

Abstract
Aims:
Hippocampal sclerosis (HS) is long-recognized in association with epilepsy (HSE) and more recently in the context of cognitive decline or dementia in the elderly (HSD), in some cases as a component of neurodegenerative diseases, including Alzheimer's disease (AD) and fronto-temporal lobe dementia (FTLD). There is an increased risk of seizures in AD and spontaneous epileptiform discharges in the dentate gyrus of transgenic AD models; epilepsy can be associated with an age-accelerated increase in AD-type pathology and cognitive decline. The convergence between these disease processes could be related to hippocampal pathology. HSE typically shows re-organization of both excitatory and inhibitory neuronal networks in the dentate gyrus, and is considered to be relevant to hippocampal excitability. We sought to compare the pathology of HSE and HSD, focusing on re-organization in the dentate gyrus. Methods: In nine post mortem cases with HSE and bilateral damage, 18 HSD and 11 controls we carried out immunostaining for mossy fibres (dynorphin), and interneuronal networks (NPY, calbindin and calretinin) on sections from the mid-hippocampal body. Fibre sprouting (FS) or loss of expression in the dentate gyrus was semi-quantitatively graded from grade 0 (normal) to grade 3 (marked alteration). Results: Significantly more re-organization was seen with all four markers in the HSE than HSD group (P < 0.01). Mild alterations were noted in HSD group with dynorphin (FS in 3 cases), calretinin (FS in 6 cases), NPY (FS in 11 cases) and calbindin (loss in 10 cases). In eight HSD cases, alteration was seen with more than one antibody but in no cases were the highest grades seen. We also noted NPY and, to a lesser extent, calretinin labelling of Hirano bodies in CA1 of AD cases and some older controls, but not in HSE. Conclusion: Reorganization of excitatory and inhibitory networks in the dentate gyrus is more typical of HSE. Subtle alterations in HSD may be a result of increased hippocampal excitability, including unrecognized seizure activity. An unexpected finding was the identification of NPY-positive Hirano bodies in HSD but not HSE, which may be a consequence of the relative vulnerabilities of interneurons in these conditions.
Keywords: dementia, dentate gyrus, epilepsy, hippocampal sclerosis, Hirano bodies, synaptic reorganization
Introduction
Hippocampal sclerosis (HS) is a pathological change long-associated with epilepsy and although not specific for syndrome or seizure-type, is particularly associated with mesial temporal lobe epilepsy (TLE) 1 from benign 2 to drug-resistant types. Evidence suggests it is a relatively common pathology, identified in 25% of all TLE patients on MRI 3 and histologically confirmed in 33.6% and 30–40% of surgical 1 and post mortem (PM) epilepsy cases 4,5 respectively. The pathological alterations of HS in epilepsy (HSE) are well described 6, with consistent patterns of neuronal loss and gliosis centred on the CA1 subfield, with more variable loss from CA4 and other subfields. More recently, the associated pathology involving the dentate gyrus has been better characterized with dispersion of the granule cell layer (GCD) 7 and sprouting of the mossy fibre axons 8 noted as common alterations. In addition to loss of principal neurones, stereotypical changes to hippocampal interneurons are well documented in surgical series, particularly in the dentate gyrus, including loss and fibre sprouting 9–13. These alterations to the dentate gyrus, which tend to occur synchronously, are likely influenced by hippocampal seizures and excitability rather than neuronal loss and they have been observed in PM as well as surgical series of patients, where they may be bilateral 14.
HS has also been increasingly recognized on imaging and at PM in older and elderly patients, typically in the absence of a seizure history, but in association with memory impairment or dementia (HSD). The proposed causes of HSD are heterogenous, and include anoxic-ischaemic injury and neurodegenerative causes 15,16. HSD has recently been linked to TDP-43 associated pathology 17 in support of underlying degenerative mechanisms. The prevalence of HSD in non-epilepsy, elderly PM series is around 16% 18 and is bilateral in half of the cases 16. The pathological differences between HSE and HSD have not been directly compared, in particular if similar alterations in the dentate gyrus occur. Differences between HSE and HSD would be useful in histopathological evaluation, and may identify distinct mechanisms of cell degeneration, and alterations specific to symptoms, including seizures vs. memory loss. The aim of the current study was to compare the dentate gyrus pathology in a series of HSE with HSD PM cases, in particular with regard to the patterns of GCD, mossy fibre sprouting and interneuronal alterations.
Materials and methods
Cases were selected from the PM archives at the National Hospital for Neurology and Neurosurgery, Queen Square and from the Queen Square Brain Bank, Institute of Neurology, London (details of all cases are available in a supplemental file, Table S1). This study has been approved by the Joint Research Ethics Committee of the Institute of Neurology and appropriate consent was obtained from the patient's next of kin. We selected nine cases with bilateral hippocampal sclerosis, as confirmed at PM, from patients with epilepsy (HSE). Four patients were male and the average age of onset of epilepsy was 11 years with mean duration of disease of 53 years, and the average age of death was 62 years (range 27–87 years). All the patients had partial epilepsy syndromes, with a diagnosis of TLE in four. We selected cases with low Braak stages 5, which included six cases with stage 1 or 2, and three with stage 3. All the patients were residents at the National Society for Epilepsy, Chalfont Centre, UK. For five patients, there was a history of progressive cognitive decline reported in later life 5.
We selected 10 cases of patients with Alzheimer's disease (AD) and diagnosis of HSD, defined as pyramidal cells loss in the hippocampal formation that is out of proportion to the AD neuropathological changes 19. Seven patients were male and the average age of death was 69 years (range 66–78 years). Nine cases were Braak stage 6 and one case Braak stage 5. In addition, we included eight cases with fronto-temporal lobe dementia (FTLD) and a diagnosis of HSD, which included four males. The average age at death was 72 years (range 67–78 years) and the diagnosis was FTLD-TDP type C in three cases, type B in one case, type A in two cases, Pick's disease in one case and unspecified in one case 20. In only one patient with AD was there a history of seizure events following onset of dementia, and in no case was epilepsy documented early in life. In addition, 11 control cases were selected with no history of AD or epilepsy; four were male and the mean age was 55 years (range 36–68 years).
In all cases, one hemisphere was selected and a block taken from the hippocampal body at the level best representing the HS pathology as evaluated on H&E and cresyl violet/luxol fast blue preparations. Sections were cut at 7 μm thickness (and 14 μm for dynorphin labelling) for immunohistochemistry. In brief, endogenous peroxidase activity was blocked with 3% hydrogen peroxide (VWR International, Pennsylvania, USA), sections were microwaved for 15 min in antigen retrieval masking buffer (Vector, Burlingame, CA, USA) and washed in phosphate buffered saline (PBS, Oxiod, Hampshire, UK). The primary antibodies, anti-neuropeptide Y (NPY, 1:4000; Sigma, USA), anti-calretinin (1:4000; Swant, Switzerland), anti-calbindin (1:10 000; Swant), anti-dynorphin (1:50; AbD Serotec, UK), GFAP (1:1500; Dako, Cambridge, UK) and GFAP-δ (1:5000; Chemicon International, Temecula, CA, USA) were diluted in Dako antibody diluent (Dako, Glostrup, Denmark) and applied overnight. DAKO envision horse radish peroxidase (HRP, Dako) was used to detect labelling. Nuclei were counterstained with Haematoxylin (VWR International). The catalysed signal amplification was employed for the dynorphin staining (see Liu et al. 21 for detailed protocol).
