

Abstract
Release of mitochondrial contents often triggers inflammation and cell death, and modulating this process can be advantageous to invading pathogens. In this issue of The EMBO Journal, Andree and colleagues reveal new findings that an intracellular bacterial pathogen exploits apoptotic machinery to suppress host immune signaling, yet avoids cell death. This study emphasizes the need to expand our understanding of the roles played by pro-apoptotic proteins in non-death scenarios.
See also: M Andree et al (October 2014)
The nature of cell death during infection can have a profound effect on disease, and therefore, microbial invaders often interfere with the execution of cell death programming. The signals sent by dying cells can be stealthy or inflammatory (Fink & Cookson, 2005). Some pathogens, such as Yersinia pestis, induce cell death to eliminate immune cells, allowing the microbes to disseminate and cause disease (Ashida et al, 2011). In contrast, cell death can be an efficient defense against intracellular pathogens, like Shigella, by eliminating the replicative niche; thus, it can be advantageous to the pathogen to circumvent cell death.
The mitochondrial network, which houses the cellular metabolic machinery, is integral to programmed cell death. Mitochondria serve as central hubs for receiving and generating cellular stress signals. Release of mitochondrial components like ROS, mtDNA or intermembrane space (IMS) proteins, generally triggers inflammation and/or cell death (West et al, 2011). The canonical apoptosis program relies on pro-apoptotic proteins located in the mitochondrial IMS. Rapid decreases in mitochondrial membrane potential (Δψm) induce release of IMS proteins into the cytosol, leading to activation of caspases and other regulators of apoptosis (Tait & Green, 2010). Two IMS proteins, second mitochondria-derived activator of caspases (SMAC) and HtrA2/OMI, are critical to the death rheostat. These proteins are potent antagonists for the inhibitor of apoptosis protein (IAP) family (Tait & Green, 2010). For example, X-linked inhibitor of apoptosis (XIAP), is antagonized by SMAC/OMI in Fas-mediated cell death. While the involvement of XIAP in apoptosis has been well studied, recent evidence highlights an additional function for XIAP in immune signaling. Mice deficient in XIAP are more susceptible to infection by intracellular pathogens and exhibit reduced proinflammatory cytokine production (Bauler et al, 2008; Prakash et al, 2010). Upon activation of the Nod-like receptors, NOD1 or NOD2, XIAP facilitates NF-κB activation, leading to proinflammatory gene transcription (Krieg et al, 2009). These studies indicate a key role for XIAP in innate immunity and suggest that XIAP-dependent innate immune signaling might be targeted by intracellular pathogens.
Andree et al (2014) now demonstrate that Shigella uses the antagonistic functions of SMAC/OMI to suppress innate immune function in the absence of cell death, revealing a new mechanism to promote infection. Infection by invasive Shigella triggered XIAP-mediated NOD1 signaling, which initially resulted in NF-κB activation and cytokine production. However, NF-κB activation and cytokine production decreased as infection progressed. Concomitant with the decline of NF-κB activation, release of SMAC and OMI from the mitochondria of infected cells steadily increased. SMAC bound to XIAP during the Shigella infection, disrupting the interaction between XIAP and RIP2, which is required for NOD-dependent NF-κB activation. Release of IMS proteins into the cytosol is generally indicative of apoptosis, but Shigella-infected cells did not show typical characteristics of programmed cell death. Activation of pro-apoptotic caspases and PARP cleavage were absent in Shigella-infected cells, and Δψm, which drops during apoptosis, remained intact. These observations suggest that Shigella suppresses innate immune responses, while maintaining viability of the host cell to support the replicative niche.
How could SMAC/OMI be released from the mitochondria without apparent dysfunction? Andree et al investigated the possibility that a pro-apoptotic member of the BCL2 protein family mediated the release of IMS proteins by mitochondrial outer membrane permeabilization. During Shigella infection, Bid was the only activated BCL2-family protein associated with mitochondria. Knockdown or knockout of Bid led to an increase in NF-κB activation upon Shigella infection, as well as a decrease in SMAC/OMI release into the cytosol. Activation of Bid by cleavage can be carried out by several host proteases, including caspase-8, cathepsin, and calpain, with caspases predominantly cleaving Bid during apoptosis. Bid from Shigella-infected cells was truncated at the calpain cleavage site, and inhibition of calpain in Shigella-infected cells decreased Bid cleavage and prevented SMAC release. Taken together, these data reinforce the pivotal role of XIAP in facilitating immune responses against intracellular pathogens and reveal a mechanism by which the mitochondrial proteins, SMAC and OMI, can be exploited to establish an immunosuppressive environment during infection.
