

Abstract
Mitochondrial dynamics and mitophagy have been linked to cardiovascular and neurodegenerative diseases. Here, we demonstrate that the mitochondrial division dynamin Drp1 and the Parkinson's disease-associated E3 ubiquitin ligase parkin synergistically maintain the integrity of mitochondrial structure and function in mouse heart and brain. Mice lacking cardiac Drp1 exhibited lethal heart defects. In Drp1KO cardiomyocytes, mitochondria increased their connectivity, accumulated ubiquitinated proteins, and decreased their respiration. In contrast to the current views of the role of parkin in ubiquitination of mitochondrial proteins, mitochondrial ubiquitination was independent of parkin in Drp1KO hearts, and simultaneous loss of Drp1 and parkin worsened cardiac defects. Drp1 and parkin also play synergistic roles in neuronal mitochondrial homeostasis and survival. Mitochondrial degradation was further decreased by combination of Drp1 and parkin deficiency, compared with their single loss. Thus, the physiological importance of parkin in mitochondrial homeostasis is revealed in the absence of mitochondrial division in mammals.
Keywords: mice, mitochondria, organelle division, respiration
Introduction
Mitochondria are highly abundant in the brain and heart. In these tissues, neurons and cardiomyocytes produce high levels of oxidative stress and live for a long period of time without proliferation; therefore, mitochondrial homeostasis is critical in these cells. Indeed, alterations in mitochondrial structure and function have been linked to many neurodegenerative and cardiovascular diseases (Nunnari & Suomalainen, 2012; Itoh et al, 2013; Ong et al, 2013).
Mitochondria grow and divide to control their number and size. Mitochondrial division is mediated by a dynamin-related GTPase, Drp1, and its receptors, which are located in the mitochondrial outer membrane (Okamoto & Shaw, 2005; Chang & Blackstone, 2010; Westermann, 2010; Kageyama et al, 2011; Tamura et al, 2011; Sesaki et al, 2013). Although Drp1 is ubiquitously expressed in many mammalian tissues, different cell types have different frequencies of mitochondrial division. In neurons, mitochondria are distributed throughout the cytoplasm, actively moving along axons and dendrites. Neuronal mitochondria also undergo frequent division and fusion and regulate their shape, size, and number. In contrast, mitochondria in cardiomyocytes are located underneath the sarcolemma, between myofibrils and around nuclei (Ong et al, 2013; Piquereau et al, 2013). Intermyofibrillar mitochondria run parallel to myofibrils to efficiently provide ATP for muscle contraction. Mitochondrial movement is limited due to this spatial organization in the cytoplasm, and mitochondrial division is estimated to only occur at an extremely low frequency in adult cardiomyocytes under physiological conditions (Beraud et al, 2009). In contrast, under pathological conditions such as ischemic reperfusion, mitochondria divide and induce apoptosis, promoting myocardial damage. In ischemic reperfusion, a pharmacological inhibitor of Drp1 decreases cell death (Ong et al, 2010). However, it is unknown whether and how Drp1 controls the morphological and functional integrity of mitochondria in cardiomyocytes.
Mitochondrial homeostasis depends on their biogenesis and degradation. Because mitochondria do not form de novo, their biogenesis is mediated by the growth of pre-existing organelles. The ubiquitin E3 ligase parkin, which is defective in Parkinson's disease, has been suggested to control the biogenesis and degradation of mitochondria (Itoh et al, 2013). During mitochondrial biogenesis, parkin regulates gene expression of mitochondrial proteins through the transcriptional coactivator PGC-1α (Shin et al, 2011). Parkin has also been shown to ubiquitinate mitochondrial proteins to stimulate mitophagy, autophagic degradation of mitochondria (Narendra et al, 2008). However, complete loss of parkin in mouse models does not result in cardiac defects or neurodegeneration, and the in vivo function of parkin in mitophagy is unclear in mammals (Dawson et al, 2010). Mitophagy also involves Drp1, which generates small mitochondria and allows autophagosomes to efficiently engulf the organelle (Twig et al, 2008; Tanaka et al, 2010; Kageyama et al, 2012). Autophagosomes then deliver the mitochondria to lysosomes for degradation. However, considering the low occurrence of mitochondrial division in cardiomyocytes, the role of Drp1 in mitophagy in the mammalian heart remains to be determined.
In this study, we characterized cardiomyocytes lacking Drp1 in mice and found that mitochondria were enlarged in the absence of Drp1 and accumulated the mitophagy adaptor protein p62, leading to decreased respiration and lethal cardiac defects. In these mitochondria, ubiquitinated proteins also accumulated; however, the loss of parkin did not decrease mitochondrial ubiquitination and exacerbated defects in both mitochondrial and cardiac functions. Synthetic impacts of the loss of Drp1 and parkin on mitochondria were found to be common in cardiomyocytes, neurons, and fibroblasts. Thus, Drp1 and parkin synergistically control the biogenesis and degradation of mitochondria to maintain organelle homeostasis.