In addition, double labelling was performed on selected HSD cases to further characterize NPY-immunopositive Hirano bodies. In brief, sections were initially incubated overnight with anti-MAP2 (1:750; Sigma), AT8 (1:1200; Autogen Bioclear, UK), or SMI 31 (1:1000; Sternberger Monoclonal Incorporation, USA) antibodies. On the following day, Dako Envision EnVision™ HRP solution was applied for 30 min before fluorescein-labelled antibody in tyramide signal amplification (TSA) buffer (1:500; Perkin Elmer, UK) was applied for 8 min. The TSA system is a sensitive detection system that has been used in previous studies on PM human tissue 22,23. Sections were thoroughly washed before anti-NPY antibodies (1:4000) were applied overnight. Rhodamine-labelled antibody in TSA buffer (1:500; Perkin Elmer) was incubated on sections for 8 min at room temperature, before sections were coverslipped in DAPI-mounting media (Vector). Immunofluorescent-labelled sections were viewed under a confocal laser scanning microscope (Zeiss LSM 710) equipped with blue diode, argon and helium/neon lasers.
Qualitative assessment
In each case, the degree of hippocampal neuronal loss in CA4 and CA1 and the pattern of granule cell dispersion were assessed on the H&E section and graded (Table 1) in addition to the distribution of astrocytic gliosis on GFAP and delta-GFAP sections. The presence of mossy fibre sprouting (MFS) and the expression patterns of calbindin, calretinin and NPY in the dentate gyrus (in particular loss of neuronal expression and abnormal fibre sprouting) were assessed and graded as previously described and validated in studies of HSE (Table 1) 12–14,23. The grading was carried out independently by two observers with good agreement (kappa scores between 0.65 and 0.74 for the four markers), and agreement reached on all cases following a joint review.
Table 1.
Grading system for assessment of neuronal loss and evaluation of mossy fibre sprouting (MFS) on dynorphin staining and alterations to inhibitory neuronal markers in the dentate gyrus
| Grade | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| CA1 | No neuronal loss | Neuronal loss present but incomplete (∼50–75%) | Majority of neurones lost (>75%) | |
| CA4 | No neuronal loss | Neuronal loss present but incomplete (∼50–75%) | Majority of neurones lost (>75%) | |
| GCD | Normal granule cell layer | Mild dispersion | Severe dispersion | |
| Dynorphin | Normal pattern: labelling of the mossy fibre pathway in CA4 and CA3 | MFS in IML | MFS in IML and OML | |
| Neuropeptide Y | Normal pattern: radial axonal sprouts inconspicuous & horizontal axonal network in OML > IML | Few radial axonal sprouts in IML but gradient between IML and OML still visible. | Radial axonal sprouts prominent and gradient between IML and OML not visible | As in grade 2 but radial axonal sprouts through IML OML and SGZ |
| Calretinin | Normal pattern: dense band of axons in immediate SGZ and IML | Loss of SGZ labelling | Diminution of dense band of axons in SGZ and IML and axonal sprouts | As in grade 2 but extensive axonal sprouts in IML and OML |
| Calbindin | Normal pattern: strong expression in granule cells, apical dendrites and mossy fibres in CA4 and CA3 | Patchy loss of expression in granule cells | Majority of granule cells negative | Loss of expression predominates in basal cell layer |
Quantitative assessment
In calbindin immunolabelled sections, quantitative analysis of the distribution of positive cell labelling was carried out as previously described 12. In brief, images were captured at ×10 magnification on calbindin stained sections with a Zeiss Axio microscope (Zeiss, Cambridge, UK). Up to 8 fields along the dentate gyrus were selected per case, avoiding angles and curvatures. Using Image-Pro software (version 6.3, Media Cybernetics, UK), the perpendicular distances of cells (μm) in the dentate gyrus was measured from a line drawn along the basal granule cell layer in each image. This was repeated for positively and negatively stained granule cells in each region. The mean distance for each group of cells and the differences between these means was recorded.
Statistical analysis of data between groups was carried out with SPSS (IBM, version 20) using the Mann-Whitney test and Pearson's correlation.
Results
Patterns of HS and GCD
Neuronal loss in the HSD group was predominantly centred on CA1, rather than CA4 subfield, compared with HSE cases (Table S1, Table 2; Figure 1A); a significant difference in the severity of neuronal loss in CA4, but not CA1, was shown for all HSD (AD + FTLD) compared to HSE (P < 0.0001). Additionally, in four HSD cases the neuronal loss in CA1 continued into the subiculum, whereas in HSE cases, there was an abrupt transition between neuronal loss in CA1 and the spared subiculum (Figure 1B). In one HSD case, the neuronal loss was restricted to a segment of CA1. GCD was less pronounced in the HSD group (Table 2) with over 80% showing no dispersion, compared to 44% with no dispersion in the HSE group (Figure 1A,B). GFAP in HSE (Figure 1D) showed a typical pattern of dense fibrous gliosis relatively restricted to CA1 and CA4, sparing CA2 and subiculum, with radial glial fibres in the dentate gyrus. In HSD cases patterns of gliosis were more variable between subfields, as previously noted 24, but in the majority of cases a cellular astrocytosis was equally or more pronounced in the subiculum than CA1, and radial gliosis was less consistently present in the dentate gyrus. In GFAP-δ immunostained sections in both HSD and HSE prominent reactive glia were noted in the subgranular zone and CA4 to a greater extent than CA1, as previously described in HSE 25 (Figure 1C,D); delta-GFAP was therefore less discriminatory than GFAP in the evaluation of HS types.
Table 2.
Results of hippocampal pathology, shown as percentage of cases with each grade in the patient groups (see text and Table 1 for details of grading scheme)
| Grade | CA1 | CA4 | Dynorphin | Calbindin | Calretinin | Neuropeptide Y | GCD | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 3 | 0 | 1 | 2 | 3 | 0 | 1 | 2 | 3 | 0 | 1 | 2 | |
| HSE (n = 9) | 0% | 33% | 67% | 11% | 44% | 44% | 37.5% | 12.5% | 50% | 0% | 11% | 33% | 56% | 29% | 14% | 0% | 57% | 12.5% | 25% | 0% | 62.5% | 44% | 11% | 45% |
| HSD AD (n = 10) | 6% | 50% | 44% | 83% | 17% | 0% | 90% | 10% | 0% | 30% | 50% | 0% | 20% | 60% | 20% | 20% | 0% | 20% | 60% | 20% | 0% | 83% | 17% | 0% |
| HSD FTLD (n = 8) | 71% | 29% | 0% | 62.5% | 25% | 12.5% | 0% | 75% | 12.5% | 12.5% | 0% | 57% | 43% | 0% | 0% | |||||||||
| Control cases (n = 11) | 91% | 9% | 0% | 91% | 9% | 0% | 100% | 0% | 0% | 67% | 22% | 11% | 0% | 100% | 0% | 0% | 0% | 100% | 0% | 0% | 0% | 100% | 0% | 0% |
| Significance* | P = 0.05 | P < 0.0001 | P = 0.01 | P = 0.005 | P = 0.002 | P = 0.01 | P = 0.05 | |||||||||||||||||
n = the number of cases in each group. With some antibodies, occasional cases were not included in the analysis due to repeated technical problems. *Significance is shown as between grades in all HSE and HSD (AD + FTLD) groups using the Mann–Whitney test. HSE, hippocampal sclerosis with epilepsy; HSD, hippocampal sclerosis with dementia; AD, Alzheimer's disease; FTLD, fronto-temporal lobe dementia; GCD, granule cell dispersion.
Figure 1.
