

Abstract
The protease activity of the paracaspase Malt1 has recently gained interest as a drug target for immunomodulation and the treatment of diffuse large B-cell lymphomas. To address the consequences of Malt1 protease inactivation on the immune response in vivo, we generated knock-in mice expressing a catalytically inactive C472A mutant of Malt1 that conserves its scaffold function. Like Malt1-deficient mice, knock-in mice had strong defects in the activation of lymphocytes, NK and dendritic cells, and the development of B1 and marginal zone B cells and were completely protected against the induction of autoimmune encephalomyelitis. Malt1 inactivation also protected the mice from experimental induction of colitis. However, Malt1 knock-in mice but not Malt1-deficient mice spontaneously developed signs of autoimmune gastritis that correlated with an absence of Treg cells, an accumulation of T cells with an activated phenotype and high serum levels of IgE and IgG1. Thus, removal of the enzymatic activity of Malt1 efficiently dampens the immune response, but favors autoimmunity through impaired Treg development, which could be relevant for therapeutic Malt1-targeting strategies.
Keywords: colitis, EAE, gastritis, NF-κB, paracaspase
See also: A Bertossi & D Krappmann (December 2014)
Introduction
The paracaspase Malt1, a signaling protein with homology to caspases, is essential for cellular activation by surface receptors with immunoreceptor tyrosine-based activation motifs (ITAMs), such as the B-cell receptor (BCR) or the T-cell receptor (TCR) (Thome, 2008; Rosebeck et al, 2011). Malt1-deficient mice show defects in the activation of B and T cells and present with a general immunodeficiency (Ruefli-Brasse et al, 2003; Ruland et al, 2003). Moreover, these animals have defects in the development of follicular, marginal zone (MZ), and B1 B cells, and a slightly altered thymic development, yet normal numbers of peripheral T cells (Ruefli-Brasse et al, 2003; Ruland et al, 2003). Malt1-deficient mice are protected against experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis (Brustle et al, 2012; McGuire et al, 2013). Malt1 acts not only downstream of the BCR and TCR, but also of other ITAM-containing immunoreceptors, such as the activating NK-cell receptors NKG2D and Ly49D, or the fungal recognition receptors Dectin-1 and Dectin-2 (Gross et al, 2006, 2008; Gringhuis et al, 2011). As a consequence, Malt1-deficient mice also show an impaired activation of NK and myeloid cells (Gross et al, 2006, 2008; Gringhuis et al, 2011).
An essential role for Malt1 downstream of these immunoreceptors is the activation of the transcription factor NF-κB, which drives the expression of cytokines and survival factors that control the inflammatory response. Malt1 also plays an important role in B-cell malignancy since it promotes the growth of cell lines derived from the activated B-cell subtype of diffuse large B-cell lymphoma (ABC DLBCL), which depend on constitutive NF-κB signaling and Malt1 activity for their survival (Ngo et al, 2006; Ferch et al, 2009; Hailfinger et al, 2009).
The mechanism by which Malt1 promotes NF-κB activation has been intensively explored and shown to rely on both, its scaffold and enzymatic functions. Antigen receptor triggering induces formation of a Carma1–Bcl-10–Malt1 (CBM) signaling complex. This promotes the activation of the IκB kinase (IKK) complex (Thome et al, 2010; Rosebeck et al, 2011), which phosphorylates the NF-κB inhibitor IκB to target it for proteasomal degradation. Malt1 has an essential scaffold function in this process, since it physically recruits the ubiquitin ligase TRAF6, which mediates the recruitment and activation of the IKK complex (Sun et al, 2004; Noels et al, 2007; Oeckinghaus et al, 2007). The protease activity of Malt1 is dispensable for IKK activation and instead promotes NF-κB activation by the cleavage of RelB, which acts as a negative regulator of canonical NF-κB activation in lymphocytes (Marienfeld et al, 2003; Hailfinger et al, 2011). Moreover, Malt1 cleaves the ubiquitin-editing enzyme A20 (Coornaert et al, 2008), and this most likely generates an A20 fragment that inhibits NF-κB activation in an IKK-independent manner.
The protease activity of Malt1 affects not only NF-κB activation, but also additional aspects of lymphocyte activation. Malt1-dependent cleavage of CYLD and Bcl-10 has been proposed to regulate antigen-induced activation of the JNK/AP-1 transcriptional pathway and β1 integrin-dependent cellular adhesion, respectively (Rebeaud et al, 2008; Staal et al, 2011). In addition, Malt1-dependent cleavage of the RNAse Regnase-1 (also known as MCPIP-1 or Zc3h12a) was recently shown to increase the stability of certain mRNAs in activated T cells (Uehata et al, 2013).
Collectively, these data have suggested that the protease activity of Malt1 is important for lymphocyte responses in vitro, and identified Malt1 as a potentially attractive target for therapeutic immunomodulation and cancer therapy. Indeed, recently developed small-molecule Malt1 inhibitors have been shown to effectively delay the growth of xenografted human ABC DLBCL (Fontan et al, 2012; Nagel et al, 2012). However, the consequences of a specific inactivation of the enzymatic function of Malt1 on the immune response, which is highly relevant for therapeutic applications, have not yet been explored.
To address these issues, we have generated genetically modified knock-in (ki) mice expressing a catalytically inactive form of Malt1 (C472A-ki mice). These mice phenocopied most of the immune defects of conventional Malt1-deficient mice and were protected from the induction of experimental autoimmune encephalomyelitis and colitis. However, the knock-in mice developed a spontaneous form of autoimmune gastritis. This correlated with high serum levels of IgG1 and IgE, an accumulation of T cells with an activated phenotype and a cell-intrinsic defect in Treg development. Our findings suggest that complete inactivation of the protease activity of Malt1 leads to impaired lymphocyte activation but also blunts Treg development, and can therefore favor the development of autoimmunity.
Results
Malt1 C472A knock-in mice express catalytically inactive Malt1
To assess the biological relevance of the Malt1 protease activity, we generated genetically modified mice by homologous recombination with a targeting vector carrying a TGTCGG to GCCAGA mutation (nucleotides 168–173 of exon 11) encoding a catalytically inactive C472A mutant of the Malt1 protein (Supplementary Fig S1A). The resulting modified Malt1 gene can be distinguished from the natural Malt1 gene by PCR-based genotyping (Supplementary Fig S1B). The mice were born healthy and at Mendelian ratios, suggesting that Malt1 activity is dispensable for embryonic development. In the spleen, thymus, and isolated peripheral T and B cells, homozygous knock-in (ki/ki) mice expressed similar levels of Malt1 as wild-type (+ / +) mice, in contrast to Malt1-deficient mice (ko/ko) that completely lack Malt1 protein expression (Fig 1A and Supplementary Fig S1C). To test whether the Malt1 mutant expressed in the knock-in mice had indeed lost catalytic activity, we stimulated splenocytes of these mice with PMA and ionomycin, a combination of drugs that efficiently activates Malt1 in lymphocytes (Coornaert et al, 2008; Rebeaud et al, 2008). This stimulation led to efficient cleavage of the Malt1 substrates Bcl-10, Regnase-1, RelB, and CYLD in Malt1-proficient, but not in Malt1-deficient nor in Malt1 C472A mutant cells (Fig1B). Similar results were obtained using isolated purified splenic B and T cells stimulated with PMA and ionomycin or with antigen receptor-triggering anti-IgM or anti-CD3 and anti-CD28 antibodies (Fig1C). These experiments also revealed the presence of a shorter isoform of RelB, which is preferentially expressed in T cells and efficiently cleaved by Malt1 (Fig1B and C). Collectively, these findings show that lymphocytes of Malt1 C472A knock-in mice express normal levels of a catalytically inactive form of Malt1.
Figure 1. Malt1 C472A knock-in mice express a catalytically inactive form of Malt1.
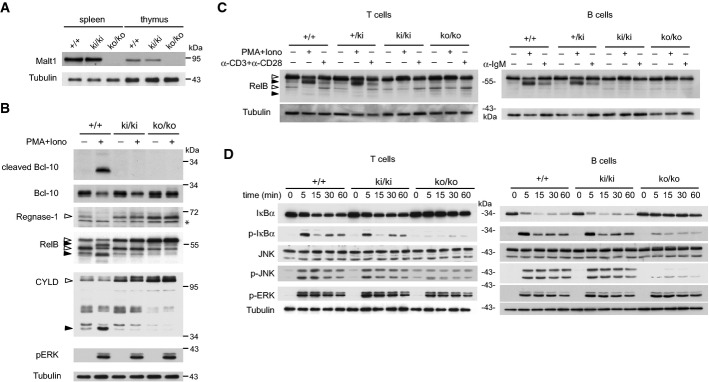
- Immunoblot analysis of spleen and thymus of Malt1-proficient (+/+), Malt1-deficient (ko/ko), or knock-in mice expressing a C472A mutant form of Malt1 (ki/ki).
- Immunoblot analysis of splenocytes, incubated with or without PMA and ionomycin (PMA+Iono) for 30 min, for the presence and cleavage of Malt1 substrates. Cleaved Bcl-10 was detected using an antibody directed against the cleavage-specific neoepitope.
- Immunoblot analysis of purified splenic T or B cells, stimulated with or without PMA and ionomycin (PMA+Iono) for 30 min or the indicated antibodies (anti-IgM, α-IgM; anti-CD3 and anti-CD28, α-CD3+α-CD28) for 60 min, for the presence and cleavage of RelB.
