

Abstract
To assess the discriminating power of multiple cerebrospinal fluid (CSF) biomarkers for Parkinson's disease (PD), we measured several proteins playing an important role in the disease pathogenesis. The activities of β-glucocerebrosidase and other lysosomal enzymes, together with total and oligomeric α-synuclein, and total and phosphorylated tau, were thus assessed in CSF of 71 PD patients and compared to 45 neurological controls. Activities of β-glucocerebrosidase, β-mannosidase, β-hexosaminidase, and β-galactosidase were measured with established enzymatic assays, while α-synuclein and tau biomarkers were evaluated with immunoassays. A subset of PD patients (n = 44) was also screened for mutations in the β-glucocerebrosidase-encoding gene (GBA1). In the PD group, β-glucocerebrosidase activity was reduced (P < 0.05) and patients at earlier stages showed lower enzymatic activity (P < 0.05); conversely, β-hexosaminidase activity was significantly increased (P < 0.05). Eight PD patients (18%) presented GBA1 sequence variations; 3 of them were heterozygous for the N370S mutation. Levels of total α-synuclein were significantly reduced (P < 0.05) in PD, in contrast to increased levels of α-synuclein oligomers, with a higher oligomeric/total α-synuclein ratio in PD patients when compared with controls (P < 0.001). A combination of β-glucocerebrosidase activity, oligomeric/total α-synuclein ratio, and age gave the best performance in discriminating PD from neurological controls (sensitivity 82%; specificity 71%, area under the receiver operating characteristic curve = 0.87). These results demonstrate the possibility of detecting lysosomal dysfunction in CSF and further support the need to combine different biomarkers for improving the diagnostic accuracy of PD.
Keywords: Parkinson's disease, CSF biomarkers, lysosomal enzymes, beta-glucocerebrosidase, alpha-synuclein
Aggregation of monomeric α-synuclein (α-syn) into fibrillar or soluble oligomeric forms represents a pivotal step in Parkinson's disease (PD) pathogenesis.1–4 The aggregated form of α-syn is the most abundant protein in Lewy bodies, the pathogenic hallmark of PD.5 Likewise, evidence indicates a fundamental role of lysosomes in α-syn degradation,5–8 linking their possible involvement in PD pathogenesis.9,10 Mutations in the GBA1 gene, encoding the lysosomal hydrolase β-glucocerebrosidase (GCase) and causing Gaucher disease,11 represent a recognized risk factor for PD.12 In another lysosomal storage disorder, GM1 gangliosidosis, caused by β-galactosidase deficiency, patients present parkinsonian features.13 Furthermore, in neurons of β-hexosaminidase–deficient mice, α-syn accumulation has been observed.14
Cerebrospinal fluid (CSF) represents a potential mirror of the complex dynamic molecular events taking place in the brain. CSF total α-syn levels (t-α-syn) are reduced in PD patients with respect to control subjects,15–19 while the oligomeric forms (o-α-syn) are increased.20,21 Two recent studies reported that the combination of α-syn and tau protein species can discriminate PD from other neurodegenerative disorders.17,19 Moreover, in small-scale studies, the activity of GCase has been found to be reduced in CSF of patients with PD22 and dementia with Lewy bodies.23
The aim of our study was to analyze whether a combination of different CSF biomarkers linked to PD pathogenesis may better discriminate PD patients from neurological controls. Thus, in a prospective consecutive series of PD patients and neurological control subjects, we evaluated CSF activity of GCase and other lysosomal enzymes (α-mannosidase, β-mannosidase, β-hexosaminidase, and β-galactosidase), total and oligomeric α-syn, and total and phosphorylated tau levels. In addition, GBA1 gene was sequenced in a large subset of PD patients.
Patients and Methods
Patients
This study included in total 86 PD patients and 45 neurological controls, prospectively and consecutively recruited between 2009 and 2011 in 2 Italian neurological centers. All patients underwent a thorough clinical assessment, including formal neuropsychological evaluation and neuroimaging (brain computed tomography/magnetic resonance imaging). Lumbar puncture was performed on 71 PD patients after written informed consent. Forty-four of 86 PD patients gave consent to genetic assessment for GBA mutations; and for 29 of them CSF was also available. Supporting Figure 1 shows the experimental scheme for PD patients subgroups. Idiopathic PD was diagnosed according to the United Kingdom Parkinson's Disease Society Brain Bank. All patients were treated either with levodopa alone or in combination with dopamine agonists, with good and stable control of motor symptoms (mean Unified Parkinson's Disease Rating Scale–Motor score [UPRDS-III] 23.69 ± 13.97), and were functionally independent (mean Hoehn and Yahr [H&Y], 2.03 ± 0.72). A neuropsychological evaluation was carried out to assess cognitive dysfunction in the diagnostic groups and included Mini Mental State Examination (MMSE) and Milan Overall Dementia Assessment (MODA). Clinical details of the 71 PD patients in which CSF enzyme activities were measured are reported in Table 1, while Supporting Table 1 reports the same data for all the PD patients included in this study. As control group, 45 subjects without cognitive or motor impairments, who underwent lumbar puncture as a part of diagnostic workup for other neurological conditions (ONDs), namely headache, peripheral neuropathies, epilepsy and postural instability, were recruited in this study. CSF was collected according to a common standard protocol following published international guidelines.24 The study was approved by the local Ethical Committees and informed written consent was signed by all patients enrolled. The work was carried out in accordance with Declaration of Helsinki.
fig 1.