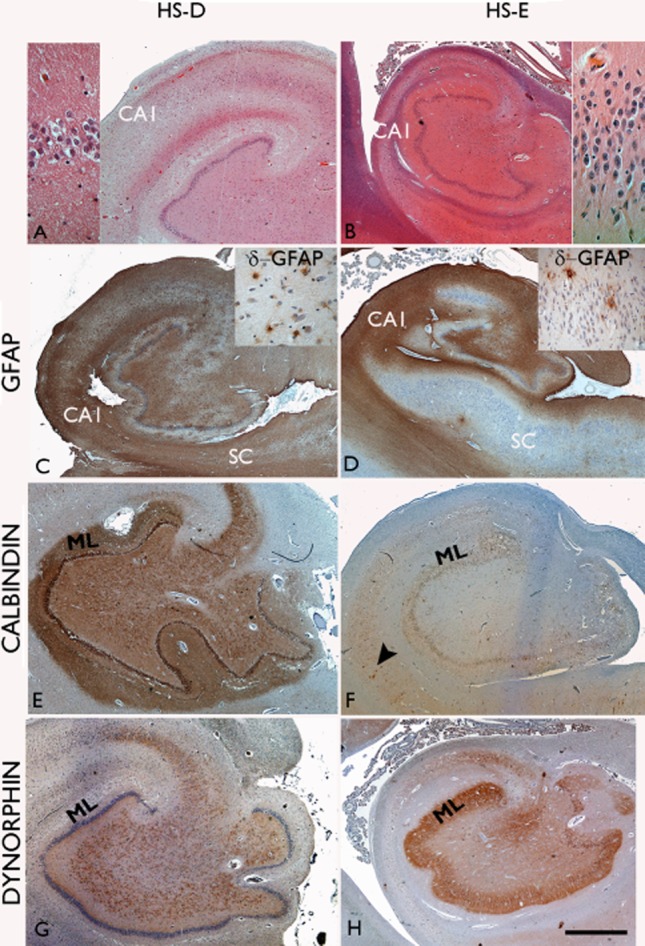
Comparison of hippocampal pathology in hippocampal sclerosis with dementia (HSD; A,C,E,G) and with epilepsy (HSE; B,D,F,H). In both HSD and HSE neuronal loss in CA1 is the predominant finding (A,B); dispersion of the granule cells is more commonly a feature accompanying HSE (B, insert) than HSD (A, insert). Astrocytic gliosis with GFAP immunostaining is more variable and cellular, but can be extensive in HSD and can be equally severe in the subiculum (SC) as in CA1 (C); in HSE there is abrupt, subfield-specific fibrous gliosis demarcating CA1 and CA4 with sparing of the subiculum (SC) and CA2 region. With delta-GFAP immunostaining in HSD and HSE (insets in C,D respectively), scattered multi-polar glial cells are prominent in CA4 and near the basal granule cell layer. (E) Normal calbindin staining is shown in the hippocampus with labelling of granule cell layer, apical dendrites in the molecular layer (ML) and axonal projection in the mossy fibre pathway to CA4 and CA3. (F) In HSE loss of calbindin expression in the granule cells and their processes is a frequent finding but with residual interneurones normally labelling (arrow). (G) A normal dynorphin immunostaining pattern in HSD highlighting the mossy fibre pathway and (H) mossy fibre sprouting in the molecular layer (ML) in HSE. Bar in all approximately equivalent to 2500 μm.
Dynorphin (Figure 1G,H; Figure 2A–C)
Figure 2.
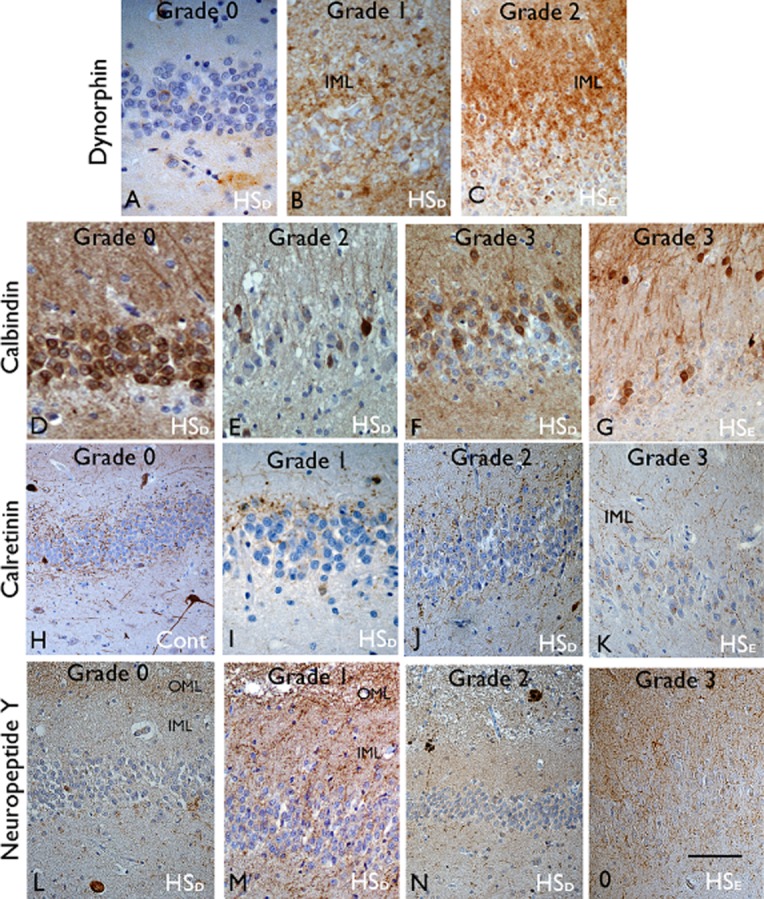
Dentate gyrus alterations in hippocampal sclerosis in epilepsy (HSE) and dementia (HSD) and control (cont). Dynorphin: (A) In the majority of HSD cases a normal mossy fibre pathway (grade 0) was visible with labelling of axons in CA4 and no immunopositivity visible in the molecular layer. (B) Occasional focal sprouting of dynorphin-positive fibres was noted in the inner molecular layer (IML) in three HSD, (grade 1). (C) Severe and extensive mossy fibre sprouting (grade 2) in the molecular layer (IML) was seen only in HSE cases. Calbindin: (D) Normal expression in granule cells, dendrites and mossy fibre axons was noted in controls and HSD cases (grade 0) with, (E) loss of expression in some cases (grade 2). (F) Loss of expression restricted to basal granule cells (grade 3) was noted in three HSD cases without dispersion and (G) was the commonest pattern in HSE cases with granule cell dispersion. Calretinin: (H) a normal pattern of calretinin labelling of the axonal plexus on either side of the granule cell layer is shown in controls (grade 0). (I) Loss of the plexus in the subgranular zone (grade I) and (J) sprouting of fibres in the IML (grade 2) was seen in some HSD cases. (K) In HSE, marked sprouting of fibres in the IML was a prominent finding (grade 3). Neuropeptide Y: (L) Normal pattern (grade 0) with an axonal plexus in the outer molecular layer (OML) was seen in controls. Progressively more prominent sprouting of fibres was noted in the inner molecular layer (IML) in HSD cases (grade 1 to 2; I,J). (O) Extensive NPY fibre sprouting through the molecular layer (grade 3) was only observed in HSE. See also Table 1 for details of descriptions of grading. Bar is equivalent to 300 μm.