- Immunoblot analysis of purified T cells or B cells of wild-type (+/+), knock-in (ki/ki), and knock-out (ko/ko) mice, stimulated with PMA and ionomycin (PMA+Iono) for the indicated times.
Data information: Samples in (B) and (C) were treated with MG-132 prior to stimulation to prevent proteasomal degradation of the RelB cleavage fragments. Immunoblotting for tubulin served as a loading control. Open and black arrowheads indicate uncleaved and cleaved Malt1 substrates, respectively. Asterisk (*) indicates an unspecific band. Cells of three mice per genotype were pooled in (C) and (D). Data are representative of three (A), four (B, D), and two (C) experiments.
Source data are available online for this figure.
IKK and JNK activation is normal in lymphocytes of Malt1 C472A knock-in mice
To test whether other parameters of lymphocyte activation were affected by the catalytic inactivation of Malt1, we next assessed purified B and T cells of knock-in mice for the inducible activation of the MAPKs ERK and JNK, and the IKK-dependent phosphorylation of the NF-κB inhibitor IκBα. ERK activation, which is Malt1-independent (Rebeaud et al, 2008), was comparable in Malt1-proficient, Malt1-deficient and C472A mutant mice (Fig1D). The IKK-dependent phosphorylation of IκBα, on the other hand, was unaltered in Malt1 knock-in T cells but strongly impaired in T cells of Malt1-deficient mice, consistent with previous reports (Ruefli-Brasse et al, 2003; Ruland et al, 2003). Malt1 C472A mutation also did not affect inducible IκBα phosphorylation in B cells, while we noticed a partial defect of IκBα phosphorylation and degradation in Malt1-deficient B cells, as described before (Ruefli-Brasse et al, 2003; Ruland et al, 2003). Controversial findings have been reported concerning the relevance of Malt1 for JNK activation in T cells (Ruefli-Brasse et al, 2003; Ruland et al, 2003). We noticed a strong or partial reduction of JNK activation in B or T cells, respectively, of Malt1-deficient mice, while JNK activation was fully preserved in Malt1 C472A mutant mice (Fig1D). This suggests that the Malt1-dependent activation of JNK and IKK depends on the scaffold, and not the enzyme function of Malt1.
Lymphocyte activation is impaired in Malt1 C472A knock-in mice
Despite normal IKK activation, naïve T cells of both knock-in and knock-out mice showed a dramatic reduction of anti-CD3- and anti-CD28-induced IL-2 production and proliferation, a defect that was only minimally stronger in knock-out compared to knock-in cells (Fig 2A and B). Nevertheless, knock-in mice had normal numbers of total thymocytes and normal numbers of total, CD4 and CD8 double-negative (DN), CD4 and CD8 double-positive (DP), and of CD4 or CD8 single-positive (SP) thymocytes (Supplementary Fig S2A). Malt1-deficient mice have strongly increased total and relative numbers of CD25−CD44− (DN4) thymocytes (Ruefli-Brasse et al, 2003). Malt1 knock-in mice showed only a mild increase in this subset, but share with Malt1-deficient mice the presence of DN4 cells that have strongly increased surface levels of CD3 and intracellular TCRαβ chains (Supplementary Fig S2B–D). Thus, the protease activity of Malt1 is required for T-cell activation and contributes to the Malt1-dependent abnormality in thymocyte development.
Figure 2. Lymphocyte activation and B-cell development are impaired in Malt1 C472A knock-in mice.

- A, B IL-2 production (A) and proliferation (B) and of naïve T cells stimulated with the indicated concentrations of plate-bound anti-CD3 and anti-CD28 antibodies (pooled from three mice and stimulated in triplicates).
- C, D Analysis of the presence of peritoneal CD5+IgM+ B1 B cells and splenic marginal zone CD19+CD23loCD21hi B cells in wild-type (+/+), knock-in (ki/ki), or knock-out (ko/ko) mice (n = 3).
- E Fluorescence staining of spleen sections for the presence of CD21/35+ marginal zone B cells (cyan) and IgD+ follicular B cells (red). Scale bar, 100 μm.
- F Analysis of total basal immunoglobulin (Ig) levels in the serum of wild-type (+/+), knock-in (ki/ki), or knock-out (ko/ko) mice (n = 8).
- G, H NP-specific immunoglobulin (Ig) levels in the serum after immunization with NP-ficoll (G) or NP-CGG (H) (n = 8).
Malt1-deficient mice also have defects in B-cell development, in particular in the generation of peritoneal B1 B cells and splenic marginal zone (MZ) B cells (Ruefli-Brasse et al, 2003; Ruland et al, 2003). Interestingly, Malt1 knock-in mice also showed an almost complete lack of peritoneal B1 B cells, and dramatically reduced numbers of MZ B cells (Fig 2C and D), which were confirmed by histological examination (Fig2E). Additionally, we noticed a clear increase in the levels of IgM surface expression on total and follicular splenic B cells that is common to Malt1-deficient and Malt1 knock-in mice (Supplementary Fig S3A). These changes were accompanied by a significant reduction in the numbers of total and B220+ cells in the spleen but not the bone marrow, which did not affect the percentages of the CD19+, follicular, and T1 and T2 subsets (Supplementary Fig S3B). Analysis of serum Ig levels of Malt1 knock-in mice revealed normal levels of IgA and slightly elevated levels of IgG1, but a strong reduction in the levels of IgM, IgG2a, and IgG3 and a partial reduction of the levels of IgG2b, all of which were slightly more pronounced in Malt1-deficient mice (Fig 2F). Moreover, both Malt1-deficient and Malt1 knock-in mice showed a complete block in the production of serum IgM and IgG3 in response to immunization with the T-independent antigen NP-ficoll (Fig2G). In addition, Malt1 knock-in mice showed strongly reduced production of serum IgM and IgG1 in response to the T-dependent antigen NP-CGG (Fig2H), including the germinal center-dependent generation of high-affinity antibodies (Supplementary Fig S4), while these responses were completely blocked in Malt1-deficient mice (Fig2H). Collectively, these findings show that the protease activity of Malt1 is essential for the activation of B and T cells, for normal development of thymocytes, B1 and MZ cells and for the generation of efficient humoral immune responses.
Malt1 activity is required for the activation of NK and dendritic cells
Malt1-deficient mice also show defects in the activation of NK and myeloid cells by ITAM-containing receptors (Gross et al, 2006, 2008). Compared to wild-type mice, knock-in mice had similar numbers and percentages of NK1.1+ splenic NK cells (Fig 3A), but isolated NK cells had impaired IFN-γ and MIP-1α responses to stimulation with PMA and ionomycin or agonistic antibodies directed against the activating receptors NK1.1, NKG2D, and Ly49D (Fig3B). Stimulation-induced LAMP-1 upregulation, which is indicative of the release of cytotoxic granules, was not significantly altered (Supplementary Fig S5). Thus, the protease activity of Malt1 is required for efficient cytokine responses, but not the degranulation of NK cells.
Figure 3. Malt1 activity is required for the activation of NK and dendritic cells.

- A Analysis of the number and percentage of NK1.1+ NK cells in the spleen of wild-type (+/+) and Malt1 knock-in mice (ki/ki) (n = 3).
- B Analysis of the percentage of NK cells producing IFN-γ or MIP-1α following stimulation with PMA and ionomycin (PMA+Iono) or agonistic antibodies directed against NKG2D, NK1.1, or Ly49D.
- C Analysis of the number and percentage of CD11c+ dendritic cells in the spleen of wild-type (+/+) and Malt1 knock-in mice (ki/ki) (n = 3).
- D Immunoblot analysis of BMDCs stimulated with or without zymosan (100 μg/ml) or LPS (10 ng/ml), for the cleavage of the Malt1 substrate Bcl-10. Immunoblotting for tubulin served as a loading control.
- E, F Analysis of TNF-α and IL-6 cytokine secretion (E) or gene transcription (F) by BMDCs of wild-type (+/+), knock-in (ki/ki), or knock-out (ko/ko) mice, stimulated with or without the indicated concentrations of zymosan or LPS for 24 h, and with 100 μg/ml zymosan and 10 ng/ml LPS in for 6 h (F).
Data information: Bars represent mean ± SD, *P < 0.05; **P < 0.01 (unpaired t-test) in (A), (B), and (F); ***P < 0.001 (two-way ANOVA test) in (E). Data are representative of three (A, E, F) or two (B–D) experiments.
Source data are available online for this figure.
Next, we assessed the mice for the presence of dendritic cells (DCs) in the spleen by flow cytometry, which revealed relatively normal numbers of CD11c+ DCs (Fig 3C). To assess whether Malt1 activity was relevant for DC activation, we stimulated bone marrow-derived DCs with zymosan, which activates the ITAM-containing receptor Dectin-1 and signals via Malt1 (Brown & Gordon, 2001; Gross et al, 2006), or with LPS, which triggers DC activation via a distinct, TLR4- and MyD88-dependent yet Malt1-independent signaling pathway (Poltorak et al, 1998; Kawai et al, 1999; Zhang et al, 1999). Zymosan, but not LPS treatment, efficiently activated Malt1, leading to cleavage of the Malt1 substrates Bcl-10, RelB, and CYLD (Fig3D and Supplementary Fig S5B). No Malt1-dependent Bcl-10 cleavage was detected in DCs of knock-in or knock-out mice (Fig3D). Compared to wild-type cells, Malt1 knock-in BMDCs also showed strongly impaired TNF-α and partially reduced IL-6 responses to zymosan. These responses were almost absent in Malt1-deficient cells (Fig3E and F), consistent with previous findings (Gross et al, 2006). In contrast, Malt1 inactivation or Malt1 deficiency did not compromise the LPS-induced secretion of these cytokines. Collectively, these finding suggest that the protease activity of Malt1 is required for the full activation of NK cells and DCs by ITAM-containing receptors.
