [Google Scholar]
- Mougeot F, Evans SA, Redpath SM. Interactions between population processes in a cyclic species: parasites reduce autumn territorial behaviour of male red grouse. Oecologia. 2005a;144:289–298. doi: 10.1007/s00442-005-0080-x. [DOI] [PubMed] [Google Scholar]
- Mougeot F, Piertney SB, Leckie F, et al. Experimentally increased aggressiveness reduces population kin structure and subsequent recruitment in red grouse Lagopus lagopus scoticus. Journal of Animal Ecology. 2005b;74:488–497. [Google Scholar]
- Mougeot F, Martínez-Padilla J, Webster LMI, Blount JD, Pérez-Rodr´guez L, Piertney SB. Honest sexual signalling mediated by parasite and testosterone effects on oxidative balance. Proceedings of the Royal Society of London. Series B. 2009;276:1093–1100. doi: 10.1098/rspb.2008.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougeot F, Martínez-Padilla J, Blount JD, Pérez-Rodr´guez L, Webster LMI, Piertney SB. Oxidative stress and the effect of parasites on a carotenoid-based ornament. Journal of Experimental Biology. 2010a;213:400–407. doi: 10.1242/jeb.037101. [DOI] [PubMed] [Google Scholar]
- Mougeot F, Martínez-Padilla J, Bortolotti GR, Webster LMI, Piertney SB. Physiological stress links parasites to carotenoid-based colour signals. Journal of Evolutionary Biology. 2010b;23:643–650. doi: 10.1111/j.1420-9101.2009.01926.x. [DOI] [PubMed] [Google Scholar]
- Paradis E. pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics. 2010;26:419–420. doi: 10.1093/bioinformatics/btp696. [DOI] [PubMed] [Google Scholar]
- Paun O, Bateman RM, Fay MF, Hedrén M, Civeyrel L, Chase MW. Stable epigenetic effects impact adaptation in allopolyploid orchids (Dactylorhiza: Orchidaceae) Molecular Biology and Evolution. 2010;27:2465–2473. doi: 10.1093/molbev/msq150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Figueroa A. msap: a tool for the statistical analysis of methylation-sensitive amplified polymorphism data. Molecular Ecology Resources. 2013;13:522–527. doi: 10.1111/1755-0998.12064. [DOI] [PubMed] [Google Scholar]
- Pérez-Figueroa A, Garc´a-Pereira M, Saura M, Rolán-Alvarez E, Caballero A. Comparing three different methods to detect selective loci using dominant markers. Journal of Evolutionary Biology. 2010;23:2267–2276. doi: 10.1111/j.1420-9101.2010.02093.x. [DOI] [PubMed] [Google Scholar]
- Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- Piertney SB, MacColl ADC, Bacon PJ, Dallas JF. Local genetic structure in red grouse (Lagopus lagopus scoticus): evidence from microsatellite DNA markers. Molecular Ecology. 1998;7:1645–1654. doi: 10.1046/j.1365-294x.1998.00493.x. [DOI] [PubMed] [Google Scholar]
- Piertney SB, MacColl ADC, Lambin X, Moss R, Dallas JF. Spatial distribution of genetic relatedness in a moorland population of red grouse (Lagopus lagopus scoticus. Biological Journal of the Linnean Society. 1999;68:317–331. [Google Scholar]
- Piertney SB, MacColl ADC, Bacon PJ, Racey PA, Lambin X, Dallas JF. Matrilineal genetic structure and female-mediated gene flow in red grouse (Lagopus lagopus scoticus): an analysis using mitochondrial DNA. Evolution. 2000;54:279–289. doi: 10.1111/j.0014-3820.2000.tb00028.x. [DOI] [PubMed] [Google Scholar]
- Piertney SB, Lambin X, MacColl ADC, et al. Temporal changes in kin structure through a population cycle in a territorial bird, the red grouse Lagopus lagopus scoticus. Molecular Ecology. 2008;17:2544–2551. doi: 10.1111/j.1365-294X.2008.03778.x. [DOI] [PubMed] [Google Scholar]
- Poole AM, Phillips MJ, Penny D. Prokaryote and eukaryote evolvability. Biosystems. 2003;69:163–185. doi: 10.1016/s0303-2647(02)00131-4. [DOI] [PubMed] [Google Scholar]
- Poulin R. Parasite manipulation of host personality and behavioural syndromes. The Journal of Experimental Biology. 2013;216:18–26. doi: 10.1242/jeb.073353. [DOI] [PubMed] [Google Scholar]
- Poulin R, Thomas F. Epigenetic effects of infection on the phenotype of host offspring: parasites reaching across host generations. Oikos. 2008;117:331–335. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. [Google Scholar]
- Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- Raymond M, Rousset F. genepop (version 1.2): population genetics software for exact tests and ecumenicism. Journal of Heredity. 1995;86:248–249. [Google Scholar]