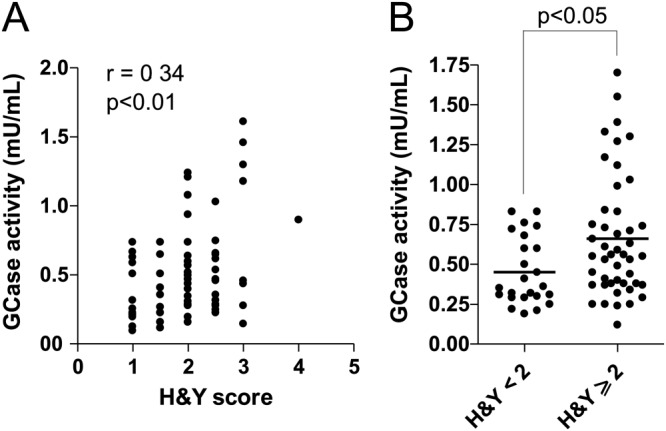
Relationship between CSF GCase activity and clinical scores. A: Correlation of GCase activity with H&Y score was calculated according to Spearman. A weak though significant positive correlation was observed (r = 0.34, P < 0.01). B: Scatter plot of GCase activity in PD patients according to H&Y staging. PD patients with an H&Y stage <2 showed decreased levels of GCase activity with respect to PD patients with H&Y score ≥2 (P < 0.05, Mann-Whitney test). CSF, cerebrospinal fluid; GCase, β-glucocerebrosidase; H&Y, Hoehn and Yahr; PD, Parkinson's disease.
TABLE 1.
Demographic and clinical details of subjects who underwent CSF analysis
| Patient groups | OND | PD |
|---|---|---|
| Patients by gender, n (M/F) | 45 (17/28) | 71 (46/25) |
| Age at examination (y) | 54.5 ± 18.8 | 64.5 ± 9.9a |
| Age at disease onset (y) | – | 58.2 ± 11.2 |
| Disease duration (y) | – | 5.2 ± 4.5 |
| UPDRS-III | – | 23.7 ± 14.0 |
| H&Y score | – | 2.0 ± 0.7 |
| Education (y) | 7.5 ± 3.2 | 8.2 ± 4.5 |
| MMSE | 27.5 ± 2.3 | 26.6 ± 2.7 |
Values are mean ± SD.
= P < 0.01.
CSF, cerebrospinal fluid; OND, other neurological diseases; PD, Parkinson's disease; UPDRS-III, Unified Parkinson's Disease Rating Scale–Motor score; H&Y, Hoehn and Yahr staging; MMSE, Mini Mental State Examination.
CSF Collection
Lumbar puncture was performed between 8:00 am and 10:00 am, after an overnight fast. CSF (10 mL) was collected in sterile polypropylene tubes, centrifuged for 10 minutes at 4000g, and 0.5-mL aliquots were immediately frozen at −80°C. Care was taken so that none of the samples was contamined by blood during the procedure. Samples showing an erythrocyte count>500/mm3 were not included in the study. For CSF α-syn, 0.5-mL samples were thawed on ice and then divided into aliquots in siliconized tubes containing a cocktail of protease inhibitors, including 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), aprotinin, E-64, ethylenediaminetetraacetic acid (EDTA), and leupeptin (Calbiochem-Novabiochem Corporation, San Diego, CA, USA), and 0.05% Tween 20. The samples were then stored at −80°C until required.
Lysosomal Enzymatic Activities
We measured the CSF activity of the following lysosomal enzymes: GCase, α-mannosidase, β-mannosidase, β-hexosaminidase, and β-galactosidase, according to previously published procedures22,23 and within 6 months of the lumbar puncture.
Briefly, for the β-mannosidase and β-hexosaminidase enzyme assays, 10 μL of CSF were incubated with 100 μL of substrate solution for 1 hour at 37°C. The reactions were stopped by adding ice-cold 0.2 M glycine-NaOH buffer, pH 10.4, to a final volume of 3 mL. To measure α-mannosidase and β-galactosidase activities, 50 μL of CSF were incubated in the presence of 100 μL of the corresponding substrate solution for 3 hour. GCase was assayed by incubating 50 μL CSF with 100 μL substrate solution for 24 hours. Fluorescence of the liberated 4-methylumbelliferone was measured on a Perkin-Elmer LS-3 fluorimeter (Waltham, MA, USA) (excitation, 360 nm; emission, 446 nm).