Malt1 inactivation prevents development of autoimmune encephalomyelitis and colitis
The observation that the Malt1 knock-in mice showed strongly impaired T-cell responses in vitro suggested that T-cell responses to autoantigens should also be compromised in vivo and that specific inhibition of the Malt1 protease activity might have potential for therapeutic immunomodulation. To test this hypothesis, we first evaluated the response of Malt1 knock-in mice to the induction of experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis induced by immunization with myelin oligodendrocyte glycoprotein (MOG). Using this protocol, control mice developed signs of EAE starting at day 9 after immunization, which gradually increased in severity over several days until mice were sacrificed (Fig4A). Interestingly, both Malt1 knock-in and Malt1-deficient animals were completely protected against EAE induction (Fig4A). This correlated with a dramatic reduction of CNS-infiltrating CD4+ cells (Fig4B) and a complete absence of IFN-γ, IL-17A, or GM-CSF-producing CD4+ cells in the CNS of knock-in mice (Fig4C). Consistent with these findings, splenic CD4+ T cells isolated from immunized mice showed strongly impaired cytokine secretion upon in vitro restimulation with increasing doses of MOG (Fig4D). Thus, mice expressing catalytically inactive Malt1 are fully protected from T-cell-mediated EAE.
Figure 4. Inactivation of the Malt1 protease activity prevents development of autoimmune encephalomyelitis and attenuates T-cell-induced colitis.

- A Development of clinical disease in wild-type (+/+, n = 5), knock-in (ki/ki, n = 6), and knock-out (ko/ko, n = 4) mice after immunization with MOG.
- B Immunohistochemical detection of CD3+ T cells in the spinal cord of mice of the indicated genotypes at day 14 after immunization with MOG. Scale bar, 200 μm. The lower panel shows insets in higher magnification.
- C Flow cytometric analysis of CD3+CD4+ T cells expressing IFN-γ, IL-17A, or GM-CSF in the spinal cord of wild-type (+/+) and knock-in (ki/ki) mice at day 14 after immunization with MOG.
- D Analysis of cytokine production of splenic CD4+ T cells of the indicated genotypes at day 10 after immunization, restimulated in vitro with the indicated concentrations of MOG peptide and CD4-depleted splenocytes for 3 days.
- E Immunohistochemical analysis of Rag2-deficient mice 8 weeks after adoptive transfer of naïve CD4+ T cells of wild-type (+/+, n = 3), knock-in (ki/ki, n = 4), and knock-out (ko/ko, n = 4) mice, for the presence of CD3+ T cells in the colon. Scale bar, 200 μm.
- F Flow cytometric analysis of the numbers of total, CD3+CD4+ T cells, and CD3+CD4+IFN-γ+ cells in mesenteric lymph nodes of Rag2-deficient mice 8 weeks after transfer of naïve CD4+ T cells of the indicated genotypes.
Data information: Bars in (C) and (D) represent means ± SD. Each symbol in (F) represents an individual mouse, and horizontal lines represent means. *P < 0.05; **P < 0.005 (unpaired t-test). Data are representative of three (A, B) and two (C–F) independent experiments.
Next, we assessed the role of the Malt1 protease activity in a T-cell-dependent mouse model of colitis that is initiated by intraperitoneal transfer of purified naïve T cells from wild-type mice into Rag2−/− mice, which lack endogenous B and T cells. In this lymphopenic host, the transferred T cells expand and trigger a T-cell-dependent form of colitis that is characterized by T-cell infiltration in the intestinal mucosa. Such signs of colitis became apparent in Rag2−/− mice when receiving naïve FACS-sorted T cells isolated from Malt1-proficient mice, but not from Malt1-deficient mice (Fig4E). T cells isolated from Malt1 knock-in mice induced colitis with lower penetrance, since in each of two independent experiments, only 1 out of 4 mice tested showed T-cell infiltration in the colon (Fig4E and F). These findings correlated with a partial and strong reduction of the numbers of total and CD4+IFN-γ+ cells in the mesenteric lymph nodes of Malt1-inactive and Malt1-deficient mice, respectively (Fig4F). Collectively, these observations support an essential role for the protease activity of Malt1 in two independent models of T-cell-dependent autoimmune diseases.
Mice expressing catalytically inactive Malt1 have an activated T-cell phenotype
When analyzing the immune status of Malt1 C472A knock-in mice beyond 6 weeks, we noticed the appearance of strongly swollen lymph nodes (Fig 5A). This feature was not present in Malt1-proficient littermates or heterozygous animals, and much less pronounced in Malt1-deficient mice (Fig5B and Supplementary Fig S6A). In contrast, Malt1 C472A knock-in mice had a reduction in the total numbers of splenocytes, similar to knock-out mice (Fig 5B). Flow cytometric analysis of the peripheral lymph nodes of the knock-in animals revealed a massive increase in the total numbers of B and T cells (Supplementary Fig S6B) and an increased percentage and number of T cells with an activated/memory phenotype, that is characterized by high surface expression of CD44 and low levels of surface CD62L (Fig 5C and D). In the spleen, the total number of CD4+ T cells was strongly reduced in both, knock-in and knock-out mice (Supplementary Fig S6C), and cells with a CD62Llo CD44hi phenotype were over-represented only in the knock-in mice (Fig5C and D). T cells from Malt1 knock-in mice showed an increased production of the Th1 cytokine IFN-γ and the Th2 cytokine IL-4, but no increase in IL-17 production after in vitro stimulation with PMA and ionomycin (Fig5E). Collectively, these data demonstrate an excessive lymphocyte expansion and an accumulation of T cells with an activated phenotype in the lymph nodes of Malt1 knock-in mice.
Figure 5. Mice expressing catalytically inactive Malt1 have enlarged lymph nodes and accumulate T cells with an activated phenotype.

- A, B Analysis of the anatomical size (A) and cellularity (B) of lymph nodes (LN; c: superficial cervical, i: inguinal, a: axillary, m: mesenteric) and spleens (SP) of wild-type (+/+, n = 4) and Malt1 knock-in (ki/ki, n = 4) mice at the age of 8 (A) and 6 (B) weeks.
- C Analysis of the expression of CD62L and CD44 on CD4+ cells isolated from lymph nodes (LN) or spleens (SP) of 6-week-old mice of the indicated genotypes.
- D Quantification of data shown in (C), obtained with wild-type (+/+, n = 4), Malt1 knock-in (ki/ki, n = 4), and knock-out (ko/ko, n = 3) mice.
- E Intracellular cytokine expression in CD4+ cells isolated from the lymph nodes (LN) or spleens (SP) of 6-week-old mice of wild-type (+/+, n = 4), Malt1 knock-in (ki/ki, n = 4), and knock-out (ko/ko, n = 3) mice.
Data information: Bars represent means ± SD; *P < 0.05; **P < 0.005; ***P < 0.0005; n.s., not significant (unpaired t-test). Data are representative of six (A, B), four (C, D), and two (E) experiments.
Malt1 C472A knock-in mice develop autoimmune gastritis
Beyond the age of 6 weeks, we also noticed a loss of weight of Malt1 knock-in mice that was not apparent in control or Malt1-deficient mice (Fig6A). Histological analysis of the gastrointestinal tract of knock-in mice demonstrated the presence of numerous CD3+ cells in the mucosa of the stomach (Fig6B), but not in the lung, liver, pancreas, kidney, heart, and skin (M. Jaworski, M. Thome, unpublished observations). No or only very few CD3+ cells were present in the stomach mucosa of control or knock-out mice, respectively (Fig6B). We also saw no obvious increase in CD3+ cells in the small or large intestine (M. Jaworski, M. Thome, unpublished observations). Compared to control and knock-out mice, the serum of Malt1 knock-in mice contained increased levels of IgE and IgG1, which are typically associated with autoimmunity (Liston et al, 2008), and these became more severe with age (Fig6C). Interestingly, the serum of 16-week-old Malt1 knock-in mice, but not of Malt1-proficient littermates, showed strong immunoreactivity toward the mucosa of the stomach, in particular the parietal cells of the tubular glands (Fig6D). Collectively, these findings suggest that mice expressing a catalytically inactive form of Malt1 develop an autoimmunity that preferentially targets the stomach.
Figure 6. Mice expressing catalytically inactive Malt1 develop autoimmune gastritis.

- Weight curves of heterozygous (+/ki, n = 4), knock-in (ki/ki, n = 4) and knock-out (ko/ko, n = 6) mice after week 6 of age.
- Immunohistochemical analysis of the gastric mucosa of 12-week-old mice of the indicated genotypes for the presence of CD3+ lymphocytes. Morphological staining was performed using hematoxylin and eosin (H&E). Scale bar, 100 μm.
- Comparison of the basal serum levels of IgE and IgG1 of wild-type and knock-in (ki/ki) littermates or knock-out (ko/ko) mice of the indicated ages (between 3 and 7 mice were analyzed for each time point and genotype).