Results
Loss of Drp1 in cardiomyocytes leads to lethal cardiac dysfunction in mice
We deleted cardiac Drp1 using its floxed allele (Wakabayashi et al, 2009; Kageyama et al, 2012) and a Myh6-Cre transgenic line, which expresses Cre recombinase in cardiomyocytes in the early postnatal days (Gaussin et al, 2002). By breeding, we generated Drp1flox/flox::Myh6-Cre (Myh6-Drp1KO) mice, Drp1flox/+::Myh6-Cre (Het) mice, and Drp1flox/+ and Drp1flox/flox (collectively controls) mice. All of the mice were born at normal Mendelian frequency (Supplementary Table S1). Drp1 expression was reduced to ∼20–30% in heart isolated from postnatal day 7 (P7) Myh6-Drp1KO mice compared with littermate controls (Fig 1A). Residual Drp1 expression in Myh6-Drp1KO hearts may be due to incomplete recombination of the Drp1 floxed allele and/or cardiac valves, coronary vessels, and the aorta, none of which express Myh6-Cre (Gaussin et al, 2002). There was no difference in the abundance of proteins used as loading controls, including the mitochondrial protein Tim23 and actin. P7 Myh6-Drp1KO mice were indistinguishable from littermate controls in terms of body size, heart size, and heart morphology (Fig1B–D). However, all Myh6-Drp1KO mice died between P9 and P11 (Fig1E). Echocardiography at P7 revealed that left ventricular (LV) function was significantly compromised, with decreased contraction and heart rate, in Myh6-Drp1KO mice (Fig1F–J and Supplementary Table S2). Similarly, LV function was reduced in P1 Myh6-Drp1KO mice (Supplementary Table S2), in which Drp1 levels were decreased to ∼50% of those in control mice (Supplementary Fig S1). In contrast, no cardiac dysfunction was observed in P7 Het mice (Fig 1F–J and Supplementary Table S2), in which Drp1 levels were reduced to ∼75% of those in control mice (Fig1A).
Figure 1. Heart function and postnatal survival require cardiomyocyte Drp1.

- A Immunoblotting of hearts isolated from control, Het, and Myh6-Drp1KO (KO) mice was performed using antibodies to Drp1, Tim23 (a mitochondrial protein), and actin.
- B Body weights of mice are shown (n = 3).
- C Heart weights were normalized relative to tibial length (n = 3).
- D Morphology and H&E histology of P7 hearts.
- E Survival curve of mice (n ≥ 8).
- F M-mode echocardiograms of P7 mice.
- G–J Echocardiographic parameters in P7 mice. Left ventricular end-diastolic dimension (LVEDD), left ventricular end-systolic dimension (LVESD), fractional shortening (FS), and heart rate (HR) are shown. Values are mean ± SEM (n = 6 for control, 7 for Het, and 5 for KO). P-values were determined using ANOVA followed by Tukey's post hoc test. **P < 0.01, ***P < 0.001.
- K–O Electrocardiogram of P7 mice. HR, P wave amplitude, PR interval, and QRS complex width are shown. P-values were determined using t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
To further examine cardiac function, we performed electrocardiography at P7. We first confirmed a decreased heart rate in Myh6-Drp1KO mice (Fig 1K and L). Although decreased, the heart rate responded normally to isoproterenol injection, suggesting that the decreased heart rate is not due to alteration in adrenergic modulation of the heart (Supplementary Fig S2). In addition, the amplitude of the P wave was decreased (Fig 1M). Approximately 30% of P7 Myh6-Drp1KO mice had polymorphic P waves with negative values for P amplitude, suggesting loss of the original function of the sinoatrial node and induction of wandering pacemaker activities. Around P11, the cardiac phenotypes became more severe and we did not observe P waves in ∼70% of Myh6-Drp1KO mice (Supplementary Fig S3). For P11 Myh6-Drp1KO mice in which P waves were detected, the PR interval was further prolonged compared with P7 Myh6-Drp1KO mice (Fig 1N and Supplementary Fig S3D). Moreover, QRS complexes became dramatically wider in Myh6-Drp1KO mice at P11, but not at P7 (Fig 1O and Supplementary Fig S3C), suggesting that the conduction system was also progressively compromised toward the death of Myh6-Drp1KO mice.
Mitochondrial respiration requires Drp1 in cardiomyocytes
To assess the impact of Drp1 loss on mitochondrial function in cardiomyocytes, we histologically analyzed the activity of electron transport chain complexes using fresh frozen sections of hearts. NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), and cytochrome c oxidase (complex IV) showed decreased activity staining in the hearts of P7 Myh6-Drp1KO mice (Fig2A). Similarly, activities of NADH dehydrogenase and cytochrome c oxidase were lower in cell lysates prepared from Myh6-Drp1KO hearts (Fig2B and C). Consistent with these findings, immunoblotting showed that the amounts of NDUFB8 (a subunit of complex I) and subunit I (a subunit of complex IV) were modestly decreased in Myh6-Drp1KO, although other electron transport chain components tested were not grossly affected (Fig2D). Moreover, to measure mitochondrial respiration, we isolated neonatal cardiomyocytes from control and Myh6-Drp1KO mice and quantitated their oxygen consumption rates (OCR). Basal respiration was significantly reduced in Drp1KO cardiomyocytes (Fig2E).
Figure 2. Decreased respiratory activities in hearts of Myh6-Drp1KO mice.