One unit (U) of enzyme activity was defined as the amount of enzyme that hydrolyzes 1 nmol of substrate per minute at 37°C. All the activities were measured in triplicate. Acceptance specification for within-run coefficient of variation (CV) was fixed at less than 10%. In order to test the stability of lysosomal enzymes in CSF after long-term storage, enzymatic activities were measured immediately after the collection (fresh CSF), while aliquots were flash-frozen in liquid nitrogen and stored either at −80°C or −20°C. Enzymatic activities were determined after 1, 2, 3, 4, 8, and 16 weeks. Total protein content was measured spectroscopically using the Bradford's method with bovine serum albumin as the standard.
Immunoassays for CSF Biomarkers
Total and oligomeric CSF α-syn were measured as reported.15,20 Briefly, for CSF total α-syn, an anti-human α-syn monoclonal antibody (clone Syn211) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as primary antibody to capture a-syn species, while the anti-human α-syn polyclonal antibody FL-140 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used for antigen detection. The standard curve for the enzyme-linked immunosorbent assay (ELISA) was constructed using recombinant human α-syn solution at different concentrations diluted in blocking buffer.
For α-syn oligomers, the antibody clone Syn211 was used for capturing, while biotinylated Syn211 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used for antigen detection. The plate was incubated with 50 μL/well of ExtrAvidin-Peroxidase (Sigma-Aldrich, Dorset, UK) and with the enhanced chemiluminescent substrate. For both immunoassays, the samples were screened in blind fashion and were randomly tested. A series of internal controls were run to check for run-to-run variations.
Total tau and phosphorylated tau 181 were measured with commercially available ELISA kits (Innotest hTAU-Ag, p-TAU 181 Ag; Innogenetics NV, Ghent, Belgium).
DNA Amplification and Sequencing
Genomic DNA was extracted from peripheral blood leukocytes of a subset of 44 PD patients using a QIAamp DNA blood Mini Kit (Qiagen GmbH, Hilden, Germany), following the manufacturer's instructions. The exonic and most intronic sequences of the GBA1 gene (Reference sequence J03059.1) were polymerase chain reaction (PCR)-amplified in 3 fragments using primers designed to selectively amplify the gene and not the highly homologous pseudogene, as described by Koprivica et al.25
Following purification of the PCR, each exon and the intronic flanking sequences were sequenced in the forward and reverse directions using the ABI PRISM Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems, Warrington, UK). The sequences were analyzed on an Applied Biosystem 3500xL sequence analyzer. Putative mutations were confirmed by sequencing duplicate PCR products. Mutations are described as recommended, considering nucleotide +1 the A of the first ATG translation initiation codon (http://www.hgvs.org/mutnomen). Nucleotide numbers are derived from the GBA1 cDNA (GenBank reference sequence NM_000157.1). Traditional protein mutation nomenclature does not start with the first ATG codon due to a processed leader sequence (reference sequence AAC63056.1). These mutations are presented without “p.” in the mutation name.
Statistical Analysis
Statistical analyses were performed using SPSS v.15.0, Graphpad Prism v. 5.0, and R software v. 2.14. Biomarker data were described by median and ranges because data distributions were skewed. Mann-Whitney test was initially used for comparisons between the 2 diagnostic groups (P < 0.05). Since the control group and PD group had a significant difference in age (P < 0.01; Table 1), an analysis of variance on ranked variables using age and sex as covariate was conducted (Table 2). Correlations were calculated using Spearman's rho (r), where the accuracy of the analysis was assessed by the area under the curve (AUC) of the receiver operating characteristic (ROC) curve. Cutoff values were calculated using sensitivity and specificity values that maximized Youden's index. A logistic regression approach was used for multiple biomarker evaluation. As the global performance measure of the final model we chose the diagnostic odds ratio (DOR) index,26 instead of sensitivity and specificity, considering that, at the moment, there is no gold standard for CSF biomarkers in PD. With the aim to find the best predictors of PD to be included in the final model, we considered all the CSF enzyme activities that had already shown significant differences between OND and PD groups after the univariate analysis.
TABLE 2.
CSF biomarkers in diagnostic groups
| Biomarkers | OND | PD | P |
|---|---|---|---|
| GCase (mU/mL) | 0.78 (0.34–1.61) | 0.51 (0.32–0.74) | 0.0181 |
| β-mannosidase (mU/mL) | 652.51 (546.55–737.51) | 644.62 (544.84–766.65) | 0.3223 |
| β-hexosaminidase (mU/mL) | 3458.15 (3094.93–4006.33) | 4013.01 (3494.48–4536.56) | 0.0290 |
| β-galactosidase (mU/mL) | 5.39 (2.71–7.26) | 5.33 (1.58–6.76) | 0.5037 |
| t-α-syn (ng/mL) | 39.67 (27.72–64.60) | 22.00 (13.50–38.60) | 0.0146 |
| o-α-syn (RLU) | 3284.50 (1664.50–11719.60) | 4838.50 (2746.00–11196.50) | 0.1110 |
| t-tau (pg/mL) | 185.00 (146.00–285.00) | 176.00 (138.00–266.00) | 0.1380 |
| p-tau (pg/mL) | 36.00 (29.00–43.00) | 42.00 (32.50–53.50) | 0.9032 |
| o/t α-syn ratio | 2.15 (1.34–4.33) | 5.81 (2.26–11.34) | 0.0008 |
Values are median (range). P values of all the biomarkers studied are given after correction for age and sex.