- Immunofluorescence analysis of sections of the gastric mucosa of wild-type mice stained with serum of 16-week-old mice of the indicated genotypes (green, Ig; blue, DAPI). Scale bar, 100 μm.
Data information: Bars represent means ± SD; *P < 0.05; **P < 0.005; ***P < 0.0005; n.s., not significant (unpaired t-test). Data are representative of two (A, C, D) and four (B) experiments.
Malt1 C472A knock-in mice have a cell-intrinsic defect in Treg development
Next, we tested whether the observed autoimmunity phenotype of the knock-in mice was due to an impaired development CD4+CD25+FoxP3+ regulatory T (Treg) cells, which are required to limit immune responses and prevent autoimmunity. Indeed, FoxP3+ Treg cells were virtually absent from the thymus and strongly reduced in the spleen and lymph nodes from knock-in and knock-out mice (Fig 7A). The upregulation of FoxP3, which is required for the development and function of regulatory T cells (Fontenot et al, 2003; Hori et al, 2003), has been shown to depend on the NF-κB subunits RelA and c-Rel (Isomura et al, 2009; Ruan et al, 2009; Deenick et al, 2010; Visekruna et al, 2010), and in particular on a c-Rel-dependent modification of the chromatin conformation in the CNS3 enhancer region of the FoxP3 locus (Long et al, 2009; Zheng et al, 2010). The Malt1 protease activity controls RelA and c-Rel activation independently of IκBα, via the cleavage of RelB (Hailfinger et al, 2011), which acts as an inhibitor of canonical NF-κB activation in T cells (Ruben et al, 1992; Weih et al, 1995, 1996; Ishimaru et al, 2006). Therefore, we next assessed the levels of RelB in thymocytes of Malt1 knock-in and wild-type mice. The absence of thymic Treg cells in the knock-in mice correlated with increased thymic RelB levels (Fig7B), suggesting that Malt1-dependent RelB downregulation might be required for the efficient NF-κB-dependent generation of FoxP3+ Treg cells. Consistent with this idea, we observed that anti-CD3- and anti-CD28-induced upregulation of FoxP3 on naïve T cells in vitro, which at least partially depends on the NF-κB pathway (Long et al, 2009; Ruan et al, 2009), was also significantly impaired in Malt1 knock-in mice compared to wild-type mice (Fig7C). In contrast, we saw no effect of the Malt1 inhibitor z-VRPR-fmk nor of genetic inactivation or loss of Malt1 on the stability of FoxP3 mRNA in this system (Supplementary Fig S7A and B). To assess whether the lack of Treg cells in the knock-in mice was due to a cell-intrinsic defect, we reconstituted CD45.1+ lethally irradiated wild-type mice with a 1:1 mixture of CD45.1+ wild-type and CD45.2+ wild-type or Malt1 knock-in bone marrow cells. Analysis of the lymph nodes, spleen, and thymus of the mice 8 weeks after bone marrow transfer showed that equal ratios of total single-positive CD4+ T cells developed from wild-type and knock-in bone marrow (Fig7D). However, thymic and peripheral Treg cells could develop from wild-type bone marrow, but no Treg development was detectable from Malt1 knock-in bone marrow (Fig7E). To exclude that the thymic stroma of Malt1 knock-in mice had a defect in supporting Treg development, we also performed reciprocal bone marrow chimeras. These experiments showed that Treg cells developed normally from bone marrow cells of wild-type origin upon transfer into either knock-in or wild-type hosts (Fig7F). Thus, normal development of Treg cells depends on the protease activity of Malt1 in a cell-intrinsic manner.
Figure 7. Malt1 C472A knock-in mice have a cell-intrinsic defect in Treg development.

- A Analysis of wild-type (+/+, n = 4), knock-in (ki/ki, n = 4), and knock-out (ko/ko, n = 3) mice for the presence of thymic (TH) and peripheral CD4+CD25+Fop3+ Treg cells. SP, spleen; LN, lymph node.
- B Western blot analysis of RelB expression in total thymocytes of wild-type and knock-in mice.
- C FoxP3 mRNA levels in naïve CD4+ peripheral T cells after stimulation with anti-CD3 and anti-CD28 in the presence of TGF-β and IL-2.
- D, E Analysis of CD45.1 wild-type mice that were lethally irradiated and reconstituted with a 1:1 ratio of wild-type CD45.1+ to CD45.2+ wild-type (+/+, n = 3) or knock-in (ki/ki, n = 4) bone marrow cells, 8 weeks after transfer. Cells from lymph nodes (LN), spleen (SP), and thymus (TH) were analyzed. The CD45.1/CD45.2 ratio was determined in total CD4+ T cells (D) and in CD4+CD25+FoxP3+ Treg cells (E).
- F CD45.2 control (+/ki) or Malt1 knock-in mice (ki/ki) were lethally irradiated and reconstituted with wild-type CD45.1+ bone marrow cells (reciprocal chimeras) and T-cell development analyzed 8 weeks after transfer (n = 3).
Data information: Bars represent means ± SD; *P < 0.05; **P < 0.005; n.s., not significant (unpaired t-test). Results are representative of four (A), two (B, C, F), and three (D, E) experiments.
Source data are available online for this figure.
Treg transfer rescues autoimmune symptoms in Malt1 C472A knock-in mice
To assess whether the lack of Treg cells accounted for the autoimmune symptoms of Malt1 knock-in mice, we adoptively transferred FACS-purified CD4+FoxP3+GFP+ Treg cells into newborn Malt1 knock-in mice. Total Treg cell numbers remained lower than in the non-reconstituted Malt1 knock-in mice, but transferred Treg cells represented more than 40% of the total Treg cells in the lymph node and spleen of the reconstituted mice (Supplementary Fig S8A and B). In contrast to the endogenous Treg cells, these low numbers of transferred Tregs were sufficient to suppress autoimmunity, since adoptive Treg transfer restored a normal weight gain of the mice for at least 15 weeks (Fig8A) and led to a normalization of the size of the lymph nodes (Fig8B). In contrast to untreated knock-in mice, the mice receiving an adoptive Treg transfer had reduced signs of T-cell activation (Fig8C), showed lower levels of IFN-γ, IL-4, and IL-17 production (Fig8D), had lower serum IgE levels than untreated knock-in mice (Fig8E), and showed an absence of CD3+ T cells in the stomach mucosa (Fig8F). Thus, the lack of functional Treg cells in the Malt1 C472A knock-in mice plays a major role in the development of autoimmune symptoms.
Figure 8. Treg transfer rescues autoimmune symptoms in Malt1 C472A knock-in mice.

- A Weight curves of untreated wild-type and heterozygous mice (control, n = 3 for each) and of knock-in mice (ki/ki) with (n = 3) or without (n = 4) transfer of 106 Treg cells at the age of 8 days.
- B–E Analysis of the total lymph node (LN) cellularity (per gram body weight) (B), the percentage of CD4+ cells with a CD62LloCD44hi phenotype (C), the percentage of cytokine-positive CD4+ cells isolated from lymph nodes (LN) or spleens (SP) (D) and the serum IgE levels (E) of 5-week-old wild-type (+/+) and knock-in (ki/ki) mice, and of knock-in mice having received an adoptive transfer of 106 Treg cells (ki/ki + Treg) at the age of 3 days (n = 3).
- F Immunohistochemical analysis of CD3+ cells in stomach sections of mice described in (B–E). Scale bar, 100 μm.
Data information: Bars represent means ± SD; *P < 0.05; **P < 0.005; n.s., not significant (unpaired t-test). Results are representative of two experiments (A-F).
Discussion
Here, we provide several lines of evidence for a differential role of the Malt1 protease and scaffold function in the control of the immune response. First, Malt1 protease activity was required for cellular immune responses of lymphocytes, NK cells and dendritic cells, and the development of follicular, marginal zone, and B1 B cells. Second, the protease activity was required for the induction of T-cell-dependent autoimmune diseases, such as EAE and colitis. Most importantly, we show that inactivation of the protease activity of Malt1 impaired the development of Foxp3+ Treg cells in a cell-intrinsic manner and favored the development of an early onset autoimmune gastritis. This autoimmunity was clearly dependent on the Malt1 scaffold function, since Malt1-deficient mice did not show signs of autoimmunity despite absence of Treg cells. Interestingly, autoimmunity features were also absent in heterozygous (+ /ki) mice, arguing against a potential dominant-negative effect of the catalytically inactive Malt1 mutant.
The comparison of Malt1-deficient and Malt1 knock-in animals has also allowed us to clearly dissect the relevance of the scaffold and the enzymatic function of Malt1 for signaling. The protease activity was not required for the antigen receptor-induced activation of the IKK complex, which required only the scaffold function of Malt1. These findings are consistent with previous studies using the Malt1 tetrapeptide inhibitor z-VRPR-fmk, which does not impair IKK activation (Duwel et al, 2009; Hailfinger et al, 2011). Additionally, the protease activity of Malt1 was dispensable for antigen receptor-induced JNK activation. A previous study had reported that Malt1 protease activity controls T-cell receptor-induced JNK activation by the cleavage of the deubiquitinase CYLD (Staal et al, 2011). The authors observed a strong effect of a non-cleavable CYLD mutant on the upregulation of the JNK target c-Jun, despite the fact that Malt1 inhibition with z-VRPR-fmk had only a minimal impact on JNK activation (Staal et al, 2011). Together with our findings, this suggests that CYLD cleavage may regulate c-Jun expression in a JNK-independent manner that remains to be explored.