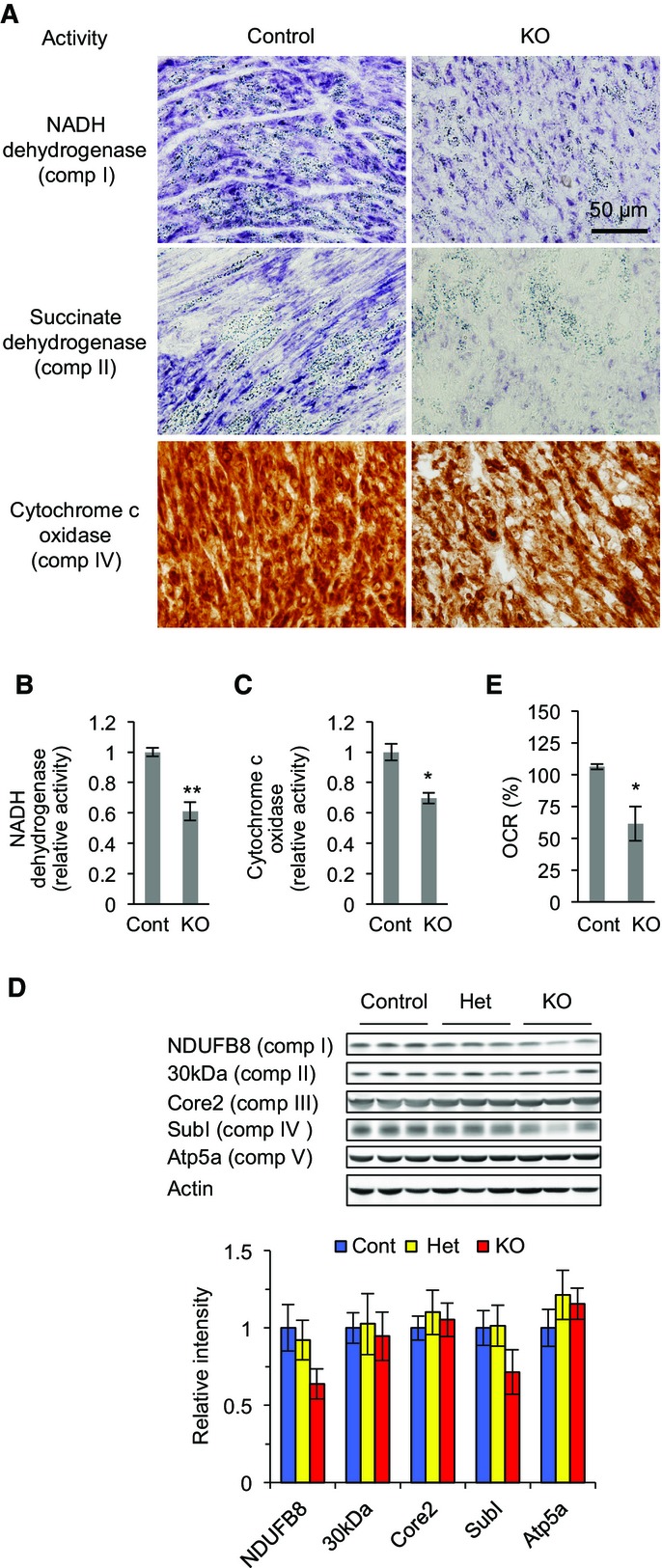
- A Fresh frozen sections of heart from P7 control and Myh6-Drp1KO mice were histologically stained for activities of NADH dehydrogenase, succinate dehydrogenase, and cytochrome c oxidase.
- B, C NADH dehydrogenase (B) and cytochrome c oxidase (C) were measured in P7 hearts using complex I and complex VI activity microplate assays. Values are mean ± SEM (n = 3). *P < 0.05, **P < 0.01. P-values were determined using t-test.
- D Immunoblotting of hearts isolated from P7 control, Het, and Myh6-Drp1KO mice using antibodies to actin and the indicated subunits of electron transport chain complexes.
- E The oxygen consumption rate (OCR) was measured in isolated cardiomyocytes. OCRs in the control were set to 100%. Values are mean ± SEM (n = 4). *P < 0.05. P-values were determined using t-test.
Mitochondria are connected and enlarged in Drp1KO cardiomyocytes
Movements of intermyofibrillar mitochondria are restricted due to highly packed myofibrils in cardiomyocytes, and rates of mitochondrial division and fusion have been estimated to be very low in adult cardiomyocytes (Beraud et al, 2009; Piquereau et al, 2013). It has also been shown that mitochondria only transiently fuse by extending nanotubules, without complete mixing of contents (Huang et al, 2013). Therefore, we wanted to test whether the lack of mitochondrial division affects organelle morphology. We performed transmission electron microscopy (TEM) on P7 left ventricles. In longitudinal, ultrathin sections of control cardiomyocytes, mitochondria were visualized as short tubular structures (Fig3A). In contrast, mitochondria in Myh6-Drp1KO cardiomyocytes displayed heterogeneous morphology with branched tubules and large ovals (Fig3A). To further analyze mitochondrial morphology, we carried out electron microscopic tomography (Fig3B and C). While mitochondria were again observed as short tubules in control cardiomyocytes, mitochondria that appeared to be separated in Myh6-Drp1KO cardiomyocytes by TEM were found to be interconnected (Fig3B and C). Consistent with their morphology, quantification of mitochondrial volume showed a significant increase in Myh6-Drp1KO (Fig3D). In addition, we found large vacuolar structures that contained remnants of mitochondria in Myh6-Drp1KO cardiomyocytes (Fig3A, right image). These structures were not observed in control littermate mice and were suggestive of autophagosomes containing mitochondria. Although mitochondria were enlarged and showed decreased respiration, the organization of myofibrils appeared to be unaffected in Myh6-Drp1KO cardiomyocytes (Fig3E).