This study provokes many questions about the roles and regulation of mitochondrial proteins during conditions of cellular stress. Although Andree et al demonstrated that Bid was important for SMAC/OMI release, it remains unclear how these proteins escape the mitochondrial IMS. The three mechanisms by which IMS proteins are known to be released from mitochondria are formation of Bax/Bak pores, opening of the mitochondrial permeability transition pore (MPTP) or outer membrane rupture (Tait & Green, 2010) (Fig1). SMAC/OMI release during Shigella infection did not appear to be dependent on any of these routes, suggesting an unknown mechanism that selectively releases SMAC/OMI into the cytosol. The release of IMS proteins is often associated with cellular damage, resulting in cell death; however, in this study, SMAC/OMI release was not coincident with other indicators of mitochondrial stress and was not lethal. It may be that low levels of stress trigger selective release of IMS proteins, which induce immunosuppressive or ‘safe’ responses, whereas high stress levels, for example, all IMS proteins released, induce proinflammatory or ‘danger’ signals that eventually result in cell death. If so, it is to the advantage of intracellular pathogens to trigger minimal stress in the infected host cell. Lastly, this study illuminates additional roles for ‘pro-apoptotic’ proteins that may be distinct from their function in programmed cell death. In addition to the work by Andree et al, other studies provide evidence that pro-apoptotic proteins like Bid, Bcl-XL, and caspases, modulate signaling independently of cell death (Bruey et al, 2007; Yeretssian et al, 2011). Thus, regulators of host cell death machinery may be important targets for future investigation into mechanisms of immune modulation in infectious and inflammatory disease.
Figure 1. Mechanisms of mitochondrial outer membrane permeabilization and release of intermembrane space (IMS) proteins.
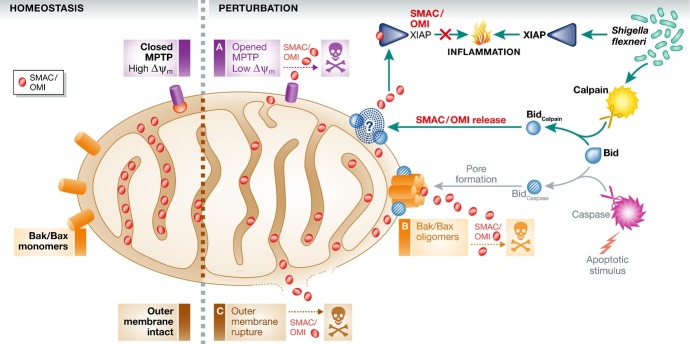
During homeostasis, mitochondria maintain high membrane potential (Δψm), an intact outer membrane, and IMS proteins, such as SMAC/OMI, are retained. In the presence of an apoptotic signal, Δψm drops significantly, leading to the release of IMS proteins by: (A) sustained opening of the mitochondrial permeability transition pore (MPTP), (B) Bid-mediated oligomerization of Bak and Bax on the outer mitochondrial membrane or (C) rupture of the outer mitochondrial membrane. In the case of Shigella infection, mitochondria selectively release the IMS proteins, SMAC and OMI, by a mechanism that remains uncharacterized. This mechanism requires Bid cleavage by calpain, rather than caspases, as would generally occur during apoptosis. Binding of SMAC/OMI to the X-linked inhibitor of apoptosis protein (XIAP) prevents the XIAP-dependent upregulation of proinflammatory cytokines that is stimulated by infection.
References
- Andree M, Seeger JM, Schull S, Coutelle O, Wagner-Stippich D, Wiegmann K, Wunderlich CM, Brinkmann K, Broxtermann P, Witt A, Fritsch M, Martinelli P, Bielig H, Lamkemeyer T, Rugarli EI, Kaufmann T, Sterner-Kock A, Wunderlich FT, Villunger A, Martins LM, et al. BID-dependent release of mitochondrial SMAC dampens XIAP-mediated immunity against Shigella. EMBO J. 2014;33:2171–2187. doi: 10.15252/embj.201387244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, Sasakawa C. Cell death and infection: a double-edged sword for host and pathogen survival. J Cell Biol. 2011;195:931–942. doi: 10.1083/jcb.201108081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauler LD, Duckett CS, O'Riordan MX. XIAP regulates cytosol-specific innate immunity to Listeria infection. PLoS Pathog. 2008;4:e1000142. doi: 10.1371/journal.ppat.1000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, Matsuzawa S, Terskikh AV, Faustin B, Reed JC. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007;129:45–56. doi: 10.1016/j.cell.2007.01.045. [DOI] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z, Knoefel WT, Reed JC. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA. 2009;106:14524–14529. doi: 10.1073/pnas.0907131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash H, Albrecht M, Becker D, Kuhlmann T, Rudel T. Deficiency of XIAP leads to sensitization for Chlamydophila pneumoniae pulmonary infection and dysregulation of innate immune response in mice. J Biol Chem. 2010;285:20291–20302. doi: 10.1074/jbc.M109.096297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeretssian G, Correa RG, Doiron K, Fitzgerald P, Dillon CP, Green DR, Reed JC, Saleh M. Non-apoptotic role of BID in inflammation and innate immunity. Nature. 2011;474:96–99. doi: 10.1038/nature09982. [DOI] [PubMed] [Google Scholar]