An interesting discovery of the present study is that both Malt1 knock-in and Malt1-deficient mice showed an almost complete absence of thymic and a strong reduction of peripheral FoxP3+ regulatory T cells that was due to a cell-intrinsic defect, since FoxP3+ Treg cells could not develop from Malt1 knock-in bone marrow in a mixed chimera setting. A loss of the development of thymic Treg cells and strongly reduced peripheral Treg numbers had been previously noticed in mice with defects in the Carma1 and Bcl-10 genes (Schmidt-Supprian et al, 2004; Barnes et al, 2009; Medoff et al, 2009; Molinero et al, 2009; Salisbury et al, 2014), and more generally in animals with a genetic defect in the TCR-NF-κB signaling axis (Schmidt-Supprian et al, 2003, 2004; Gupta et al, 2008). This has been attributed to a requirement of NF-κB for the expression of FoxP3 (Isomura et al, 2009; Long et al, 2009; Ruan et al, 2009; Deenick et al, 2010; Visekruna et al, 2010; Zheng et al, 2010). We have previously shown that the protease activity of Malt1 controls NF-κB activation by the cleavage and subsequent degradation of RelB (Hailfinger et al, 2011), which acts as an inhibitor of canonical NF-κB activation in T cells (Ruben et al, 1992; Weih et al, 1995, 1996; Ishimaru et al, 2006). Together with our present findings, this suggests a role for Malt1-dependent cleavage of RelB in controlling the TCR–Carma1–Bcl-10–Malt1–NF-κB signaling pathway that is essential for the development of FoxP3+ thymic and peripheral Treg cells. We also explored a possible involvement of the Malt1 substrate Regnase-1, which might affect FoxP3 mRNA stability (Uehata et al, 2013). However, we did not observe an effect of inhibition or genetic inactivation of Malt1 on the mRNA stability of FoxP3 in the in vitro iTreg induction system. Nevertheless, it remains likely that a combined absence of cleavage of multiple Malt1 substrates accounts for the observed defect in Treg cell development.
An unexpected finding of the present study was that mice expressing catalytically inactive Malt1 developed an early onset autoimmune gastritis despite strongly compromised immune responses. This phenotype was rescued by the transfer of highly purified Treg cells. Moreover, it required a minimal capacity of lymphocyte activation through the scaffold function of Malt1, since Malt1-deficient mice did not develop autoimmunity despite an even stronger Treg deficiency. Indeed, Malt1 knock-in mice retained minimal capacities for proliferation and cytokine responses of T cells and Ig production of B cells, while these responses were completely blunted in the Malt1-deficient mice. Of note, mice with a point mutation in Carma1 (unmodulated mice), which show a partial defect in T-cell stimulation, also develop enlarged lymph nodes and increased serum levels of IgE with age, and progress to atopic dermatitis, although they do not seem to manifest other features of autoimmunity (Jun et al, 2003). It is possible that Malt1 knock-in mice would acquire additional symptoms of autoimmunity upon aging, but that these are occluded by the fact that the mice succumb early to gastritis.
Interestingly, autoimmunity has been reported as a common but poorly understood feature of mice and humans with partial TCR signaling defects (Toyabe et al, 2001; Aguado et al, 2002; Sommers et al, 2002; Siggs et al, 2007; Holst et al, 2008). The autoimmune phenotype has been attributed to an impaired balance between immunogenic and tolerogenic TCR signals, including an impairment of Treg development or function and compromised tolerogenic signaling due to a change in the TCR signaling threshold required for the negative selection of autoreactive T cells in the thymus (Liston et al, 2008). Malt1 knock-in mice have reduced numbers of FoxP3+ Treg cells, but the mechanisms involved in breaking tolerance in these mice are unclear. Malt1 has been previously shown to be dispensable for thymic negative selection (Jost et al, 2007), nevertheless, it remains possible that compromised TCR signaling in the Malt1 knock-in thymocytes promotes subtle changes in the TCR repertoire that allow the escape of autoreactive T cells from negative selection in the thymus.
The fact that knock-in mice expressing catalytically inactive Malt1 have impaired Treg development raises the concern that treatment of patients with Malt1 inhibitors may also lead to a decrease in Treg cell numbers. This may be of concern in the context of organ transplantation and the treatment of autoimmune diseases; however, it may possibly be an advantage in the treatment of cancer patients, where a reduction in Treg numbers may actually favor the anti-tumor immune response. Since the requirement of Malt1 protease activity for the maintenance and function of peripheral Treg cells is not yet known, Treg numbers should be carefully monitored in preclinical trials with Malt1 inhibitors.
Despite possible concerns about compromised Treg development, our mouse model of a genetic inactivation of the protease activity of Malt1 provides support to the concept that therapeutic Malt1 inhibition could be useful in the treatment of multiple sclerosis, since mice expressing catalytically inactive Malt1 were completely resistant to the induction of EAE. Moreover, naïve T cells from Malt1 knock-in mice were strongly impaired in their capacity to induce autoimmune colitis. Together with our result that the knock-in mice had dramatically reduced responses to immunization and that Malt1 knock-in lymphocytes show an almost complete inhibition of activation-induced cytokine and proliferative responses in vitro, this demonstrates that the protease activity of Malt1 accounts for most of its immunological functions. Malt1 inhibitors might therefore be useful for immunomodulation in selected autoimmune diseases and more generally in the treatment of inflammatory conditions. Finally, therapeutic Malt1 inhibition remains highly attractive as a pathway-specific therapy to treat lymphomas with constitutive Malt1 activity, such as ABC DLBCL and MALT lymphomas.
Materials and Methods
Mice
To generate a targeting vector, an approximately 8.9-kb region was first subcloned from a positively identified C57BL/6 BAC clone (RPCI23 405H16). The region was designed so that the long homology arm extended approximately 5.8 kb 3′ to exon 11 which contains the TGTCGG to GCCAGA mutation (nucleotides 168–173 of exon 11), leading to a Cys472Ala (C472A) mutation in the Malt1 protein. The additional base changes were engineered to create an additional restriction site for MscI. A neomycin resistance cassette flanked by FRT sites was inserted on the 5′ side of exon 11. The short homology arm extended approximately 2.1 kb 5′ to exon 11 and the neomycin resistance cassette. The linearized targeting vector was electroporated into C57BL/6 embryonic stem cells, and positive ES clones were used for injection into BALB/c blastocysts (inGenious Targeting Laboratory). To delete the neomycin resistance cassette in the resulting Malt1-KI-neo mice, they were bred to C57BL/6 FLP mice (The Jackson laboratory), and Neo deletion was confirmed by PCR. Malt1 knock-in mice were genotyped by PCR-mediated amplification of a 500-bp fragment of exon11 (F: 5′-TATTATGCAGGGCACGGTTA-3′, R: 5′-ACTTCTGACCCTTGGGGAAT-3′), which upon digestion by MscI gives rise to two fragments of 340 and 160 bp, whereas the Malt1 wild-type fragment is not cut. Homozygous Malt1 knock-in mice (C57BL/6-Malt1tmC472A) have a poor breeding performance, and mating was therefore routinely done with heterozygous mice.
Bernard Malissen (Centre d'Immunologie de Marseille-Luminy, Marseille, France) kindly provided FoxP3-eGFP B6 mice (Wang et al, 2008), and Vishva Dixit (Genentech, San Francisco, USA) kindly provided Malt1-deficient mice (Ruefli-Brasse et al, 2003). Rag2-deficient mice, B6 mice, and CD45.1 (Ly5.1+) mice were from The Jackson Laboratory. Mixed bone marrow chimeras were generated by mixture of bone marrow from CD45.1+ wild-type and CD45.2+ Malt1 wild-type or Malt1 knock-in mice at a ratio of 1:1 and injection into CD45.1+ wild-type recipients after lethal irradiation. Chimeras were analyzed 8 weeks after reconstitution. All mice were maintained in the specific pathogen-free animal facility of the University of Lausanne. All mouse experiments were authorized by the Swiss Federal Veterinary Office.
FACS analysis
Single-cell suspensions from thymus, spleen, lymph nodes, and BM were prepared by mechanical dissociation of respective tissues and passage of cells through a 70-μm filter. Spleen cells were counted after erythrocyte lysis in hypotonic Tris-NH4Cl buffer. Fc receptors were blocked by incubating cells in staining buffer (PBS supplemented with 2% heat-inactivated FCS) with anti-CD16/CD32 antibodies (2.4G2, hybridoma supernatant). Staining was performed in staining buffer on ice for 30 min with optimal dilutions. Intracellular staining for FoxP3, IFN-γ, and IL-17A was performed using the Cytofix/Cytoperm kit (BD) according to the manufacturer's instruction. 7AAD and DAPI were routinely used to exclude dead cells. For intracellular cytokine staining, cells were re-stimulated in vitro with PMA (40 ng/ml; Alexis) and ionomycin (1 μM; Calbiochem) in the presence of Brefeldin A (10 μg/ml, eBioscience) for 4–6 h at 37°C. Dead cells were excluded using fixable viability dye (eBioscience). Data were acquired on a FACSCanto or LSR II flow cytometer (both BectonDickinson) and were analyzed with FlowJo software (TreeStar).