In HSE, MFS was confirmed in five cases, with grade 2 sprouting observed in four; there was no MFS in any of the controls. Dynorphin immunolabelling confirmed a normal mossy fibre pathway in the majority of HSD cases, with only three cases showing MFS, one in the AD group (grade 1) and two in the FTLD group (grade 1) (Table S1, Table 2); no grade 2 MFS was noted in the HSD group. Of note, in contrast to the loss of dynorphin labelling of axons and terminals in the normal mossy fibre projection to CA4 and CA3 in HSE cases with MFS, in HSD these pathways appeared better preserved. In addition, in one AD case, plaque-like aggregates of dynorphin were noted in the CA4 region (Figure 3A), likely corresponding to neuritic plaques.
Figure 3.
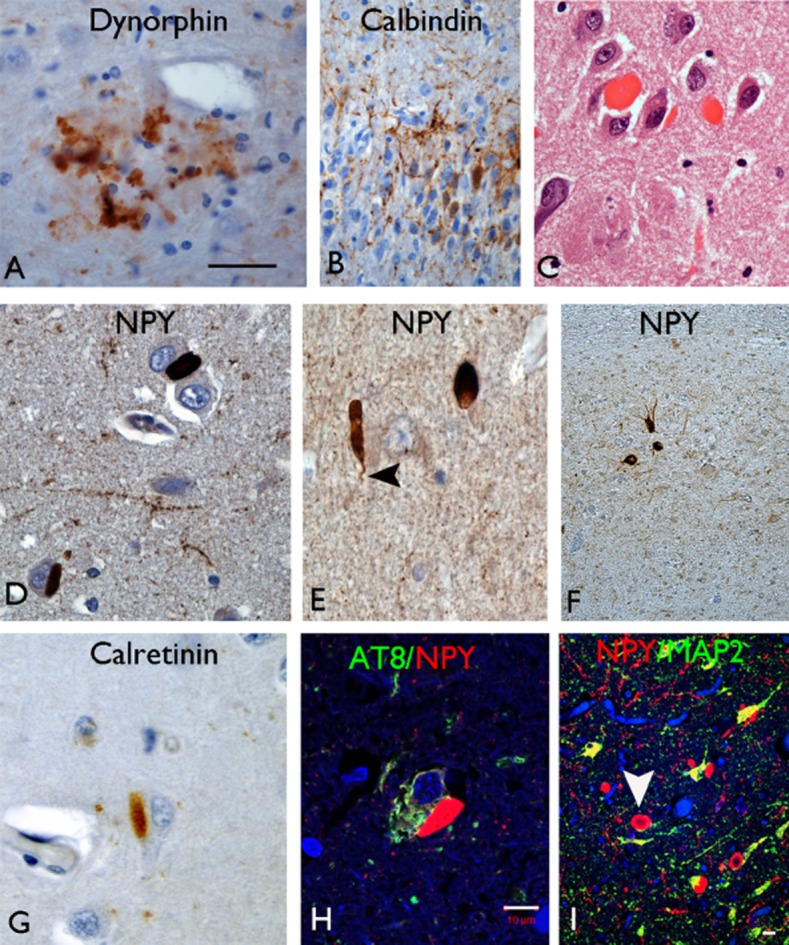
(A) Dynorphin occasionally labelled neuritic plaques in CA4 in Alzheimer's disease cases. (B) Sprouting of calbindin-positive fibres was noted in the dentate gyrus in HSE cases but not in HSD. (C) Hirano bodies were noted in CA1 in the control and HSD groups but not in HSE on H&E preparations. (D) NPY showed intense labelling of numerous Hirano body-like structures in CA1 in HSD in addition to labelling axons and residual interneurons in this region. They were seen in proximity to negatively labelled pyramidal neurones (D) or free in the neuropil (E) with occasional body showing a tail like process (arrowhead in E). (F) In HSE cases, only interneurons and extensive fibre networks were labelled in CA1 with NPY. (G) Weaker labelling of Hirano bodies was noted with calretinin. (H) Con-focal microscopy showed no co-localization of NPY-positive Hirano-like bodies with AT8 positive pyramidal cells in CA1; double-labelling with dendritic marker MAP2 (I) showed approximation of NPY Hirano bodies to neuronal process and dendrites. In addition, a peripheral rim of NPY positivity was appreciated in some Hirano bodies with a hollow central core (arrowhead). Bars in A,C,D,E,G = 75 μm, B,F = 140 μm, H = 20 μm, I = 20 μm.
Calbindin (Figure 1E,F; Figure 2D–G)
Reduced calbindin expression was noted in the granule cells in all HSE cases, the predominant pattern being grade 3, with loss of expression restricted to the more basal cells, as reported previously 12. In addition, prominent sprouting of calbindin-positive fibres through the dentate gyrus was noted in four cases (Figure 3B) as previously described in epilepsy 11. Loss of calbindin was statistically less frequent in HSD (Figure 2D–E), observed in 10 of the 18 cases (seven from the AD group); Only two cases had a grade 3 pattern with the loss of calbindin in the more basal cells compared to the distal cells (Figure 2F), but in contrast to HSE this was not accompanied by GCD (Table 2). Measurements confirmed greater mean differences in the position of positive vs. negatively labelled granule cells from the basal layer in HSE than HSD or control groups (mean values 30.2, 10.1 and −3.9 μm respectively; P < 0.05). Sprouting of calbindin-positive axon fibres was not observed in any HSD cases. The loss of calbindin in granule cells was also present in three controls. In all cases with reduced expression of calbindin in the hippocampal granule cells, normal interneuronal staining was preserved in the adjacent cortex.
Calretinin (Figure 2H–K)
The predominant pattern in HSE was grade 3 (4 cases), with prominent sprouting of fibres and loss of the normal organization (Table 2, Figure 2K). All controls showed a normal calretinin pattern (Figure 2H). Abnormal calretinin pattern was seen in six of the 18 HSD cases (grade 2 (three cases) and grade 1 (three cases); 4 in the AD group) (Figure 2I–J) but no grade 3 patterns were observed. In a further case, loss of calretinin labelling in the dentate gyrus was noted. Calretinin interneurons appeared generally preserved and normal in appearance in the adjacent temporal cortex in AD and FTLD cases.
Neuropeptide Y (Figure 2L–O)
The predominant pattern in the HSE was grade 3, with prominent sprouting of fibres through the dentate gyrus (Figure 1O); a normal NPY pattern was seen in only one case. Controls all showed a normal NPY pattern. In HSD cases, NPY patterns were abnormal in 11 cases [grade 2 (two cases) and grade 1 (nine cases)] (Figure 1L–N); no grade 3 pattern was observed. Occasional NPY processes were seen within senile plaques (Figure 2N).
Statistical analysis confirmed significantly higher grades of reorganization of calbindin, calretinin, NPY and dynorphin labelling in the dentate gyrus in HSE compared to all HSD cases (Table S1 and Table 2). There were strong correlations between calretinin, NPY and dynorphin grades (but not calbindin) in the HSE group (all P < 0.001), but this was less evident in the HSD group (all P > 0.01). In eight cases in the HSD group, abnormal patterns were observed in the dentate gyrus with two or more of the four markers and in two cases (one AD, one FTLD) with all four. In one HSD patient with a history of seizures, grade 1 patterns were seen with calbindin and NPY but dynorphin and calretinin patterns were normal. There were no statistical differences in any of the pathological measures between HSE patients with or without cognitive decline, but the numbers in each group were small; similarly no differences were noted between the HSD AD and FTLD groups.