CSF, cerebrospinal fluid; OND, other neurological diseases; PD, Parkinson's disease; GCase, β-glucocerebrosidase; t-α-syn, total α-synuclein; o-α-syn, oligomeric α-synuclein; RLU, Relative Luminescence Units; t-tau, total tau; p-tau, phosphorylated tau 181; o/t α-syn ratio, oligomeric/total α-synuclein ratio.
Results
Demographic data are listed in Table 1. While PD patients were older than controls, no differences in education, MMSE scores, and MODA items were observed between the 2 groups.
Stability of Lysosomal Enzymatic Activities After Long-Term Storage
Data obtained by analyzing 4 samples for each storage temperature have been reported in Supporting Figure 2. Alpha-mannosidase activity showed a dramatic reduction (−60%) after 1 week of storage at −20°C, and a 20% reduction after 1 week of storage at −80°C. Therefore, this enzyme was considered unstable and it was excluded from our study.
fig 2.
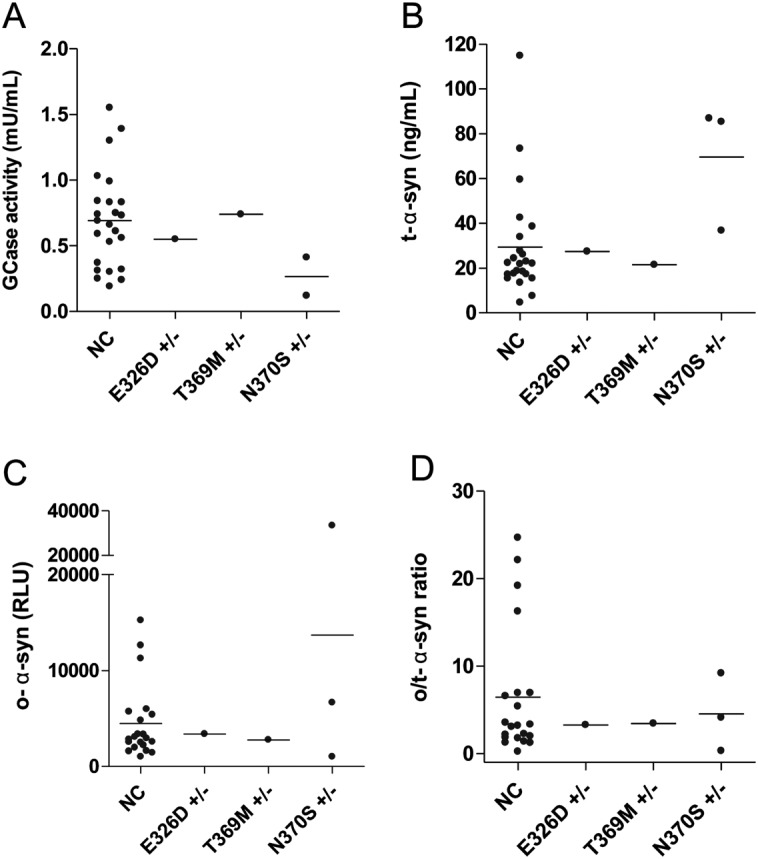
GCase activity and α-syn species levels in PD patients carrying GBA1 mutations and NCs. A: Levels of GCase activity in NC PD patients and in patients carrying different GBA1 mutations. B: Total α-syn levels in CSF of NC and PD patients with GBA1 mutations. C: CSF o-α-syn levels in NC and PD patients with GBA1 mutations. D: CSF o/t-α-syn ratio levels in NC and PD patients with GBA1 mutations. GCase, β-glucocerebrosidase; α-syn, α-synuclein; PD, Parkinson's disease; NC, noncarrier; CSF, cerebrospinal fluid; o-α-syn, oligomeric α-synuclein; o/t-α-syn ratio, oligomeric/total α-synuclein ratio.
Activities of Lysosomal Enzymes in Diagnostic Groups
CSF total protein levels showed no significant differences between PD and OND groups (PD = 0.34 ± 0.15 mg/mL; OND = 0.33 ± 0.18 mg/mL; P = 0.75). Thus, normalization of the enzymatic activities for protein content did not influence the results obtained when comparing the 2 groups.