Stimulation and Western blotting
Splenocytes and thymocytes were isolated by mechanical tissue separation and subsequent red blood cell lysis in hypotonic Tris-NH4Cl buffer. Cells were washed with PBS and resuspended in prewarmed RPMI 1640 supplemented with 10% FCS for 10 min at 37°C before stimulation. Lymphocyte stimulation was initiated by addition of PMA (40 ng/ml; Alexis) and ionomycin (1 μM; Calbiochem), by immobilized anti-CD3 (145-2C11) and anti-CD28 (37.51) (eBioscience), or by addition of anti-IgM (AffiniPure F(ab')2; Jackson Immunoresearch) for the indicated times at 37°C at the indicated concentrations. Where indicated, cells were pretreated with MG-132 (5 μM; Calbiochem) for 30 min prior to stimulation.
Pelleted cells were lysed in lysis buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 1% Triton X-100, protease inhibitors (Complete; Roche), and phosphatase inhibitors (NaF, Na4P2O7, and Na3VO4), and cell lysates were fractionated by SDS–PAGE and subjected to immunoblot assays with the indicated antibodies.
Primary antibodies used in this study were as follows: rabbit anti-RelB (4954), rabbit anti-IκBα (L35A5), rabbit anti-phospho-IκBα (5A5), anti-SAPK/JNK, rabbit anti-CYLD (D1A10) (all Cell Signaling), rabbit anti-IκBα (Sigma), mouse anti-phospho ERK (MAPK-YT) (Sigma), mouse anti-tubulin (B-5-1-2) (Sigma), rabbit anti-Bcl-10 (H-197; Santa Cruz), p-JNK (Bioscource), Regnase-1 (Clone 604421) (R&D Systems), and rabbit anti-Malt1 (H-300) (Santa Cruz). An antibody specific for cleaved Bcl-10 was generated as described (Rebeaud et al, 2008). Western blots were revealed using HRP-coupled goat anti-mouse or anti-rabbit antibodies (Jackson Immunoresearch).
Primary mouse CD4+ T cells and B cells were isolated from spleens and lymph nodes, using anti-CD4-coated MACS beads or the B-cell isolation kit, respectively, according to the manufacturer's description (Miltenyi). Naïve T cells were sorted by flow cytometry from CD4+-enriched populations as CD4+CD62LhiCD44loCD25− and were typically more than 99% pure.
T-cell proliferation and differentiation
Sorted CFSE-labeled naïve T cells were stimulated by plate-bound anti-CD3 and anti-CD28 for 48 h, and CFSE dilution was analyzed by flow cytometry. For in vitro induction of FoxP3+ Treg cells, purified peripheral naïve CD4+ T cells were stimulated for the indicated times with plate-bound anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) in the presence of TGF-β (5 ng/ml) and human IL-2 (100 U/ml). To analyze FoxP3 mRNA stability, cells were treated with actinomycin D (5 μg/ml) for the indicated times.
BMDC generation and activation
Bone marrow-derived DCs (BMDCs) were generated by differentiation by GM-CSF. Briefly, BM cells were obtained from femurs of B6 mice by crushing bones with a mortar. After filtering through a 70-μm mesh, BM cells were cultured for 10 days in complete Iscove modified Eagle medium (IMDM; Invitrogen) containing HEPES (10 mM), penicillin (50 IU/ml), streptomycin (50 μg/ml), β-mercaptoethanol (50 μM), 10% FCS, and 10% (vol/vol) GM-CSF-containing culture supernatant (A02 line donated by Fabienne Tacchini-Cottier, Lausanne, Switzerland) in untreated 100-mm plastic dishes. Medium was changed on days 3, 6, and 9 of culture. BMDCs (day 10) were stimulated with LPS (LPS-EK Ultrapure, Invivogen) or zymosan (hot alkali treated, depleted; Invivogen) at the indicated concentrations. Supernatants were harvested after 24 h for ELISA.
mRNA expression
For analyzing RNA expression, total cellular RNA was isolated by TRIzol reagent (Life technologies) and cDNA was synthesized from 0.1 to 1 μg total RNA with the GoScript Reverse transcriptase kit (Promega) using random hexamers. Quantitative real-time PCR with SYBR green was done with the LightCycler480 system (Roche) and expression normalized to SDHA expression. Primer sequences were as follows: mouse TNF-α, F: 5′-CTGTAGCCCACGTCGTAGC-3′, R: 5′-TTGAGATCCATGCCGTTG-3′; mouse IL-6, F: 5′-GCTACCAAACTGGATATAATCAGGA-3′, R: 5′-CCAGGTAGCTATGGTACTCCAGAA-3′; mouse SDHA: F: 5′-AAGTTGAGATTTGCCGATGG-3′, R: 5′-TGGTTCTGCATCGACTTCTG-3′; and mouse FoxP3: F: 5′-GGAAACACCCAGCCACTC-3′, R: 5′-CTTCCAAGTCTCGTCTGAAGG-3′.
NK-cell isolation and activation
Splenic NK cells were purified by non-adherence to nylon wool followed by depleting non-NK cells using the NK Cell Isolation Kit II and a MACS separator (Miltenyi Biotec). Purified NK cells were cultured in DMEM with 10% FCS, gentamicin (10 μg/ml), β-mercaptoethanol (50 μM), MEM NEAA (0.1 μM), l-glutamine (4 μM), and 500 μg/ml recombinant human IL-2 (a gift from N. Rufer, University of Lausanne). After 5 days, cells were harvested and stimulated with plastic-immobilized mAbs to NK1.1 (PK136, BioXCell) (5 μg/ml), NKG2D (191,004, R&D Systems) (5 μg/ml), Ly49D (4E5, BD Pharmingen) (2.5 μg/ml), or by the addition of PMA (50 ng/ml) and ionomycin (1 μg/ml). GolgiPlug and GolgiStop were added after 1 h of culture, and the cells were harvested 4 h later. Cells were incubated with Aquadead (Invitrogen) (to exclude dead cells) and anti-CD16/32 (2.4G2) hybridoma supernatant before staining with fluorescent mAbs to NK1.1 (PK136), NKp46 (29A1.4), CD3ε (17A2), CD19 (6D5) (all from BD Biosciences). Intracellular staining for IFN-γ (XMG1.2, eBioscience) was performed using the BD Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences). Cells were analyzed on a LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo (Tree Star).
Mouse immunization
Wild-type, Malt1 ki/ki littermates, and sex- and age-matched Malt1 ko/ko mice (6–8 weeks old) were immunized by subcutaneous injection of 50 μg NP-CGG (Biosearch Technology) in alum or intraperitoneal injection of 25 μg NP-Ficoll and bled at the indicated times to analyze serum antibody concentration.
ELISA
For NP-specific antibody detection, Nunc Immuno Plate MaxiSorp plates (Thermo Fisher Scientific) were coated with 50 μg/ml NP26-BSA and incubated overnight at 4°C. After blocking with 1% BSA in PBS, the plates were incubated with serially diluted serum samples and then with biotin-conjugated detection antibodies (Southern Biotech) followed by streptavidin-conjugated HRP (Jackson Immunoresearch). After colorimetric reaction (SigmaFAST OPD tablets), the absorbance at 490 nm was measured using an ELISA plate reader and absolute values were calculated with an internal standard (wild-type serum at day 14 of immunization). For IgG detection, Nunc plates were coated with 5 μg/ml anti-Ig capture antibody (Southern Biotech) in 50 mM sodium bicarbonate and incubated overnight at 4°C. The coated plates were incubated with serially diluted serum samples and detected with isotype-specific antibodies (Southern Biotech), and developed as described above. IL-6, TNF-α, IL-2, IL-17A, GM-CSF, and IFN-γ concentrations in culture supernatants were determined using ready-set go kits (eBioscience) according to the manufacturer's protocols.
Induction and assessment of experimental autoimmune encephalomyelitis
Malt1 wild-type, Malt1 knock-in, and Malt1 knock-out mice were immunized s.c. with 200 μg of MOG 35–55 peptide (Gutcher et al, 2006) in CFA (Chondrex, Inc), and two i.p. injections of 200 ng of pertussis toxin (Enzo Life Sciences) at days 0 and 1. Starting from day 4, mice were monitored daily for the appearance of clinical signs of disease. The following clinical score was used: 0, no detectable signs of EAE; 0.5, distal limp tail; 1, complete limp tail; 1.5, limp tail and hind limb weakness; 2, unilateral partial hind limb paralysis; 2.5, bilateral partial hind limb paralysis; 3, complete bilateral hind limb paralysis; 3.5, complete hind limb paralysis and unilateral forelimb paralysis; 4, total paralysis of fore and hind limbs; and 5, death. Mice were killed between 10 and 18 days after immunization. CD4+ splenocytes isolated at day 10 after immunization were restimulated with titrated doses of MOG 35–55 peptide and CD4-depleted and mitomycin C-treated (50 μg/ml, 30 min) splenocytes. After 72 h, cytokine content in supernatants was assessed by ELISA.
Colitis
For induction of colitis, Rag2−/− mice were injected i.p. with 5 × 105 sorted naïve purified T cells (CD4+CD62LhiCD44loCD25−) isolated from Malt1 wild-type, knock-in, and knock-out mice. Mice were weighed, assessed for clinical symptoms of colitis weekly, and killed 8 weeks after transfer. The frequency of CD4+ cells in the mesenteric lymph nodes expressing IFN-γ was assessed by intracellular staining after 5 h of PMA and ionomycin stimulation in the presence of Brefeldin A.