An additional finding on NPY sections was the prominent labelling of structures resembling Hirano bodies in 12 HSD cases (8 AD, 4 FTLD) (Figure 3D–E). They were regionally restricted to CA1 sector, not seen in other subfields or the neocortex, and their presence was confirmed on corresponding H&E sections (Figure 3C). NPY-positive Hirano bodies were observed adjacent to remaining pyramidal cell somata or ‘free’ in the neuropil (Figure 3D–E). NPY-positive processes were visible as a ‘tail'-like extension from some Hirano bodies (Figure 3E) and a central unlabelled core was noted for some bodies imparting ring-like structures. In seven cases, Hirano body-like structures were visible on calretinin-labelled sections but not with calbindin or dynorphin labelling (Figure 3G). Double-labelling immunofluorescence of NPY with dendritic marker, anti-MAP2, confirmed focal co-localization in some Hirano bodies (Figure 3I), whereas others appeared lying adjacent to, but distinct from, phosphorylated tau (AT8)-positive pyramidal cells (Figure 2H). We did not see Hirano bodies co-localizing with neurofilament-positive axons (not shown). NPY (or calretinin)-labelled Hirano bodies were not seen with any of the epilepsy cases (even up to age 87 years), where only normal NPY interneurons were observed (Figure 3F). NPY-positive Hirano bodies were noted in CA1 in three controls (57–68 years).
Discussion
Hippocampal sclerosis is a generic term used for the description of neuronal loss and gliosis, preferentially involving hippocampal subfields. Although historically associated with epilepsy, there has been increasing recognition in recent years of a similar pathological change arising as a ‘pure’ lesion in elderly, or in association with a neurodegenerative condition. These different clinical groups of HS have not been directly compared histologically. Identification of different cellular alterations would be helpful in their diagnostic evaluation, would allow a deeper understanding of different mechanisms of cell degeneration that may be operating, and would enable the identification of pathological alterations that correlate with specific clinical symptoms, for example cognitive dysfunction vs. hippocampal epileptogenicity.
HSE and HSD may both be bilateral or asymmetrical between hemispheres 23,26, with no clear predilection for left or right and may vary in the extent of its longitudinal involvement along the hippocampal axis (Table 3). In this present study we focused on one level of the hippocampal body from each case, representative of the sclerosis, for detailed analysis. We observed differences between HSD and HSE in the distribution of cell loss between subfields at this coronal level. The neuronal loss in HSD predominantly involved CA1 sector, typically extending into the subiculum in several cases, but sparing CA4. This distribution of neuronal loss has been previously reported in studies of HSD 17,26. This contrasts to the classical pattern of atrophy in HSE, where CA1 damage is accompanied by CA4 neuronal loss with an abrupt transition between the neuronal loss and gliosis in CA1 and the adjacent well-preserved subiculum 1. We also showed that GCD, a common alteration in HSE 27,28, was rare and mild in HSD. GCD has been associated with both an early age of onset of seizures and with the severity of CA4 neuronal loss 27,29. An atypical pattern of HSE shows predominant involvement of CA1 only, and has been associated with a later age of onset of seizures 30,31. These observations could indicate an age-dependent vulnerability of the dentate gyrus and CA4, which is spared in older onset HSE and HSD cases.
Table 3.
Summary of the clinical, pathological and aetiological features that enable the distinction of HSE from HSD
| Feature | HS in epilepsy (HSE) | HS in dementia and ageing (HSD) |
|---|---|---|
| Clinical context | ||
| Typical presentation | Childhood to young adulthood Seizure syndromes, particularly mTLE |
Adult to the ‘oldest-old’ Dementia, cognitive decline. |
| Prevalence | Surgical epilepsy series ∼35% PM epilepsy series ∼30–40% |
In elderly between ∼3–24% ‘Pure’ HS in 23% of dementia cases |
| Distribution | ||
| Bilaterality | Bilateral in ∼48–56% in epilepsy PM series (all syndromes including TLE) | Bilateral in ∼45–60% |
| Longitudinal extent | Localized or extensive along rostro-caudal length 14 | Localized or extensive along rostro-caudal length 26 |
| Pattern of HS | ||
| Distribution of neuronal loss and gliosis | CA1: Typically severe cell loss Subiculum: Spared CA4/3: Neuronal loss Granule cell dispersion in ∼50% |
CA1: Extensive to patchy loss Subiculum: Often cell loss CA4: Spared Granule cell dispersion less evident |
| Circuitry reorganization | ||
| Mossy fibre system | Mossy fibre sprouting typically present and extensive | Mossy fibre sprouting usually absent If present mild and focal |
| Calbindin expression | Reduced expression in granule cells, particularly basal cells | Reduced expression in granule cells may occur |
| NPY expression | Resistance of CA1 interneurones Prominent sprouting in DG |
Loss of CA1 interneurones Mild sprouting in DG in some |
| Calretinin | Reorganization of DG networks | Mild reorganization of DG networks may be present |
| Patho-mechanisms | ||
| Causes of neuronal loss | Seizure mediated/excitotoxic neuronal injury | Heterogenous causes: ‘Ageing’ Vascular brain injury AD, FTLD Synucleinopathies with HS |
| Contribution of TDP-43, ApoE | TDP-43 not identified in surgical unilateral cases 17,59 or bilateral HSE at PM 5 ApoE ε4 associated with earlier onset 60 in some but not all studies 61 |
TDP-43 inclusions in up to 93% of pure HSD
17,47
26 HSD not associated with ApoE ε4 17,47 |
mTLE, mesial temporal lobe epilepsy; DG, dentate gyrus; HS, hippocampal sclerosis; PM, post mortem; AD, Alzheimer's disease; FTLD, fronto-temporal lobe dementia.
Studies of HS in epilepsy series have focussed on cellular alterations that could render this region hyper-excitable. MFS was first observed in experimental seizure models as an early event 8 and has been suggested to be critical to the development of recurrent seizure activity associated with HSE, through enhanced excitability and/or synchronization of granule cells. We have previously shown, in a PM series of HSE cases, that MFS can persist into the ninth decade, even with the remission of seizures 23. Therefore, there is an alternative argument that MFS is an epiphenomenon or response to seizures, rather than a primary epileptogenic process. In one HSD patient in the present series in whom occasional clinical seizures were documented, MFS was not present and in other HSD cases, MFS was rare and mild. In elderly patients and those with cognitive decline, partial seizures may be unrecognized 32,33 because EEG may not be performed as a routine test, therefore we cannot entirely exclude ‘sub-clinical’ seizure activity as the mechanism for the focal MFS seen in HSD. Notwithstanding, the findings favour the view that severe MFS is linked with hippocampal excitability and is therefore a relatively reliable histological marker for HSE.
Alterations to dentate gyrus interneurones have been extensively studied in HSE, in surgical and PM tissues. Cell loss, cyto-morphological alterations and axonal sprouting represent the more common findings (for review see 34) and these stereotypical alterations parallel the principal neuronal loss in established cases 14. Interneuronal alterations are not exclusive to HSE associated with mTLE, and can be seen with other epilepsy syndromes 14,23. It is the axonal sprouting of interneurons that is considered a more functionally significant process in relation to network changes, correlating with synaptic re-organization 9,11. These alterations are easily recognized and reliably graded in tissue sections, as in the present study, and sprouting of these inhibitory networks in the dentate gyrus tend to occur in synchrony with the sprouting of excitatory networks (the mossy fibres) 23. We identified significantly more cases with interneuronal sprouting using calretinin and NPY labelling in HSE than HSD cases with more extensive sprouting (corresponding to the higher grades) as well as sprouting of calbindin-positive fibres, which was exclusive to HSE cases.