Figure 3. Mitochondria are interconnected in Myh6-Drp1KO cardiomyocytes.

- A Ultrastructure was examined using conventional transmission electron microscopy in P7 control and Myh6-Drp1KO mice. An asterisk indicates a vacuolar structure that contains remnants of mitochondria in Myh6-Drp1KO cardiomyocytes.
- B, C EM tomography (xyz, 6.3 μm × 6.3 μm × 0.45 μm). Three-dimensional surface-mesh models of individual mitochondria are shown in different colors. Ultrathin slice images are computed from tomograms and merged with mesh models. (C, top images) Each tomogram was computed from 242 tilted transmission electron micrograms. Boundary boxes (yellow) and arbitrary positioned ultrathin slices of tomograms are displayed. (C, middle images) Three-dimensional surface-mesh models of the mitochondrial outer membrane were created. Different colors indicate different mitochondria. (C, bottom images) Two representative mitochondria are shown at high magnification in each tomogram.
- D Mitochondrial volume was quantified using tomography. Values are mean ± SD (n = 34 for control and 47 for KO). P-values were determined using the Mann–Whitney U-test.
- E Immunofluorescence microscopy of sections of P7 control and Myh6-Drp1KO hearts using anti-actin antibodies. Nuclei are stained with DAPI.
- F Immunofluorescence of neonatal cardiomyocytes at day 4 after isolation using antibodies to a mitochondrial protein, pyruvate dehydrogenase (PDH). F-actin was visualized with rhodamine–phalloidin.
- G Quantification of cardiomyocytes that contain enlarged mitochondria. Values are mean ± SEM (n = 3, ˜50 cells were examined in each experiment). ***P < 0.001. P-values were determined using t-test.
- H Immunofluorescence of isolated neonatal cardiomyocytes using antibodies to Tom20 and an oxidative marker for both proteins and lipids, 4-hydroxynonenal (HNE). Cardiomyocytes with positive staining for HNE were scored. Values are mean ± SEM (n = 3, ˜50 cells were examined in each experiment). *P < 0.05. P-values were determined using t-test.
Consistent with the in vivo data, neonatal cardiomyocytes isolated from Myh6-Drp1KO mice contained enlarged mitochondria and normal myofibrils while control and Het cardiomyocytes had short tubular mitochondria (Fig3F and G). These mitochondria in Drp1KO cardiomyocytes look morphologically similar to those in Drp1KO fibroblasts treated with oxidative stress in culture (Kageyama et al, 2012), suggesting increased amounts of oxidative damage. Indeed, immunofluorescence staining with antibodies to 4-hydroxynonenal (HNE), which recognizes oxidized proteins and lipids, strongly labeled the inside of large spherical mitochondria in Drp1KO cardiomyocytes (Fig3H).
Although mitochondrial structure and function were altered, apoptosis was not induced in Drp1KO cardiomyocytes. Immunoblotting showed no proteolytic activation of caspase-3 or cleavage of its substrate PARP in Myh6-Drp1KO hearts (Fig4A).
Figure 4. Accumulation of p62 and ubiquitinated proteins on mitochondria in Myh6-Drp1KO cardiomyocytes.

- A Immunoblotting of hearts isolated from P7 control, Het, and Myh6-Drp1KO mice using antibodies to p62, LC3, a mitochondrial protein (Tim23), caspase-3, and PARP. An active form of caspase-3 (17 kDa), which is generated by cleavage of full-length protein (35 kDa), was not observed in control, Het, or Myh6-Drp1KO hearts. Similarly, cleavage of PARP was not detected. Band intensity was quantified and normalized to amount of protein loaded in each lane. Values are mean ± SEM (n = 3). **P < 0.01. P-values were determined using t-test.
- B–D Immunofluorescence microscopy was performed on heart sections from P7 control and Myh6-Drp1KO mice using the indicated antibodies. Boxed regions show magnified images in (B) and (C).