Adoptive Treg transfer
GFP+ Treg cells were isolated from spleens and lymph nodes of FoxP3-EGFP mice, by enrichment using a Treg isolation kit, according to the manufacturer's description (Miltenyi), and further purified by flow cytometric sorting of GFP+CD4+CD25+ cells, to obtain Treg cells that were typically more than 99% pure. 106 purified Treg cells were adoptively transferred by i.p. injection into 3- to 8-day-old neonates.
Immunohistochemical and immunofluorescence stainings
Organs (spinal cord, colon, stomach) were removed from sacrificed mice, fixed in 10% neutral buffered formalin, embedded in paraffin, and sectioned. 4- to 5-μm sections were stained with hematoxylin and eosine or anti-CD3 using standard methods.
For detection of autoantibodies in serum, wild-type stomach tissue was embedded in OCT (Sakura Tissue-Tek) and sections of 5 μm were cut with a Leica cryostat and fixed in acetone. Sections were blocked with 0.1% BSA and 1% normal mouse serum in PBS and then incubated with 1/300 diluted serum from 16-week-old Malt1 + / + and Malt1 ki/ki mice. After washing with PBS, the sections were incubated with biotinylated goat anti-mouse IgE + IgG1 (Invitrogen and Southern Biotech) in blocking buffer followed by Streptavidin-Alexa 488 (Molecular Probes). After a wash with PBS, slides were mounted using DAPI-containing ProLong antifade kit (Molecular Probes). For immunofluorescence staining of OCT-embedded spleen tissues, acetone-fixed 5- to 8-μm cryosections were preblocked with normal mouse and normal donkey serum. Then, they were labeled with the following primary antibodies: sheep anti-IgD (Binding Site, clone 1/70) and rat anti-CD35 (CR1, clone 8C12). The secondary and tertiary reagents used were donkey anti-rat Ig Cy3, donkey anti-sheep Ig Alexa 488, Streptavidin-Alexa 488 (Invitrogen). After DAPI staining, slides were mounted using DABCO. All images were captured with an Axio Imager.Z1 microscope (Zeiss) and analyzed using Adobe Photoshop.
Statistical analysis
Statistical significance was determined by two-tailed unpaired Student's t-test or two-way ANOVA using GraphPad Prism (GraphPad Software).
Acknowledgments
We would like to thank G. Guarda, S. Hailfinger, F. Martinon, and F. Tacchini-Cottier for comments on the manuscript, V. Philippe for discussions, D. Labes for cell sorting, and J. Dudda and A. Wilson for technical advice. This work was supported by grants from the Swiss National Science Foundation (SNSF), the Leenaards and Emma Muschamp Foundations, the Swiss Multiple Sclerosis Society and by a collaboration agreement with Ono Pharmaceuticals. We also acknowledge support by the SNSF (to W.H. and S.L.), the Max Cloëtta Foundation (to B.J.M.), the Pierre Mercier Foundation and an unrestricted grant from Astellas (to D.G).
Author contributions
MJ designed and did experiments, analyzed data, assembled the figures, and wrote the manuscript. BJM contributed to the material and design of experiments in Fig4A–D. JG and WH performed and analyzed the experiments in Fig3B. MJ and SAL designed and analyzed experiments in Figs2E and 6D. DG contributed to the design of Figs5C, D and 7A, OG contributed to the design of the knock-in mouse model, SF and MP provided technical assistance. MT designed and organized the study and wrote the paper. All authors contributed with discussions and critical comments to the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
Supplementary Figures
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 7
References
- Aguado E, Richelme S, Nunez-Cruz S, Miazek A, Mura AM, Richelme M, Guo XJ, Sainty D, He HT, Malissen B, Malissen M. Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science. 2002;296:2036–2040. doi: 10.1126/science.1069057. [DOI] [PubMed] [Google Scholar]
- Barnes MJ, Krebs P, Harris N, Eidenschenk C, Gonzalez-Quintial R, Arnold CN, Crozat K, Sovath S, Moresco EM, Theofilopoulos AN, Beutler B, Hoebe K. Commitment to the regulatory T cell lineage requires CARMA1 in the thymus but not in the periphery. PLoS Biol. 2009;7:e51. doi: 10.1371/journal.pbio.1000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Gordon S. Immune recognition. A new receptor for beta-glucans. Nature. 2001;413:36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- Brustle A, Brenner D, Knobbe CB, Lang PA, Virtanen C, Hershenfield BM, Reardon C, Lacher SM, Ruland J, Ohashi PS, Mak TW. The NF-kappaB regulator MALT1 determines the encephalitogenic potential of Th17 cells. J Clin Invest. 2012;122:4698–4709. doi: 10.1172/JCI63528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, Sun L, Chen ZJ, Marynen P, Beyaert R. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol. 2008;9:263–271. doi: 10.1038/ni1561. [DOI] [PubMed] [Google Scholar]
- Deenick EK, Elford AR, Pellegrini M, Hall H, Mak TW, Ohashi PS. c-Rel but not NF-kappaB1 is important for T regulatory cell development. Eur J Immunol. 2010;40:677–681. doi: 10.1002/eji.201040298. [DOI] [PubMed] [Google Scholar]
- Duwel M, Welteke V, Oeckinghaus A, Baens M, Kloo B, Ferch U, Darnay BG, Ruland J, Marynen P, Krappmann D. A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol. 2009;182:7718–7728. doi: 10.4049/jimmunol.0803313. [DOI] [PubMed] [Google Scholar]
- Ferch U, Buschenfelde CM, Gewies A, Wegener E, Rauser S, Peschel C, Krappmann D, Ruland J. MALT1 directs B cell receptor-induced canonical nuclear factor-kappaB signaling selectively to the c-Rel subunit. Nat Immunol. 2007;8:984–991. doi: 10.1038/ni1493. [DOI] [PubMed] [Google Scholar]
- Ferch U, Kloo B, Gewies A, Pfander V, Duwel M, Peschel C, Krappmann D, Ruland J. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2009;206:2313–2320. doi: 10.1084/jem.20091167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontan L, Yang C, Kabaleeswaran V, Volpon L, Osborne MJ, Beltran E, Garcia M, Cerchietti L, Shaknovich R, Yang SN, Fang F, Gascoyne RD, Martinez-Climent JA, Glickman JF, Borden K, Wu H, Melnick A. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell. 2012;22:812–824. doi: 10.1016/j.ccr.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Gringhuis SI, Wevers BA, Kaptein TM, van Capel TM, Theelen B, Boekhout T, de Jong EC, Geijtenbeek TB. Selective C-Rel activation via Malt1 controls anti-fungal T(H)-17 immunity by dectin-1 and dectin-2. PLoS Pathog. 2011;7:e1001259. doi: 10.1371/journal.ppat.1001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, Forster I, Ruland J. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. 2006;442:651–656. doi: 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- Gross O, Grupp C, Steinberg C, Zimmermann S, Strasser D, Hannesschlager N, Reindl W, Jonsson H, Huo H, Littman DR, Peschel C, Yokoyama WM, Krug A, Ruland J. Multiple ITAM-coupled NK cell receptors engage the Bcl10/Malt1 complex via Carma1 for NF-{kappa}B and MAPK activation to selectively control cytokine production. Blood. 2008;112:2421–2428. doi: 10.1182/blood-2007-11-123513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Manicassamy S, Vasu C, Kumar A, Shang W, Sun Z. Differential requirement of PKC-theta in the development and function of natural regulatory T cells. Mol Immunol. 2008;46:213–224. doi: 10.1016/j.molimm.2008.08.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutcher I, Urich E, Wolter K, Prinz M, Becher B. Interleukin 18-independent engagement of interleukin 18 receptor-alpha is required for autoimmune inflammation. Nat Immunol. 2006;7:946–953. doi: 10.1038/ni1377. [DOI] [PubMed] [Google Scholar]
- Hailfinger S, Lenz G, Ngo V, Posvitz-Fejfar A, Rebeaud F, Guzzardi M, Penas EM, Dierlamm J, Chan WC, Staudt LM, Thome M. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 2009;106:19946–19951. doi: 10.1073/pnas.0907511106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailfinger S, Nogai H, Pelzer C, Jaworski M, Cabalzar K, Charton JE, Guzzardi M, Decaillet C, Grau M, Dorken B, Lenz P, Lenz G, Thome M. Malt1-dependent RelB cleavage promotes canonical NF-kappaB activation in lymphocytes and lymphoma cell lines. Proc Natl Acad Sci USA. 2011;108:14596–14601. doi: 10.1073/pnas.1105020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst J, Wang H, Eder KD, Workman CJ, Boyd KL, Baquet Z, Singh H, Forbes K, Chruscinski A, Smeyne R, van Oers NS, Utz PJ, Vignali DA. Scalable signaling mediated by T cell antigen receptor-CD3 ITAMs ensures effective negative selection and prevents autoimmunity. Nat Immunol. 2008;9:658–666. doi: 10.1038/ni.1611. [DOI] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- Ishimaru N, Kishimoto H, Hayashi Y, Sprent J. Regulation of naive T cell function by the NF-kappaB2 pathway. Nat Immunol. 2006;7:763–772. doi: 10.1038/ni1351. [DOI] [PubMed] [Google Scholar]
- Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, Banerjee A, Proietto A, Gugasyan R, Wu L, McNally A, Steptoe RJ, Thomas R, Shannon MF, Gerondakis S. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med. 2009;206:3001–3014. doi: 10.1084/jem.20091411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost PJ, Weiss S, Ferch U, Gross O, Mak TW, Peschel C, Ruland J. Bcl10/Malt1 signaling is essential for TCR-induced NF-kappaB activation in thymocytes but dispensable for positive or negative selection. J Immunol. 2007;178:953–960. doi: 10.4049/jimmunol.178.2.953. [DOI] [PubMed] [Google Scholar]
- Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, Cook MC, Kucharska EM, Hara H, Penninger JM, Domashenz H, Hong NA, Glynne RJ, Nelms KA, Goodnow CC. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 2003;18:751–762. doi: 10.1016/s1074-7613(03)00141-9. [DOI] [PubMed] [Google Scholar]
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- Liston A, Enders A, Siggs OM. Unravelling the association of partial T-cell immunodeficiency and immune dysregulation. Nat Rev Immunol. 2008;8:545–558. doi: 10.1038/nri2336. [DOI] [PubMed] [Google Scholar]
- Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. 2009;31:921–931. doi: 10.1016/j.immuni.2009.09.022. [DOI] [PubMed] [Google Scholar]
- Marienfeld R, May MJ, Berberich I, Serfling E, Ghosh S, Neumann M. RelB forms transcriptionally inactive complexes with RelA/p65. J Biol Chem. 2003;278:19852–19860. doi: 10.1074/jbc.M301945200. [DOI] [PubMed] [Google Scholar]
- McGuire C, Wieghofer P, Elton L, Muylaert D, Prinz M, Beyaert R, van Loo G. Paracaspase MALT1 deficiency protects mice from autoimmune-mediated demyelination. J Immunol. 2013;190:2896–2903. doi: 10.4049/jimmunol.1201351. [DOI] [PubMed] [Google Scholar]
- Medoff BD, Sandall BP, Landry A, Nagahama K, Mizoguchi A, Luster AD, Xavier RJ. Differential requirement for CARMA1 in agonist-selected T-cell development. Eur J Immunol. 2009;39:78–84. doi: 10.1002/eji.200838734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinero LL, Yang J, Gajewski T, Abraham C, Farrar MA, Alegre ML. CARMA1 controls an early checkpoint in the thymic development of FoxP3+ regulatory T cells. J Immunol. 2009;182:6736–6743. doi: 10.4049/jimmunol.0900498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel D, Spranger S, Vincendeau M, Grau M, Raffegerst S, Kloo B, Hlahla D, Neuenschwander M, Peter von Kries J, Hadian K, Dorken B, Lenz P, Lenz G, Schendel DJ, Krappmann D. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell. 2012;22:825–837. doi: 10.1016/j.ccr.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- Noels H, van Loo G, Hagens S, Broeckx V, Beyaert R, Marynen P, Baens M. A novel TRAF6 binding site in MALT1 defines distinct mechanisms of NF-kappaB activation by API2-MALT1 fusions. J Biol Chem. 2007;282:10180–10189. doi: 10.1074/jbc.M611038200. [DOI] [PubMed] [Google Scholar]
- Oeckinghaus A, Wegener E, Welteke V, Ferch U, Arslan SC, Ruland J, Scheidereit C, Krappmann D. Malt1 ubiquitination triggers NF-kappaB signaling upon T-cell activation. EMBO J. 2007;26:4634–4645. doi: 10.1038/sj.emboj.7601897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Rebeaud F, Hailfinger S, Posevitz-Fejfar A, Tapernoux M, Moser R, Rueda D, Gaide O, Guzzardi M, Iancu EM, Rufer N, Fasel N, Thome M. The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat Immunol. 2008;9:272–281. doi: 10.1038/ni1568. [DOI] [PubMed] [Google Scholar]
- Rosebeck S, Rehman AO, Lucas PC, McAllister-Lucas LM. From MALT lymphoma to the CBM signalosome: three decades of discovery. Cell Cycle. 2011;10:2485–2496. doi: 10.4161/cc.10.15.16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, Tone M, Chen YH. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–940. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben SM, Klement JF, Coleman TA, Maher M, Chen CH, Rosen CA. I-Rel: a novel rel-related protein that inhibits NF-kappa B transcriptional activity. Genes Dev. 1992;6:745–760. doi: 10.1101/gad.6.5.745. [DOI] [PubMed] [Google Scholar]
- Ruefli-Brasse AA, French DM, Dixit VM. Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science. 2003;302:1581–1584. doi: 10.1126/science.1090769. [DOI] [PubMed] [Google Scholar]
- Ruland J, Duncan GS, Wakeham A, Mak TW. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity. 2003;19:749–758. doi: 10.1016/s1074-7613(03)00293-0. [DOI] [PubMed] [Google Scholar]
- Salisbury EM, Wang L, Choi O, Rutschmann S, Ashton-Rickardt PG. N-Ethyl-N-nitrosourea mutagenesis in the mouse provides strong genetic and in vivo evidence for the role of the Caspase Recruitment Domain (CARD) of CARD-MAGUK1 in T regulatory cell development. Immunology. 2014;141:446–456. doi: 10.1111/imm.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Supprian M, Courtois G, Tian J, Coyle AJ, Israel A, Rajewsky K, Pasparakis M. Mature T cells depend on signaling through the IKK complex. Immunity. 2003;19:377–389. doi: 10.1016/s1074-7613(03)00237-1. [DOI] [PubMed] [Google Scholar]
- Schmidt-Supprian M, Tian J, Grant EP, Pasparakis M, Maehr R, Ovaa H, Ploegh HL, Coyle AJ, Rajewsky K. Differential dependence of CD4+CD25+ regulatory and natural killer-like T cells on signals leading to NF-kappaB activation. Proc Natl Acad Sci USA. 2004;101:4566–4571. doi: 10.1073/pnas.0400885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggs OM, Miosge LA, Yates AL, Kucharska EM, Sheahan D, Brdicka T, Weiss A, Liston A, Goodnow CC. Opposing functions of the T cell receptor kinase ZAP-70 in immunity and tolerance differentially titrate in response to nucleotide substitutions. Immunity. 2007;27:912–926. doi: 10.1016/j.immuni.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommers CL, Park CS, Lee J, Feng C, Fuller CL, Grinberg A, Hildebrand JA, Lacana E, Menon RK, Shores EW, Samelson LE, Love PE. A LAT mutation that inhibits T cell development yet induces lymphoproliferation. Science. 2002;296:2040–2043. doi: 10.1126/science.1069066. [DOI] [PubMed] [Google Scholar]
- Staal J, Driege Y, Bekaert T, Demeyer A, Muyllaert D, Van Damme P, Gevaert K, Beyaert R. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. 2011;30:1742–1752. doi: 10.1038/emboj.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- Thome M. Multifunctional roles for MALT1 in T-cell activation. Nat Rev Immunol. 2008;8:495–500. doi: 10.1038/nri2338. [DOI] [PubMed] [Google Scholar]
- Thome M, Charton JE, Pelzer C, Hailfinger S. Antigen receptor signaling to NF-kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb Perspect Biol. 2010;2:a003004. doi: 10.1101/cshperspect.a003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyabe S, Watanabe A, Harada W, Karasawa T, Uchiyama M. Specific immunoglobulin E responses in ZAP-70-deficient patients are mediated by Syk-dependent T-cell receptor signalling. Immunology. 2001;103:164–171. doi: 10.1046/j.1365-2567.2001.01246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehata T, Iwasaki H, Vandenbon A, Matsushita K, Hernandez-Cuellar E, Kuniyoshi K, Satoh T, Mino T, Suzuki Y, Standley DM, Tsujimura T, Rakugi H, Isaka Y, Takeuchi O, Akira S. Malt1-induced cleavage of regnase-1 in CD4(+) helper T cells regulates immune activation. Cell. 2013;153:1036–1049. doi: 10.1016/j.cell.2013.04.034. [DOI] [PubMed] [Google Scholar]
- Visekruna A, Huber M, Hellhund A, Bothur E, Reinhard K, Bollig N, Schmidt N, Joeris T, Lohoff M, Steinhoff U. c-Rel is crucial for the induction of Foxp3(+) regulatory CD4(+) T cells but not T(H)17 cells. Eur J Immunol. 2010;40:671–676. doi: 10.1002/eji.200940260. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kissenpfennig A, Mingueneau M, Richelme S, Perrin P, Chevrier S, Genton C, Lucas B, DiSanto JP, Acha-Orbea H, Malissen B, Malissen M. Th2 lymphoproliferative disorder of LatY136F mutant mice unfolds independently of TCR-MHC engagement and is insensitive to the action of Foxp3+ regulatory T cells. J Immunol. 2008;180:1565–1575. doi: 10.4049/jimmunol.180.3.1565. [DOI] [PubMed] [Google Scholar]
- Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- Weih F, Durham SK, Barton DS, Sha WC, Baltimore D, Bravo R. Both multiorgan inflammation and myeloid hyperplasia in RelB-deficient mice are T cell dependent. J Immunol. 1996;157:3974–3979. [PubMed] [Google Scholar]
- Zhang FX, Kirschning CJ, Mancinelli R, Xu XP, Jin Y, Faure E, Mantovani A, Rothe M, Muzio M, Arditi M. Bacterial lipopolysaccharide activates nuclear factor-kappaB through interleukin-1 signaling mediators in cultured human dermal endothelial cells and mononuclear phagocytes. J Biol Chem. 1999;274:7611–7614. doi: 10.1074/jbc.274.12.7611. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 7