In previous studies of the hippocampus in AD, a reduction of NPY axons in the dentate gyrus was demonstrated, with clusters or beaded clumps of axons, some associated with neuritic plaques 35. We also confirmed occasional association of NPY fibres in addition to dynorphin fibres, associated with neuritic plaques in AD. In an APP-mutant model of AD, ectopic expression of NPY was shown in granule cells as well as sprouting of NPY fibres in the molecular layer from hilar NPY neurones 36. The authors proposed that this represented amyloid β-mediated neuronal excitatory activity and subsequent re-modelling of inhibitory circuits that resemble alterations in experimental epilepsy models 36. Our study supports the notion that subtle inhibitory neuronal network alterations may occur in HSD, similar to HSE. As for our observations with MFS in HSD, we cannot exclude subclinical epileptic activity or un-recognized seizures and local hippocampal excitability as a cause for these alterations. Furthermore, in eight of the HSD cases with MFS and interneuronal alterations, parallel changes were seen with more than one marker, supporting systematic network re-organization occurring in the dentate gyrus in some cases. A further consideration is that the cellular pathology occurring in HS with neurodegeneration could inhibit these synaptic reorganizational changes that are more profuse in HS with epilepsy.
Loss of calbindin expression in the dentate granule cells has been shown in experimental AD models (human APP transgenic mutant) as well as in PM series 37–39 and correlated with both cognitive deficits and the abundance of amyloid β protein 40. Loss of calbindin in AD was the most frequent abnormality, observed in 70% of AD HSD compared to 37% in the FTLD group in this study. It is also known that an age-related decline of calbindin in granule cells occurs, which has been associated with impaired hippocampal function 41, and was noted in three controls in the current study. We recently reported a pattern of calbindin loss from the basal granule cells in HSE associated with early onset of seizures, which was the most prominent pattern also in the present HSE cases. This pattern was noted in only two of the HSD cases. It has been proposed that loss of calbindin causes changes to the intrinsic excitability of granule cells in addition to its roles in memory impairment 42. A reduction of CA4 calretinin positive neurones has also been shown in experimental 43 and AD cases 44, although less is known about hippocampal interneuronal alterations in FTLD 45. Calretinin axonal networks have not been previously studied in HSD; they were mildly altered in a minority of cases in the present series.
HSD may be encountered as a ‘pure’ or isolated pathology in the elderly, associated with dementia or an amnestic syndrome and in the absence of typical neuronal inclusions associated with AD or FTLD or vascular disease. This may represent a relatively unrecognized cause of dementia, in the oldest age groups 46 and is associated with TDP-43 immunoreactive inclusions in the granule cells 17,26,47. The present study was limited by not having ‘pure’ HSD cases available for comparison to HSE. Nevertheless from a practical viewpoint, when HS is encountered at PM examination in the elderly, the pattern of neuronal loss in addition to assessment of any neurodegenerative features (including TDP-43), interneuronal markers and mossy fibre alterations may help to identify the cause of the sclerosis (as summarized in Table 3).
A serendipitous finding in the study was the labelling of Hirano bodies in AD and controls for NPY and, to a lesser extent, calretinin. The origin and physiological function of Hirano bodies in ageing and AD has remained elusive since their first recognition 40 year ago. They are filamentous, paracrystalline inclusions, comprising many proteins including actin, co-filin and alpha-actinin 48,49. The focal overlap of expression of the NPY-positive Hirano body with MAP2 supports the view that some are within dendrites, as previously reported 48,50, whereas others may be axonal. It is estimated that CA1 pyramidal cells are supported by 16 types of GABA-ergic neurones 51, including NPY interneurons, many forming a connected ‘pair’ with a single CA1 pyramidal cell 52. NPY-positive interneurons in CA1 are bi-stratified cells with axon terminals on the dendritic domains of CA1 pyramidal cells, mediating inhibition via GABAA receptors 51,53. The NPY cell dendrites extend into the stratum oriens and radiatum and, in turn, receive excitatory input from pyramidal cell collaterals.
Previous studies of CA1 NPY interneurones in AD described the most dramatic changes occurring in this subfield with surviving cells bearing shortened dendrites (‘dendritic stumps’) and damaged thick axons, although labelling of Hirano bodies was not described 35,54. In the PS1/APP transgenic model of AD, loss of NPY-somatostatin co-labelled neurones in CA1 was an early finding, demonstrating a specific vulnerability of these cells in AD, which preceded the pyramidal cell loss 55. In HSE by contrast, relative resistance of NPY-positive interneurons in CA1 has been shown 56 in epilepsy models and in human MTLE/HS cases, despite the extensive loss of CA1 pyramidal cells and hilar NPY cells 31. These observations highlight a different time-course for loss of CA1 NPY interneurons in HSD due to AD vs. HSE. Recent studies have shown PSD-95, a structural protein of excitatory synapses, to label a proportion of Hirano bodies 50; a further study showed co-localization of Hirano bodies with GluR1 and R2 AMPA receptors in the vicinity of pyramidal cells 57. It has been postulated Hirano bodies represent aberrant formation of post-synaptic structures in response to distal synaptic destruction 50. In experimental systems, Hirano bodies have been shown to be both protective against neurodegeneration mediated by tau or APP 58 as well as to inhibit normal cellular function 49. One explanation is that Hirano bodies arise in residual NPY neurones in AD as a compensatory, but cellular degenerative response, to the ongoing imbalances between reciprocal inhibitory and excitatory networks as the disease progresses. The potential functional significance of Hirano bodies is therefore of interest and requires further study.
In summary, reorganization of excitatory and inhibitory networks in the dentate gyrus is more typical of HSE. Subtle alterations may occur in HSD, which could be a result of increased hippocampal excitability, including unrecognized seizure activity. An unexpected finding was the identification of NPY-positive Hirano bodies in HSD but not HSE, which may be a consequence of the relative vulnerabilities of interneurons in these conditions.
Acknowledgments
We are grateful to Queen Square Brain Bank for the donation of tissues, in particular to Linda Parsons and Hilary Ailing. This work was undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. JL is supported by a grant from the MRC (G0600934). The authors have no conflicts of interest to declare. RB carried out the laboratory work, quantitative analysis and contributed to the manuscript preparation; JL carried out all double labelling and confocal analysis and developed the immunohistochemistry protocols; SMS contributed to the clinical data collection; MT contributed with the study design, data analysis and the manuscript preparation. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site
Clinical and pathology data of individual cases.