Median, ranges, and P-values for all enzyme activities in the diagnostic groups are reported in Table 2. As a general comment, values of enzymatic activities showed a large overlap between the 2 groups. However, in the PD group a significant decrease of GCase activity was found, analogously to what was previously observed.22,23 Also, a significant increase in β-hexosaminidase activity was seen, further indicating a derangement of lysosomal function. The correlation matrix among CSF biomarkers for PD and OND groups is reported in Supporting Table 2, and Supporting Table 3 reports the correlations with clinical parameters and functional scores in PD patients. In the PD group, a positive correlation of GCase with H&Y stage (r = 0.34, P < 0.01; Fig. 1A) and with UPDRS-III (r = 0.27, P < 0.01) was found. Interestingly, when the PD cohort was divided into 2 subgroups according to H&Y stage (H&Y < 2 vs H&Y ≥ 2), significantly reduced GCase levels were noticed in earlier stages (Fig. 1B). However, the relatively small number of cases with high H&Y stage seemed to influence this weak correlation. When repeated, excluding the cases with H&Y stage 3 and the single case with H&Y stage 4, the correlation was only of borderline significance (P = 0.054, data not shown).
TABLE 3.
Logistic regression analysis of multiple CSF biomarkers between PD and neurological controls
| Parameter | Estimate | SE | Lower 95% CI | Upper 95% CI | P | AUC | Sensitivity | Specificity | PPV | NPV | LR+ | LR− | DOR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intercept | −3.0249 | 1.3595 | −5.6895 | −0.3603 | 0.0261 | 0.87 | 0.82 | 0.71 | 0.87 | 0.63 | 2.82 | 0.25 | 11.17 |
| Age | 0.0664 | 0.0214 | 0.0245 | 0.1083 | 0.0020 | ||||||||
| GCase | −1.8589 | 0.6339 | −3.1013 | −0.6165 | 0.0034 | ||||||||
| o/t α-syn ratio | 0.2250 | 0.0874 | 0.0537 | 0.3963 | 0.0100 |
Logistic regression was performed in order to evaluate the discriminative power of combination of multiple CSF biomarkers. For each biomarker sensitivity, specificity, PPV, NPV, positive/negative LR, and DOR for the best model, are reported. Combination of GCase, o/t α-syn ratio, and age gave the best performance in discriminating PD patients from the OND group.
CSF, cerebrospinal fluid; PD, Parkinson's disease; CI, confidence interval; AUC, area under the receiver operating characteristic curve; PPV, positive predictive value; NPV, negative predictive value; LR, likelihood ratio; DOR, diagnostic odds ratio; GCase, β-glucocerebrosidase; o/t α-syn ratio, oligomeric/total α-synuclein; OND, other neurological diseases.
The overall performance in discriminating PD from OND was assessed by means of ROC analysis. Discriminative power was quite poor when considering either GCase (specificity 47.6%, sensitivity 85.7%, DOR = 5.5) or β-hexosaminidase (specificity 52.6%, sensitivity 80.0%, DOR = 4.4) as a single parameter (Supporting Table 4).
CSF Total and Oligomeric α-syn in Diagnostic Groups
CSF levels of t-α-syn were significantly decreased in PD patients when compared to the OND group (P < 0.05; Table 2). Also for this biomarker, CSF levels showed a large overlap between the 2 groups. Only in the OND group, CSF t-α-syn level showed an inverse correlation with β-hexosaminidase (r = −0.48, P < 0.001) and with o-α-syn (r = −0.37, P < 0.01).
The median levels of CSF o-α-syn were slightly increased in the PD group (Table 3); however, when the oligomeric/total α-syn ratio (o/t-α-syn ratio) was calculated, increased values in the PD group with respect to OND subjects were highly significant (P < 0.001; Table 3). In the whole sample, CSF o-α-syn was positively associated with β-hexosaminidase activity (r = 0.37, P < 0.01); however, no significant correlations were found between α-syn related biomarkers and clinical scores in any of the groups analyzed.
ROC analysis of t-α-syn alone did not give better results than lysosomal enzymes activities (specificity 75.7%, sensitivity 63.0%, DOR = 5.3). Also o-α-syn as a single marker had a quite poor performance in distinguishing PD patients from control subjects (specificity 41.6%, sensitivity 81.2%, DOR = 3.1), while the o/t-α-syn ratio had slightly better performance (specificity 85.7%, sensitivity 56.2%, DOR = 7.7; Supporting Table 4).
CSF t-tau and p-tau Levels
CSF levels of t-tau and p-tau did not differ significantly between the 2 groups (Table 2). A positive association between tau species and β-hexosaminidase was found in the OND subjects (r = 0.63, P < 0.01 for t-tau; r = 0.53, P < 0.01 for p-tau). Interestingly, in the PD group, t-tau correlated with UPDRS-III and H&Y scores (r = 0.35, P < 0.01 for UPDRS; r = 0.33, P < 0.01 H&Y).
Evaluation of Multiple Biomarkers
In order to assess the performance of multiple biomarkers, a combination logistic regression approach was used. Table 3 shows global performance of the best model according to several measures of test effectiveness, including sensitivity and specificity, positive predictive value (PPV), negative predictive value (NPV), positive/negative likelihood ratio, AUC, and DOR. The best model included GCase, o/t α-syn ratio, and age, having a specificity of 71% and a sensitivity of 82% (PPV 87%, NPV 63%, DOR = 11.17).