Mitochondria accumulate p62 and ubiquitinated proteins in Drp1KO cardiomyocytes independent of parkin
To further explore our observation that Drp1KO cardiomyocytes showed vacuolar compartments that contain mitochondrial remnants (Fig3A, right image), we performed immunoblotting of isolated hearts with antibodies to two autophagy markers, p62/SQSTM and LC3. p62 is degraded by autophagy and inhibition of autophagy increases its abundance (Klionsky et al, 2012). During mitophagy, p62 is recruited to mitochondria, where it functions as an adaptor protein (Tanaka et al, 2010). LC3-II is a component of autophagosomes and is converted from LC3-I during autophagy (Klionsky et al, 2012). We found that the level of p62 was increased ∼fourfold in Myh6-Drp1KO compared with control and Het littermates (Fig4A). In contrast, LC3-II levels remained unchanged. Consistent with the biochemical data, immunohistochemistry also showed accumulation of p62 in Myh6-Drp1KO hearts (Fig4B). Notably, p62 signals were concentrated in punctate structures decorated by ubiquitin (Fig4B), which is conjugated to mitochondrial proteins during mitophagy (Youle & Narendra, 2011). To test whether these structures contain mitochondria, we immunostained heart sections with antibodies to p62, ubiquitin, and pyruvate dehydrogenase (PDH, a mitochondrial matrix protein). p62-positive structures were co-labeled with antibodies to ubiquitin and PDH (Fig4C) in Drp1KO cardiomyocytes. High-magnification views showed that mitochondrial PDH signals were encircled by p62 and ubiquitin signals (Fig4C). Furthermore, triple immunostaining showed that ubiquitin-decorated mitochondria are not colocalized with a lysosomal marker, Lamp1, in Drp1KO hearts. Interestingly, many lysosomes were found near ubiquitin-decorated mitochondria. It appears that mitophagy was incompletely executed in Drp1KO cardiomyocytes, generating intermediate structures with accumulated p62 and ubiquitinated proteins on mitochondria without increases in the amount of LC3-II.
To test whether the accumulation of p62, in the absence of Drp1, is due to inhibition of autophagic flux, we examined the accumulation of LC3-II in WT and Drp1KO mouse embryonic fibroblasts (MEF) in the presence of bafilomycin A, an inhibitor of lysosomal H+-ATPases that blocks autophagic degradation (Klionsky et al, 2012). We found that LC3-II similarly built up in both cells (Supplementary Fig S5), suggesting that the loss of Drp1 does not affect autophagic flux.
To determine whether the accumulation of p62 and ubiquitinated proteins on mitochondria requires parkin in Drp1KO hearts, we crossed Myh6-Drp1KO mice to parkin−/− (ParkinKO) mice and generated ParkinKO::Myh6-Drp1KO mice (ParkinDrp1KO). We isolated hearts from control, Drp1KO, ParkinKO, and ParkinDrp1KO mice and performed immunoblotting with antibodies to Drp1 and p62. While Drp1KO and ParkinDrp1KO hearts had decreased Drp1 levels (Fig5A), ParkinKO hearts showed increased Drp1 levels, consistent with a previous study showing that parkin ubiquitinates Drp1 for degradation (Wang et al, 2011). Remarkably, the abundance of p62 was similarly increased in Drp1KO and ParkinDrp1KO hearts (Fig5A). In contrast, ParkinKO hearts contained normal levels of p62. Furthermore, immunofluorescence of heart sections showed that p62 was accumulated in both Drp1KO and ParkinDrp1KO hearts, but not in control and ParkinKO hearts (Fig5B). Colocalization of p62 and ubiquitin was also unaffected in ParkinDrp1KO hearts (Fig5B). Therefore, the increased levels of p62 and mitochondrial ubiquitination are independent of parkin in Drp1KO hearts.
Figure 5. Mitochondria accumulate p62 and ubiquitinated proteins independent of parkin in Myh6-Drp1KO cardiomyocytes.

- A Immunoblotting of hearts isolated from P7 control, Myh6-Drp1KO, ParkinKO, and ParkinDrp1KO mice using the indicated antibodies. Band intensity was quantified. Values are mean ± SEM (n = 3).
- B Immunofluorescence microscopy was performed on heart sections from P7 control and Myh6-Drp1KO mice using antibodies to p62 and ubiquitin.
- C M-mode echocardiograms of P7 mice.
- D, E Echocardiographic parameters in P7 mice. Fractional shortening (FS) and heart rate (HR) are shown. Values are mean ± SEM (n = 18 for control, 12 for Myh6-Drp1KO, 14 for ParkinKO, and 3 for ParkinDrp1KO). P-values were determined using ANOVA followed by Tukey's post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001.
- F Immunoblotting of hearts isolated from P7 control, Myh6-Drp1KO, ParkinKO, and ParkinDrp1KO mouse using antibodies to the indicated subunits of electron transport chain complexes and Tim23. Band intensity was quantified. Values are mean ± SEM (n = 6).
- G, H Immunofluorescence microscopy of heart sections from control and Myh6-Drp1KO mice was performed using antibodies to p62, ubiquitin, parkin, and a mitochondrial protein, Hsp60. Arrowheads indicate p62- and ubiquitin-positive structures in (G). Arrows indicate enlarged mitochondria in (H).
Additional loss of parkin exacerbates cardiac defects in Drp1KO cardiomyocytes
To determine whether the simultaneous loss of Drp1 and parkin affects heart function, we performed echocardiography on control, Drp1KO, ParkinKO, and ParkinDrp1KO mice at P7. We found that that LV function significantly worsened in ParkinDrp1KO mice compared with Drp1KO mice, with a drop of heart contraction from ∼45% (Drp1KO) to ∼30% (ParkinDrp1KO) (Fig 5C and D, and Supplementary Table S3). Similarly, the heart rate was significantly lower in ParkinDrp1KO mice (Fig 5C and E, and Supplementary Table S3). ParkinKO mice showed no cardiac dysfunction.