- Redpath SM, Mougeot F, Leckie FM, Elston DA, Hudson PJ. Testing the role of parasites in driving the cyclic population dynamics of a gamebird. Ecology Letters. 2006;9:410–418. doi: 10.1111/j.1461-0248.2006.00895.x. [DOI] [PubMed] [Google Scholar]
- Reyna-López G, Simpson J, Ruiz-Herrera J. Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Molecular and General Genetics MGG. 1997;253:703–710. doi: 10.1007/s004380050374. [DOI] [PubMed] [Google Scholar]
- Richards EJ. Inherited epigenetic variation—revisiting soft inheritance. Nature Reviews Genetics. 2006;7:395–401. doi: 10.1038/nrg1834. [DOI] [PubMed] [Google Scholar]
- Richards CL, Bossdorf O, Pigliucci M. What role does heritable epigenetic variation play in phenotypic evolution? Bioscience, 2010;60,:232–237. [Google Scholar]
- Richards CL, Schrey AW, Pigliucci M. Invasion of diverse habitats by few Japanese knotweed genotypes is correlated with epigenetic differentiation. Ecology Letters. 2012;15:1016–1025. doi: 10.1111/j.1461-0248.2012.01824.x. [DOI] [PubMed] [Google Scholar]
- Rousset F. genepop’007: a complete reimplementation of the genepop software for Windows and Linux. Molecular Ecology Resources. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- Roux F, Colomé-Tatché M, Edelist C, et al. Genome-wide epigenetic perturbation jump-starts patterns of heritable variation found in nature. Genetics. 2011;188:1015–1017. doi: 10.1534/genetics.111.128744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon A, Clotault J, Jenczewski E, Chable V, Manzanares-Dauleux MJ. Brassica oleracea displays a high level of DNA methylation polymorphism. Plant Science. 2008;174:61–70. [Google Scholar]
- Schrey AW, Coon CA, Grispo MT, et al. Epigenetic variation may compensate for decreased genetic variation with introductions: a case study using house sparrows (Passer domesticus) on two continents. Genetics Research International. 2012;2012:979751. doi: 10.1155/2012/979751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz B, Eckstein RL, Durka W. Scoring and analysis of methylation-sensitive amplification polymorphisms for epigenetic population studies. Molecular Ecology Resources. 2013;13:642–653. doi: 10.1111/1755-0998.12100. [DOI] [PubMed] [Google Scholar]
- Seivwright LJ, Redpath SM, Mougeot F, Watt L, Hudson PJ. Faecal egg counts provide a reliable measure of Trichostrongylus tenuis intensities in free-living red grouse Lagopus lagopus scoticus. Journal Of Helminthology. 2004;78:69–76. doi: 10.1079/joh2003220. [DOI] [PubMed] [Google Scholar]
- Seivwright LJ, Redpath SM, Mougeot F, Leckie F, Hudson PJ. Interactions between intrinsic and extrinsic mechanisms in a cyclic species: testosterone increases parasite infection in red grouse. Proceedings of the Royal Society of London. Series B. 2005;272:2299–2304. doi: 10.1098/rspb.2005.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annual Review of Biochemistry. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- Shaw JL, Moss R. Factors affecting the establishment of the caecal threadworm Trichostrongylus tenuis in red grouse (Lagopus lagopus scoticus. Parasitology. 1989;99:259–264. doi: 10.1017/s0031182000058716. [DOI] [PubMed] [Google Scholar]
- Skinner M. Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics. 2011;6:838–842. doi: 10.4161/epi.6.7.16537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G, Ritchie MG. How might epigenetics contribute to ecological speciation? Current Zoology, 2013;59,:686–696. [Google Scholar]