Relationship of Lysosomal Enzyme Activity and α-syn Species With GBA1 Genotype
The complete coding region and most intronic sequences of the GBA1 gene were analyzed in 44 of 86 PD patients. Eight patients (18%) presented a sequence variation within the coding region of the GBA1 gene: N370S mutation in heterozygosis (n = 3), T369M mutation (n = 2), E326K mutation (n = 2), and E326D mutation (n = 1). The N370S, T369M, and E326K mutations have been previously described, while the E326D mutation is novel. In 29 of the 44 genotyped patients, CSF was available and enzyme activity measurements were carried out. No correlation between the presence of GBA1 alterations and GCase activity was found. However, 2 patients carrying the N370S mutation showed a lower GCase activity than the median activity of PD patients without GBA1 mutation (the enzyme activity was not available for the third patient; Fig. 2A). Interestingly, patients carrying the N370S mutation also had higher CSF levels of t-α-syn (Fig. 2B) and 1 of them showed the highest value of o-α-syn (Fig. 2C). We also repeated the analysis on performance of biomarkers after excluding N370S mutation carriers. At univariate analysis the differences were still significant, possibly indicating that lysosomal enzymes activities are also changed in the population without N370S mutation (Supporting Table 5). Logistic regression showed similar results (Supporting Table 6).
Discussion
The most relevant result of this study is the discriminative power of the combination of CSF GCase activity, o/t α-syn ratio, and age in distinguishing PD patients from neurological controls. Lysosomal dysfunction and α-syn accumulation represent major pathogenic events in PD.6,7 Therefore, putative biomarkers related to these pathways are of major interest. In this work, we found that CSF GCase activity is reduced in PD patients, and particularly in the earlier stage subgroup (H&Y ≤ 2). Also, the parallel increase of β-hexosaminidase activity, as found in Gaucher patients, is consistent with the capability of CSF to globally reflect lysosomal derangement. Mutations on the GBA1 gene have been extensively reported as a strong risk factor for PD,12,27–31 with a penetrance ranging from 7% to 30% in N370S mutation carriers aged from 50 to 80 years.32 In N370S GBA1 mutation carriers, we observed lower GCase activity compared to noncarriers, while these cases also showed higher median levels of t-α-syn with respect to PD patients not harboring GBA1 mutations. The highest value of o-α-syn observed was in one N370S GBA1 mutation carrier (Fig. 2D). It is of interest to note that after excluding PD patients carrying the N370S mutation, mean values of GCase activity remained significantly lower than those observed in neurological controls. In PD patients screened for GBA1 mutations we also noted other sequence changes, T369M and E326K, and a new one, E326D. Both T369M and E326K have been reported as single alterations in patients affected by PD.28,33–35 In a postmortem investigation carried out in brains from PD patients, either with heterozygous GBA mutations or with sporadic disease, GCase activity was significantly decreased in several brain areas of both groups, heavily affecting the substantia nigra.36 The molecular link between GCase and α-syn has been recently shown in cellular models.37,38 Accumulation of mutant and wild-type α-syn might impair lysosomal GCase targeting, additionally damaging lysosomal degradation pathways and amplifying α-syn aggregation.37 On the other hand, a recent study challenged the hypothesis of the link between GCase and α-syn, showing that, after pharmacological inhibition of GCase, α-syn cellular levels were unchanged.39
In contrast to the reduction of CSF GCase activity, we found a significant increase of CSF β-hexosaminidase activity in the PD group. This enzyme is also elevated in plasma of GD patients.40 It is worth noting that β-hexosaminidase is involved in the production of GM3 ganglioside, which plays a protective effect in neurodegenerative processes taking place in PD.41 Again, the decrease of GCase together with the increase of β-hexosaminidase activity observed in CSF of PD patients strengthen the parallelism between GD and PD.
Recently, the activity of several lysosomal enzymes has been measured in CSF from a Dutch cohort of de novo PD patients and healthy controls.42 In this series, GCase activity showed only a trend toward reduction, together with a significant increase of cathepsin E. Major differences in the study groups may be responsible for the dissimilar results. In our cohort the vast majority of PD patients had already been treated with dopaminergic drugs, while the Dutch cohort included only de novo PD patients. As a control group, patients with minor neurological conditions were enrolled while in the Dutch cohort the controls were healthy subjects. Also, CSF total protein content was significantly higher in PD patients than in healthy controls in the Dutch cohort, while no difference was observed between PD and OND in the present series, thereby also suggesting population-specific differences.