Combination of Drp1 and parkin deficiency also exacerbated the integrity of mitochondria. We examined components of the electron transport chain complexes using immunoblotting. ParkinDrp1KO mice showed further decreases in the abundance of electron transport chain subunits, compared with Drp1KO mice, although the protein import translocase Tim23 was unaffected (Fig5F). Thus, parkin is critical for cardiac function and the maintenance of the electron transport chain in cardiomyocytes when Drp1 is absent.
To examine the localization of parkin in Drp1KO cardiomyocytes, we performed immunofluorescence microscopy of control and Myh6-Drp1KO hearts. Results showed that parkin did not accumulate in p62- and ubiquitin-decorated structures (Fig5G) or enlarged mitochondria (Fig5H) in Myh6-Drp1KO hearts.
Simultaneous loss of Drp1 and parkin accelerates degeneration of Purkinje neurons and worsens morphological abnormalities of mitochondria
Considering the involvement of Drp1 and parkin in neurodegenerative diseases, the synthetic phenotypes induced by loss of Drp1 and parkin in the heart prompted us to test whether these two proteins also synergistically function in neurons. To address this, we studied Purkinje neurons (PN) in the cerebellum. We have previously shown that Drp1 loss leads to neurodegeneration of postmitotic PNs in Drp1flox/flox mice expressing Cre recombinase from the postmitotic PN-specific L7 promoter (L7-Drp1KO mice) (Fig6A and B) (Kageyama et al, 2012). By breeding L7-Drp1KO mice to ParkinKO mice, which showed no degeneration of PNs, we generated ParkinKO::L7-Drp1KO mice. We found that ParkinKO::L7-Drp1KO mice showed accelerated PN degeneration compared with L7-Drp1KO mice (Fig6A and B). Upon Drp1 loss, the mitochondria in L7-Drp1KO mice were elongated due to unopposed fusion in PNs and then further changed their morphology to large spheres, likely due to oxidative damage (Fig6B and C) (Kageyama et al, 2012). These morphological changes began prior to neurodegeneration and ∼25% of PNs carried spherical mitochondria in L7-Drp1KO mice at 1 month of age (Fig6B and C). In contrast, ∼50% of PNs had mitochondria that had already transformed into spherical structures in ParkinKO::L7-Drp1KO mice (Fig6B and C). The enlarged mitochondria in L7-Drp1KO PNs accumulated ubiquitinated proteins (Fig6D). Similar mitochondrial ubiquitination was observed in ParkinKO::L7-Drp1KO mice (Fig6D), suggesting that parkin is dispensable for mitochondrial ubiquitination in L7-Drp1KO PNs. Thus, Drp1 and parkin synergistically support the survival of PNs and the morphology of mitochondria.
Figure 6. The simultaneous loss of Drp1 and parkin accelerates mitochondrial enlargement and neurodegeneration in postmitotic Purkinje neurons (PNs).

- Immunofluorescence of cerebellar sagittal sections around the median line using antibodies to Car8, a PN marker. Control, L7-Drp1KO, ParkinKO, and Parkin::L7-Drp1KO mice were analyzed at 1, 2, and 3 months of age. Images taken at 2 months are shown.
- Quantification of PN density. The number of somas of PNs that contain tubular (filled) or large spherical (striped) mitochondria was determined and normalized relative to the length of the Purkinje cell layer. Values represent the mean ± SEM (n = 6 for each genotype). *P < 0.05, **P < 0.01, ***P < 0.001. P-values were determined using t-test.
- Mitochondrial morphology. Sagittal sections of the cerebellum at 1 month were stained using antibodies against Car8 and PDH. The somas of PNs are outlined based on Car8 staining (white line).
- Mitochondrial ubiquitination. Sections of the cerebellum at 2 months were labeled using antibodies to ubiquitin, Tom20, and Car8. The somas of PNs are outlined based on Car8 staining (white line).
ParkinDrp1KO MEFs lose integrity of the mitochondrial matrix and mitochondrial DNA
To further elucidate the role of Drp1 and parkin in the organization of mitochondria, we generated MEFs lacking both Drp1 and parkin. ParkinDrp1KO MEFs were generated by infecting Parkin−/−::Drp1flox/flox MEFs, which were used as ParkinKO MEFs, with lentiviruses expressing Cre recombinase. We performed immunofluorescence microscopy using antibodies to the outer membrane protein Tom20 and the matrix protein PDH (Fig 7A). In WT and ParkinKO MEFs, mitochondria displayed indistinguishable morphology with short tubules with occasional branches. As previously described (Ishihara et al, 2009; Wakabayashi et al, 2009), the majority of Drp1KO MEFs contained elongated tubules. We also noticed that ∼30% of Drp1KO MEFs had parts of the elongated tubules that were swollen. Additional loss of parkin exacerbated the morphological defects in the absence of Drp1, as ∼80% of ParkinDrp1KO MEFs contained large spherical mitochondria connected to long tubules (Fig7A). In these spherical mitochondria, PDH signals were encircled by Tom20 signals, showing expansion of the matrix space and changes in the surface area to volume ratio. These spherical mitochondria looked similar to those in Drp1KO MEFs treated with hydrogen peroxide (Kageyama et al, 2012), suggesting increased amounts of oxidative damage. To determine whether the formation of the spherical mitochondria in ParkinDrp1KO MEFs results from increased oxidative stress, we incubated ParkinDrp1KO MEFs with an antioxidant, N-acetylcysteine. We found that N-acetylcysteine decreased the number of cells with spherical mitochondria in ParkinDrp1KO MEFs (Supplementary Fig S6).