References
- 1.Blumcke I, Coras R, Miyata H, Ozkara C. Defining clinico-neuropathological subtypes of mesial temporal lobe epilepsy with hippocampal sclerosis. Brain Pathol. 2012;22:402–411. doi: 10.1111/j.1750-3639.2012.00583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Labate A, Gambardella A, Andermann E, Aguglia U, Cendes F, Berkovic SF, Andermann F. Benign mesial temporal lobe epilepsy. Nature reviews. Neurology. 2011;7:237–240. doi: 10.1038/nrneurol.2010.212. [DOI] [PubMed] [Google Scholar]
- 3.Semah F, Picot MC, Adam C, Broglin D, Arzimanoglou A, Bazin B, Cavalcanti D, Baulac M. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology. 1998;51:1256–1262. doi: 10.1212/wnl.51.5.1256. [DOI] [PubMed] [Google Scholar]
- 4.Bonilha L, Martz GU, Glazier SS, Edwards JC. Subtypes of medial temporal lobe epilepsy: influence on temporal lobectomy outcomes? Epilepsia. 2012;53:1–6. doi: 10.1111/j.1528-1167.2011.03298.x. [DOI] [PubMed] [Google Scholar]
- 5.Thom M, Liu JY, Thompson P, Phadke R, Narkiewicz M, Martinian L, Marsdon D, Koepp M, Caboclo L, Catarino CB, Sisodiya SM. Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: a post-mortem study. Brain. 2011;134:2969–2981. doi: 10.1093/brain/awr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thom M. Hippocampal sclerosis: progress since Sommer. Brain Pathol. 2009;19:565–572. doi: 10.1111/j.1750-3639.2008.00201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. 1990;535:195–204. doi: 10.1016/0006-8993(90)91601-c. [DOI] [PubMed] [Google Scholar]
- 8.Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- 9.Magloczky Z. Sprouting in human temporal lobe epilepsy: excitatory pathways and axons of interneurons. Epilepsy Res. 2010;89:52–59. doi: 10.1016/j.eplepsyres.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 10.Magloczky Z, Halasz P, Vajda J, Czirjak S, Freund TF. Loss of calbindin-D28K immunoreactivity from dentate granule cells in human temporal lobe epilepsy. Neuroscience. 1997;76:377–385. doi: 10.1016/s0306-4522(96)00440-x. [DOI] [PubMed] [Google Scholar]
- 11.Magloczky Z, Wittner L, Borhegyi Z, Halasz P, Vajda J, Czirjak S, Freund TF. Changes in the distribution and connectivity of interneurons in the epileptic human dentate gyrus. Neuroscience. 2000;96:7–25. doi: 10.1016/s0306-4522(99)00474-1. [DOI] [PubMed] [Google Scholar]
- 12.Martinian L, Catarino CB, Thompson P, Sisodiya SM, Thom M. Calbindin D28K expression in relation to granule cell dispersion, mossy fibre sprouting and memory impairment in hippocampal sclerosis: a surgical and post mortem series. Epilepsy Res. 2012;98:14–24. doi: 10.1016/j.eplepsyres.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 13.Mathern GW, Babb TL, Pretorius JK, Leite JP. Reactive synaptogenesis and neuron densities for neuropeptide Y, somatostatin, and glutamate decarboxylase immunoreactivity in the epileptogenic human fascia dentata. J Neurosci. 1995;15:3990–4004. doi: 10.1523/JNEUROSCI.15-05-03990.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thom M, Liagkouras I, Martinian L, Liu J, Catarino CB, Sisodiya SM. Variability of sclerosis along the longitudinal hippocampal axis in epilepsy: a post mortem study. Epilepsy Res. 2012;102:45–59. doi: 10.1016/j.eplepsyres.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Probst A, Taylor KI, Tolnay M. Hippocampal sclerosis dementia: a reappraisal. Acta Neuropathol. 2007;114:335–345. doi: 10.1007/s00401-007-0262-1. [DOI] [PubMed] [Google Scholar]
- 16.Zarow C, Sitzer TE, Chui HC. Understanding hippocampal sclerosis in the elderly: epidemiology, characterization, and diagnostic issues. Curr Neurol Neurosci Rep. 2008;8:363–370. doi: 10.1007/s11910-008-0057-3. [DOI] [PubMed] [Google Scholar]
- 17.Nelson PT, Schmitt FA, Lin Y, Abner EL, Jicha GA, Patel E, Thomason PC, Neltner JH, Smith CD, Santacruz KS, Sonnen JA, Poon LW, Gearing M, Green RC, Woodard JL, Van Eldik LJ, Kryscio RJ. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain. 2011;134:1506–1518. doi: 10.1093/brain/awr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickson DW, Davies P, Bevona C, Van Hoeven KH, Factor SM, Grober E, Aronson MK, Crystal HA. Hippocampal sclerosis: a common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathol. 1994;88:212–221. doi: 10.1007/BF00293396. [DOI] [PubMed] [Google Scholar]
- 19.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu JY, Martinian L, Thom M, Sisodiya SM. Immunolabeling recovery in archival, post-mortem, human brain tissue using modified antigen retrieval and the catalyzed signal amplification system. J Neurosci Methods. 2010;190:49–56. doi: 10.1016/j.jneumeth.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 22.Liu JY, Thom M, Catarino CB, Martinian L, Figarella-Branger D, Bartolomei F, Koepp M, Sisodiya SM. Neuropathology of the blood-brain barrier and pharmaco-resistance in human epilepsy. Brain. 2012;135:3115–3133. doi: 10.1093/brain/aws147. [DOI] [PubMed] [Google Scholar]
- 23.Thom M, Martinian L, Catarino C, Yogarajah M, Koepp MJ, Caboclo L, Sisodiya SM. Bilateral reorganization of the dentate gyrus in hippocampal sclerosis: a postmortem study. Neurology. 2009;73:1033–1040. doi: 10.1212/WNL.0b013e3181b99a07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyata H, Hori T, Vinters HV. Surgical pathology of epilepsy-associated non-neoplastic cerebral lesions: a brief introduction with special reference to hippocampal sclerosis and focal cortical dysplasia. Neuropathology. 2013;33:442–458. doi: 10.1111/neup.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinian L, Boer K, Middeldorp J, Hol EM, Sisodiya SM, Squier W, Aronica E, Thom M. Expression patterns of glial fibrillary acidic protein (GFAP)-delta in epilepsy-associated lesional pathologies. Neuropathol Appl Neurobiol. 2009;35:394–405. doi: 10.1111/j.1365-2990.2009.00996.x. [DOI] [PubMed] [Google Scholar]
- 26.Zarow C, Weiner MW, Ellis WG, Chui HC. Prevalence, laterality, and comorbidity of hippocampal sclerosis in an autopsy sample. Brain Behav. 2012;2:435–442. doi: 10.1002/brb3.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blumcke I, Kistner I, Clusmann H, Schramm J, Becker AJ, Elger CE, Bien CG, Merschhemke M, Meencke HJ, Lehmann T, Buchfelder M, Weigel D, Buslei R, Stefan H, Pauli E, Hildebrandt M. Towards a clinico-pathological classification of granule cell dispersion in human mesial temporal lobe epilepsies. Acta Neuropathol. 2009;117:535–544. doi: 10.1007/s00401-009-0512-5. [DOI] [PubMed] [Google Scholar]
- 28.Wieser HG. ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia. 2004;45:695–714. doi: 10.1111/j.0013-9580.2004.09004.x. [DOI] [PubMed] [Google Scholar]
- 29.Thom M, Liagkouras I, Elliot KJ, Martinian L, Harkness W, McEvoy A, Caboclo LO, Sisodiya SM. Reliability of patterns of hippocampal sclerosis as predictors of postsurgical outcome. Epilepsia. 2010;51:1801–1808. doi: 10.1111/j.1528-1167.2010.02681.x. [DOI] [PubMed] [Google Scholar]
- 30.Blumcke I, Pauli E, Clusmann H, Schramm J, Becker A, Elger C, Merschhemke M, Meencke HJ, Lehmann T, von Deimling A, Scheiwe C, Zentner J, Volk B, Romstock J, Stefan H, Hildebrandt M. A new clinico-pathological classification system for mesial temporal sclerosis. Acta Neuropathol. 2007;113:235–244. doi: 10.1007/s00401-006-0187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Lanerolle NC, Kim JH, Williamson A, Spencer SS, Zaveri HP, Eid T, Spencer DD. A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: evidence for distinctive patient subcategories. Epilepsia. 2003;44:677–687. doi: 10.1046/j.1528-1157.2003.32701.x. [DOI] [PubMed] [Google Scholar]