- Snell-Rood EC, Troth A, Moczek AP. DNA methylation as a mechanism of nutritional plasticity: limited support from horned beetles. Molecular and Developmental Evolution. 2013;320:22–34. doi: 10.1002/jez.b.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson TJ, Prendergast BJ. Reversible DNA methylation regulates seasonal photoperiodic time measurement. Proceedings of the National Academy of Sciences. 2013;110:16651–16656. doi: 10.1073/pnas.1310643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics. 2008;24:1461–1462. doi: 10.1093/bioinformatics/btn209. [DOI] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nature Reviews Genetics. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- Tan MP. Analysis of DNA methylation of maize in response to osmotic and salt stress based on methylation-sensitive amplified polymorphism. Plant Physiology and Biochemistry. 2010;48:21–26. doi: 10.1016/j.plaphy.2009.10.005. [DOI] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nature Genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara P, Mougeot F, Martínez-Padilla J, Leckie F, Redpath SM. The condition dependence of a secondary sexual trait is stronger under high parasite infection level. Behavioral Ecology. 2012;23:502–511. [Google Scholar]
- Vergeer P, Ouborg NJ, et al. Evidence for an epigenetic role in inbreeding depression. Biology Letters. 2012;8:798–801. doi: 10.1098/rsbl.2012.0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven KJ, Jansen JJ, van Dijk PJ, Biere A. Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytologist. 2010;185:1108–1118. doi: 10.1111/j.1469-8137.2009.03121.x. [DOI] [PubMed] [Google Scholar]
- Vos P, Hogers R, Bleeker M, et al. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Research. 1995;23:4407–4414. doi: 10.1093/nar/23.21.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson H, Lee DL, Hudson PJ. The effect of Trichostrongylus tenuis on the caecal mucosa of young, old and anthelmintic-treated wild red grouse, Lagopus lagopus scoticus. Parasitology. 1987;94:405–411. doi: 10.1017/s0031182000054056. [DOI] [PubMed] [Google Scholar]
- Watson A, Moss R, Parr R, Mountford M, Rothery P. Kin landownership, differential aggression between kin and, non-kin and population fluctuations in red grouse. Journal of Animal Ecology. 1994;63:39–50. [Google Scholar]
- Webster LMI, Mello LV, Mougeot F, Martínez-Padilla J, Paterson S, Piertney SB. Identification of genes responding to nematode infection in red grouse. Molecular Ecology Resources. 2011a;11:305–313. doi: 10.1111/j.1755-0998.2010.02912.x. [DOI] [PubMed] [Google Scholar]
- Webster LMI, Paterson S, Mougeot F, Martínez-Padilla J, Piertney SB. Transcriptomic response of red grouse to gastro-intestinal nematode parasites and testosterone: implications for population dynamics. Molecular Ecology. 2011b;20:920–931. doi: 10.1111/j.1365-294X.2010.04906.x. [DOI] [PubMed] [Google Scholar]
- Wenzel MA, Webster LM, Blanco G, et al. Pronounced genetic structure and low genetic diversity in European red-billed chough (Pyrrhocorax pyrrhocorax) populations. Conservation Genetics. 2012;13:1213–1230. [Google Scholar]
- Wenzel MA, Webster LM, Paterson S, Mougeot F, Martínez-Padilla J, Piertney SB. A transcriptomic investigation of handicap models in sexual selection. Behavioral Ecology and Sociobiology. 2013;67:221–234. [Google Scholar]
- Wilson GR. The prevalence of caecal threadworms (Trichostrongylus tenuis) in red grouse (Lagopus lagopus scoticus. Oecologia. 1983;58:265–268. doi: 10.1007/BF00399229. [DOI] [PubMed] [Google Scholar]
- Wong B, Wiley EO, Johnson WE, et al. Tissue sampling methods and standards for vertebrate genomics. Giga Science. 2012;1:1–12. doi: 10.1186/2047-217X-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, Li X, Dai F, et al. Comparative methylomics between domesticated and wild silkworms implies possible epigenetic influences on silkworm domestication. BMC Genomics. 2013;14:646. doi: 10.1186/1471-2164-14-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Li X, Korban SS. AFLP-based detection of DNA methylation. Plant Molecular Biology Reporter. 2000;18:361–368. [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Principal coordinate analysis plots of epigenetic and genetic variation in the study populations.
Clone sequence characterization following genome mapping and geneontology annotation. Contains chicken/turkey chromosome locations of mapped clone sequences, nearest ensemble gene identifiers and associated geneontology terms.
Data Availability Statement
DNA clone sequences: genbank Accession nos KJ655444–KJ655520.
Clone sequence characterization including geneontology terms uploaded as supplementary information (Table S1).
Custom R scripts with input files: Dryad 10.5061/dryad.f727j