Figure 7. Synergistic effects of loss of Drp1 and parkin in mouse embryonic fibroblasts (MEFs).

- A WT, Drp1KO, ParkinKO, and ParkinDrp1KO MEFs were subjected to immunofluorescence with antibodies to the matrix protein PDH and the outer membrane protein Tom20.
- B Cells that contained enlarged mitochondria were scored. Values represent the mean ± SEM (n = 3). More than 50 cells were examined in each experiment. **P < 0.01. P-values were determined using t-test.
- C MEFs were stained with MitoTracker and picogreen and observed using fluorescence microscopy.
- D–F The oxygen consumption rate (OCR) was measured in MEFs. Oligomycin (1 μg/ml), FCCP (1 μM), and rotenone (0.1 μM)/antimycin A (2 μM) were added at the times indicated by green lines; OCRs in WT at 0 min were set to 100%. Basal respiration (E) and respiratory capacity (F) are shown. Values are mean ± SEM (n ≥ 14). **P < 0.01, ***P < 0.001. P-values were determined using t-test.
- G, H MEFs were infected with lentivirus expressing mt-Keima and observed at day 3 after infection using the dual-excitation imaging system as described (Katayama et al, 2011). Excitation 458 nm/emission > 650 nm was used to detect mt-Keima in mitochondria in the cytosol (FLmito, green) and excitation 561 nm/emission > 650 nm for mitochondria that have been delivered to lysosomes (FLlyso, red). Fluorescence signals of mt-Keima in lysosomes and mitochondria were measured in five different positions and averaged, and their ratio (FLlyso/FLmito) determined. Values are mean ± SEM (n = 10–15 cells). **P < 0.01, ***P < 0.001. P-values were determined using t-test.
Mitochondrial DNA was also affected in MEFs that contained swollen mitochondria in live cell imaging with fluorescent dyes for mitochondria (MitoTracker) and DNA (picogreen) (Fig7C). In WT and ParkinKO MEFs, mitochondrial DNA was distributed throughout mitochondria as small punctate structures (Fig7C). Conversely, in Drp1KO and ParkinDrp1KO MEFs with spherical mitochondria, mitochondrial DNA was accumulated in these swollen portions while tubular mitochondria still contained punctate structures of mitochondrial DNA, although their number appeared to be decreased.
In addition to defects in structural integrity, mitochondrial respiration was also decreased by a combination of Drp1 and parkin deficiency. We measured OCRs in WT, Drp1KO, ParkinKO, and ParkinDrp1KO MEFs. We found that the basal respiration was significantly decreased in ParkinDrp1KO MEFs, whereas WT, Drp1KO, and ParkinKO MEFs showed similar activities (Fig7D and E). In addition, the respiratory capacity was slightly decreased in Drp1KO MEFs, but not in ParkinKO MEFs, and ParkinDrp1KO MEFs showed a further decrease in the capacity compared with Drp1KO MEFs (Fig7D and F). Thus, Drp1 loss leads to slight decreases in mitochondrial function and additional loss of parkin worsens mitochondrial dysfunction, although single loss of parkin does not affect mitochondrial respiration.
ParkinDrp1KO MEFs are defective in the delivery of mitochondria to lysosomes
To gain insight into the role of Drp1 and parkin in mitophagy, we monitored the delivery of mitochondria to lysosomes using a biosensor for this process, mt-Keima, and the dual-excitation imaging system (Katayama et al, 2011). mt-Keima is a variant of RFP that changes its fluorescent profile in response to pH and is resistant to degradation in lysosomes, fused to a presequence to the mitochondrial matrix. We infected WT and Drp1KO MEFs using lentiviruses carrying mt-Keima and observed the intracellular distribution of mt-Keima (Fig7G). While mt-Keima signals were detected almost exclusively in mitochondria at day 2 after infection, mt-Keima was also found in lysosomes at day 3. The lysosomal localization of mt-Keima was confirmed by co-transfection with a lysosomal marker Lamp3-GFP. We quantified the fluorescence intensity of mt-Keima signals in mitochondria (excitation 458 nm and emission > 650 nm) and in lysosomes (excitation 561 nm and emission > 650 nm) at day 3 and calculated the ratio of the intensity (FLlyso/FLmito) to quantitatively assess the delivery of mitochondria to lysosomes. WT and Drp1KO MEFs showed similar FLlyso/FLmito ratios, while ParkinKO MEFs showed a significant decrease to ∼60% of that in WT MEFs (Fig7H). Additional loss of Drp1 further decreased FLlyso/FLmito in ParkinDrp1KO MEFs (Fig7H). Thus, we propose that Drp1 and parkin may control mitochondrial degradation at the steady state through partially overlapping pathways.