With respect to α-syn–related biomarkers, in our PD series we confirmed the reduction of CSF t-α-syn levels.15,17,18,43–45 We also found an increase of both o-α-syn and the o-α-syn/t-α-syn ratio, in agreement with results from other groups.20,21
Recently, in a large cohort study by Mollenhauer and colleagues,17 the combination of CSF α-syn measurement together with tau protein showed a good performance in distinguishing synucleinopathies (ie, PD, Lewy body dementia [DLB], multiple system atrophy [MSA]) from other neurological disorders. The tau/α-syn ratio has also been shown to be able to discriminate PD from tauopathies and other neurodegenerative conditions.19
Tau biomarkers were not included in the final logistic regression model because of the poor discriminative power in the univariate analysis. This observation is not surprising, as these biomarkers were shown to reliably distinguish PD from other neurodegenerative disorders,17 but had limited value in discriminating PD from neurological controls.19
The present study is limited by the lack of healthy subjects as controls. Our control group was composed of patients with minor neurological conditions necessitating lumbar puncture for diagnostic purposes. In order to minimize this issue, we carefully excluded pathological conditions possibly causing CSF alteration of protein content due to inflammatory reaction, as well as any other clinically relevant non-neurological disease. Another limitation is the partial genetic assessment carried out in our PD cohort, mostly due to the unavailability of some patients to undergo genetic analysis.
In conclusion, lysosomal dysfunction can be detected in CSF of PD patients, and the combined measurement of GCase activity, o/t α-syn ratio, and age best discriminates PD patients from neurological controls, further indicating that multiple CSF biomarkers reflecting different pathogenic pathways may be of help in improving PD diagnostic accuracy.45
Acknowledgments
The authors would like to thank Mr. Cristiano Spaccatini for his technical assistance, Dr. Richard Honeywell and Professor Keith Bagnall for the English language revision.
Supporting Data
Additional supporting information may be found in the online version of this article at the publisher's web-site.
References
- 1.Trojanowski JQ, Lee VM. Aggregation of neurofilament and alpha-synuclein proteins in Lewy bodies: implications for the pathogenesis of Parkinson disease and Lewy body dementia. Arch Neurol. 1998;55:151–152. doi: 10.1001/archneur.55.2.151. [DOI] [PubMed] [Google Scholar]
- 2.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karpinar DP, Balija MB, Kugler S, et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson's disease models. EMBO J. 2009;28:3256–3268. doi: 10.1038/emboj.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Winner B, Jappelli R, Maji SK, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 6.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 7.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 8.Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci. 2004;24:1888–1896. doi: 10.1523/JNEUROSCI.3809-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain. 2008;131(Pt 8):1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- 11.Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet. 2008;372:1263–1271. doi: 10.1016/S0140-6736(08)61522-6. [DOI] [PubMed] [Google Scholar]
- 12.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roze E, Paschke E, Lopez N, et al. Dystonia and parkinsonism in GM1 type 3 gangliosidosis. Mov Disord. 2005;20:1366–1369. doi: 10.1002/mds.20593. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki K, Iseki E, Togo T, et al. Neuronal and glial accumulation of alpha- and beta-synucleins in human lipidoses. Acta Neuropathol. 2007;114:481–489. doi: 10.1007/s00401-007-0264-z. [DOI] [PubMed] [Google Scholar]
- 15.Tokuda T, Salem SA, Allsop D, et al. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson's disease. Biochem Biophys Res Commun. 2006;349:162–166. doi: 10.1016/j.bbrc.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 16.Mollenhauer B, Cullen V, Kahn I, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213:315–325. doi: 10.1016/j.expneurol.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 17.Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Doring F, Trenkwalder C, Schlossmacher MG. alpha-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol. 2011;10:230–240. doi: 10.1016/S1474-4422(11)70014-X. [DOI] [PubMed] [Google Scholar]
- 18.Hong Z, Shi M, Chung KA, et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson's disease. Brain. 2010;133(Pt 3):713–726. doi: 10.1093/brain/awq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parnetti L, Chiasserini D, Bellomo G, et al. Cerebrospinal fluid Tau/alpha-synuclein ratio in Parkinson's disease and degenerative dementias. Mov Disord. 2011;26:1428–1435. doi: 10.1002/mds.23670. [DOI] [PubMed] [Google Scholar]
- 20.Tokuda T, Qureshi MM, Ardah MT, et al. Detection of elevated levels of alpha-synuclein oligomers in CSF from patients with Parkinson disease. Neurology. 2010;75:1766–1772. doi: 10.1212/WNL.0b013e3181fd613b. [DOI] [PubMed] [Google Scholar]
- 21.Bruggink KA, Kuiperij HB, Ekholm-Pettersson F, Verbeek MM. Detection of elevated levels of alpha-synuclein oligomers in CSF from patients with Parkinson disease. Neurology. 2011;77:510–511. doi: 10.1212/WNL.0b013e318219dd92. author reply. [DOI] [PubMed] [Google Scholar]