- 32.Gaitatzis A, Sisodiya SM, Sander JW. The somatic comorbidity of epilepsy: a weighty but often unrecognized burden. Epilepsia. 2012;53:1282–1293. doi: 10.1111/j.1528-1167.2012.03528.x. [DOI] [PubMed] [Google Scholar]
- 33.Pandis D, Scarmeas N. Seizures in Alzheimer disease: clinical and epidemiological data. Epilepsy Curr. 2012;12:184–187. doi: 10.5698/1535-7511-12.5.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenfield JG, Love S, Louis DN, Ellison D. Greenfield's Neuropathology. London: Hodder Arnold; 2008. 8th edn. [Google Scholar]
- 35.Chan-Palay V, Lang W, Haesler U, Kohler C, Yasargil G. Distribution of altered hippocampal neurons and axons immunoreactive with antisera against neuropeptide Y in Alzheimer's-type dementia. J Comp Neurol. 1986;248:376–394. doi: 10.1002/cne.902480307. [DOI] [PubMed] [Google Scholar]
- 36.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dumas TC, Powers EC, Tarapore PE, Sapolsky RM. Overexpression of calbindin D(28k) in dentate gyrus granule cells alters mossy fiber presynaptic function and impairs hippocampal-dependent memory. Hippocampus. 2004;14:701–709. doi: 10.1002/hipo.10210. [DOI] [PubMed] [Google Scholar]
- 38.Jouvenceau A, Potier B, Poindessous-Jazat F, Dutar P, Slama A, Epelbaum J, Billard JM. Decrease in calbindin content significantly alters LTP but not NMDA receptor and calcium channel properties. Neuropharmacology. 2002;42:444–458. doi: 10.1016/s0028-3908(01)00202-7. [DOI] [PubMed] [Google Scholar]
- 39.Odero GL, Oikawa K, Glazner KA, Schapansky J, Grossman D, Thiessen JD, Motnenko A, Ge N, Martin M, Glazner GW, Albensi BC. Evidence for the involvement of calbindin D28k in the presenilin 1 model of Alzheimer's disease. Neuroscience. 2010;169:532–543. doi: 10.1016/j.neuroscience.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci USA. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moreno H, Burghardt NS, Vela-Duarte D, Masciotti J, Hua F, Fenton AA, Schwaller B, Small SA. The absence of the calcium-buffering protein calbindin is associated with faster age-related decline in hippocampal metabolism. Hippocampus. 2012;22:1107–1120. doi: 10.1002/hipo.20957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bouilleret V, Schwaller B, Schurmans S, Celio MR, Fritschy JM. Neurodegenerative and morphogenic changes in a mouse model of temporal lobe epilepsy do not depend on the expression of the calcium-binding proteins parvalbumin, calbindin, or calretinin. Neuroscience. 2000;97:47–58. doi: 10.1016/s0306-4522(00)00017-8. [DOI] [PubMed] [Google Scholar]
- 43.Popovic M, Caballero-Bleda M, Kadish I, Van Groen T. Subfield and layer-specific depletion in calbindin-D28K, calretinin and parvalbumin immunoreactivity in the dentate gyrus of amyloid precursor protein/presenilin 1 transgenic mice. Neuroscience. 2008;155:182–191. doi: 10.1016/j.neuroscience.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi H, Brasnjevic I, Rutten BP, Van Der Kolk N, Perl DP, Bouras C, Steinbusch HW, Schmitz C, Hof PR, Dickstein DL. Hippocampal interneuron loss in an APP/PS1 double mutant mouse and in Alzheimer's disease. Brain Struct Funct. 2010;214:145–160. doi: 10.1007/s00429-010-0242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferrer I. Neurons and their dendrites in frontotemporal dementia. Dement Geriatr Cogn Disord. 1999;10:55–60. doi: 10.1159/000051214. (Suppl. 1) [DOI] [PubMed] [Google Scholar]
- 46.Corrada MM, Berlau DJ, Kawas CH. A population-based clinicopathological study in the oldest-old: the 90+ study. Curr Alzheimer Res. 2012;9:709–717. doi: 10.2174/156720512801322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pao WC, Dickson DW, Crook JE, Finch NA, Rademakers R, Graff-Radford NR. Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord. 2011;25:364–368. doi: 10.1097/WAD.0b013e31820f8f50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peterson C, Kress Y, Vallee R, Goldman JE. High molecular weight microtubule-associated proteins bind to actin lattices (Hirano bodies) Acta Neuropathol. 1988;77:168–174. doi: 10.1007/BF00687427. [DOI] [PubMed] [Google Scholar]
- 49.Myre MA. Clues to gamma-secretase, huntingtin and Hirano body normal function using the model organism Dictyostelium discoideum. J Biomed Sci. 2012;19:41. doi: 10.1186/1423-0127-19-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shao CY, Mirra SS, Sait HB, Sacktor TC, Sigurdsson EM. Postsynaptic degeneration as revealed by PSD-95 reduction occurs after advanced Abeta and tau pathology in transgenic mouse models of Alzheimer's disease. Acta Neuropathol. 2011;122:285–292. doi: 10.1007/s00401-011-0843-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol. 2005;562:9–26. doi: 10.1113/jphysiol.2004.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maccaferri G. Stratum oriens horizontal interneurone diversity and hippocampal network dynamics. J Physiol. 2005;562:73–80. doi: 10.1113/jphysiol.2004.077081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szilagyi T, Orban-Kis K, Horvath E, Metz J, Pap Z, Pavai Z. Morphological identification of neuron types in the rat hippocampus. Rom J Morphol Embryol. 2011;52:15–20. [PubMed] [Google Scholar]
- 54.Decressac M, Barker RA. Neuropeptide Y and its role in CNS disease and repair. Exp Neurol. 2012;238:265–272. doi: 10.1016/j.expneurol.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 55.Ramos B, Baglietto-Vargas D, del Rio JC, Moreno-Gonzalez I, Santa-Maria C, Jimenez S, Caballero C, Lopez-Tellez JF, Khan ZU, Ruano D, Gutierrez A, Vitorica J. Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer's disease. Neurobiol Aging. 2006;27:1658–1672. doi: 10.1016/j.neurobiolaging.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 56.Long L, Xiao B, Feng L, Yi F, Li G, Li S, Mutasem MA, Chen S, Bi F, Li Y. Selective loss and axonal sprouting of GABAergic interneurons in the sclerotic hippocampus induced by LiCl-pilocarpine. Int J Neurosci. 2011;121:69–85. doi: 10.3109/00207454.2010.530007. [DOI] [PubMed] [Google Scholar]
- 57.Aronica E, Dickson DW, Kress Y, Morrison JH, Zukin RS. Non-plaque dystrophic dendrites in Alzheimer hippocampus: a new pathological structure revealed by glutamate receptor immunocytochemistry. Neuroscience. 1998;82:979–991. doi: 10.1016/s0306-4522(97)00260-1. [DOI] [PubMed] [Google Scholar]
- 58.Furgerson M, Fechheimer M, Furukawa R. Model Hirano bodies protect against tau-independent and tau-dependent cell death initiated by the amyloid precursor protein intracellular domain. Plos ONE. 2012;7:e44996. doi: 10.1371/journal.pone.0044996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.King A, Sweeney F, Bodi I, Troakes C, Maekawa S, Al-Sarraj S. Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer's disease. Neuropathology. 2010;30:408–419. doi: 10.1111/j.1440-1789.2009.01085.x. [DOI] [PubMed] [Google Scholar]
- 60.Kauffman MA, Consalvo D, Moron DG, Lereis VP, Kochen S. ApoE epsilon4 genotype and the age at onset of temporal lobe epilepsy: a case-control study and meta-analysis. Epilepsy Res. 2010;90:234–239. doi: 10.1016/j.eplepsyres.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 61.Yeni SN, Ozkara C, Buyru N, Baykara O, Hanoglu L, Karaagac N, Ozyurt E, Uzan M. Association between APOE polymorphisms and mesial temporal lobe epilepsy with hippocampal sclerosis. Eur J Neurol. 2005;12:103–107. doi: 10.1111/j.1468-1331.2004.00956.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinical and pathology data of individual cases.