- 22.Balducci C, Pierguidi L, Persichetti E, et al. Lysosomal hydrolases in cerebrospinal fluid from subjects with Parkinson's disease. Mov Disord. 2007;22:1481–1484. doi: 10.1002/mds.21399. [DOI] [PubMed] [Google Scholar]
- 23.Parnetti L, Balducci C, Pierguidi L, et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in dementia with Lewy bodies. Neurobiol Dise. 2009;34:484–486. doi: 10.1016/j.nbd.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Teunissen CE, Petzold A, Bennett JL, et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology. 2009;73:1914–1922. doi: 10.1212/WNL.0b013e3181c47cc2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koprivica V, Stone DL, Park JK, et al. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet. 2000;66:1777–1786. doi: 10.1086/302925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glas AS, Lijmer JG, Prins MH, Bonsel GJ, Bossuyt PM. The diagnostic odds ratio: a single indicator of test performance. J Clin Epidemiol. 2003;56:1129–1135. doi: 10.1016/s0895-4356(03)00177-x. [DOI] [PubMed] [Google Scholar]
- 27.Lesage S, Anheim M, Condroyer C, et al. French Parkinson's Disease Genetics Study Group. Large-scale screening of the Gaucher's disease-related glucocerebrosidase gene in Europeans with Parkinson's disease. Hum Mol Genet. 2011;20:202–210. doi: 10.1093/hmg/ddq454. [DOI] [PubMed] [Google Scholar]
- 28.Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab. 2004;81:70–73. doi: 10.1016/j.ymgme.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 29.Neudorfer O, Giladi N, Elstein D, et al. Occurrence of Parkinson's syndrome in type I Gaucher disease. QJM. 1996;89:691–694. doi: 10.1093/qjmed/89.9.691. [DOI] [PubMed] [Google Scholar]
- 30.Sun QY, Guo JF, Wang L, et al. Glucocerebrosidase gene L444P mutation is a risk factor for Parkinson's disease in Chinese population. Mov Disord. 2010;25:1005–1011. doi: 10.1002/mds.23009. [DOI] [PubMed] [Google Scholar]
- 31.Ziegler SG, Eblan MJ, Gutti U, et al. Glucocerebrosidase mutations in Chinese subjects from Taiwan with sporadic Parkinson disease. Mol Genet Metab. 2007;91:195–200. doi: 10.1016/j.ymgme.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anheim M, Elbaz A, Lesage S, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology. 2012;78:417–420. doi: 10.1212/WNL.0b013e318245f476. [DOI] [PubMed] [Google Scholar]
- 33.Clark LN, Ross BM, Wang Y, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology. 2007;69:1270–1277. doi: 10.1212/01.wnl.0000276989.17578.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalinderi K, Bostantjopoulou S, Paisan-Ruiz C, Katsarou Z, Hardy J, Fidani L. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Greece. Neurosci Lett. 2009;452:87–89. doi: 10.1016/j.neulet.2009.01.029. [DOI] [PubMed] [Google Scholar]
- 35.Siebert M, Donis KC, Socal M, et al. Glucocerebrosidase gene variants in parkinsonian patients with Machado Joseph/spinocerebellar ataxia 3. Parkinsonism Relat Disord. 2012;18:185–190. doi: 10.1016/j.parkreldis.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 36.Gegg ME, Burke D, Heales SJ, et al. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol. 2012;72:455–463. doi: 10.1002/ana.23614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mazzulli JR, Xu YH, Sun Y, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cullen V, Sardi SP, Ng J, et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann Neurol. 2011;69:940–953. doi: 10.1002/ana.22400. [DOI] [PubMed] [Google Scholar]
- 39.Dermentzaki G, Dimitriou E, Xilouri M, Michelakakis H, Stefanis L. Loss of beta-glucocerebrosidase activity does not affect alpha-synuclein levels or lysosomal function in neuronal cells. PloS One. 2013;8:e60674. doi: 10.1371/journal.pone.0060674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Natowicz MR, Prence EM, Cajolet A. Marked variation in blood beta-hexosaminidase in Gaucher disease. Clin Chim Acta. 1991;203:17–22. doi: 10.1016/0009-8981(91)90152-3. [DOI] [PubMed] [Google Scholar]
- 41.Di Pasquale E, Fantini J, Chahinian H, Maresca M, Taieb N, Yahi N. Altered ion channel formation by the Parkinson's-disease-linked E46K mutant of alpha-synuclein is corrected by GM3 but not by GM1 gangliosides. J Mol Biol. 2011;397:202–218. doi: 10.1016/j.jmb.2010.01.046. [DOI] [PubMed] [Google Scholar]
- 42.van Dijk KD, Persichetti E, Chiasserini D, et al. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson's disease. Mov Disord. 2013;28:747–754. doi: 10.1002/mds.25495. [DOI] [PubMed] [Google Scholar]
- 43.Parnetti L. Biochemical diagnosis of neurodegenerative diseases gets closer. Lancet Neurol. 2011;10:203–205. doi: 10.1016/S1474-4422(11)70019-9. [DOI] [PubMed] [Google Scholar]
- 44.Shi M, Bradner J, Hancock AM, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol. 2011;69:570–580. doi: 10.1002/ana.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Dijk KD, Bidinosti M, Weiss A, Raijmakers P, Berendse HW, van de Berg WD. Reduced alpha-synuclein levels in cerebrospinal fluid in Parkinson's disease are unrelated to clinical and imaging measures of disease severity. Eur J Neurol. doi: 10.1111/ene.12176. (in press) [Epub 2013 Apr 30.] 10.1111/ene.12176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web-site.