Discussion
In this study, we show the synergistic role of Drp1 and parkin in mitochondrial homeostasis in mouse heart and brain. In the absence of mitochondrial division mediated by Drp1, mitochondria showed increased connectivity and size and became defective in mitophagy. This mitophagy defect led to accumulation of the mitophagy adaptor protein p62 and ubiquitinated proteins on mitochondria in a parkin-independent manner, increases in oxidative damage, and loss of mitochondrial respiratory competence. Mice lacking Drp1 in cardiomyocytes or PNs exhibited reduced cardiac function or neurodegeneration. Although loss of parkin resulted in no cardiac defects or PN degeneration, the additional loss of Drp1 exaggerated both cardiac and neurodegenerative phenotypes. We therefore speculate that Drp1 and parkin work in different pathways, which may partially overlap, in the turnover of mitochondria. The pathway mediated by Drp1 appears to be dominant in cardiomyocytes and PNs, whereas the parkin pathway may play a major role in degradation of mitochondria at the steady state in MEFs. Since ParkinKO MEFs show decreased delivery of mitochondria to lysosomes but maintain normal mitochondrial respiration and morphology, the parkin-mediated pathway appears to be dispensable for the maintenance of mitochondrial homeostasis in MEFs. Nonetheless, the combination of loss of Drp1 and parkin aggravates defects in cardiac function and neuronal survival as well as mitochondrial structure and function in MEFs, suggesting that the mechanism by which Drp1 and parkin synergistically maintain mitochondrial homeostasis is universal among different cell types.
It has been proposed that Drp1 and parkin act at distinct steps in mitophagy; parkin stimulates degradation of mitochondria by ubiquitination of mitochondrial proteins, while Drp1-mediated mitochondrial division helps autophagosomes engulf mitochondria (Youle & Narendra, 2011). Our data suggest that other E3 ligases mediate mitochondrial ubiquitination in Drp1-dependent mitochondrial degradation although we do not rule out the possibility that parkin also functions in this pathway together with other E3 ligases. Our model also suggests another mechanism that helps the delivery of mitochondria to lysosomes independently of Drp1 in the parkin-dependent pathway. It has been reported that mitochondria still make small fragments in Drp1KO MEFs, suggesting D-independent mitochondrial division (Wakabayashi et al, 2009). Alternatively, there is another degradation mechanism in which mitochondria are delivered to lysosomes through mitochondria-derived vesicles independently of Drp1 (Soubannier et al, 2012). This Drp1-independent mechanism, whose molecular basis has not been identified yet, may be part of the parkin-dependent mitochondrial degradation process.
Both cardiomyocytes and PNs have high demands for energy production and appear to require efficient mitochondrial turnover mediated by Drp1 to protect them from oxidative damage and maintain their functionality. On the other hand, cultured MEFs, which are less dependent on mitochondrial respiration and continuously grow, show modest mitochondrial defects in the absence of Drp1. It has been shown that oxidative damage transforms mitochondrial morphology from elongated tubules, caused by Drp1 deficiency, to large spheres (Kageyama et al, 2012). The spherical morphology destroys the proper surface area to volume ratio of mitochondria and changes the intra-mitochondrial distribution of matrix proteins and mitochondrial DNA, likely compromising the electron transport chain complexes and respiratory function. We suggest that the additional loss of parkin accelerates oxidative damage due to an additional decrease in mitochondrial turnover in the absence of Drp1.
In addition to degradation, Drp1 and parkin may also play synergistic roles in the biogenesis of mitochondria as the amounts of several components of the electron transport chain were further decreased in ParkinDrp1KO hearts, compared with Drp1KO hearts. The previous finding that acute loss of parkin in adult mice, but not germline loss of parkin (e.g. ParkinKO mice), decreases the level of PGC1-α suggests a compensatory mechanism for the role of parkin in the regulation of PGC1-α (Shin et al, 2011). Drp1 may use a different mechanism to control mitochondrial biogenesis as we found similar levels of PGC1-α in Drp1, ParkinKO, and ParkinDrp1KO hearts.
Materials and Methods
Mice
All animal work was carried out according to guidelines established by the Johns Hopkins University Committee on Animal Care. Drp1flox/flox mice, Parkin−/− mice, and L7-Drp1KO mice have been described previously (Von Coelln et al, 2004; Wakabayashi et al, 2009; Kageyama et al, 2012). Myh6-Cre mice (Agah et al, 1997) were obtained from the Jackson Laboratory. By breeding, we generated Myh6-Drp1KO mice (Myh6-Cre+/−::Drp1flox/flox) and Het mice (Myh6-Cre+/−::Drp1+/flox) and control (Drp1+/flox and Drp1flox/flox).
Immunoblotting
Hearts were flash-frozen in liquid nitrogen, homogenized, and sonicated in cell lysis buffer (Cell Signaling Technology) containing complete mini protease inhibitor (Roche). The lysates were centrifuged at 14,000 × g for 10 min, and the supernatants were collected. Protein concentrations were determined using the bicinchoninic acid assay (Pierce). Proteins were separated by SDS–PAGE and transferred onto Immobilon-FL (Millipore). After blocking in 3% BSA/PBS/Tween-20 for 1 h at room temperature, proteins were incubated with antibodies to Tim23 (611222; BD Biosciences), LC3 (Novus), PARP (Cell Signaling Technology), caspase-3 (Cell Signaling Technology), actin (Santa Cruz Biotechnology), OXPHOS cocktails for NDUFB8, FeS, Core2, subunit I, and subunit α (MitoSciences). Proteins were visualized by fluorophores conjugated with secondary antibodies and analyzed using a PharosFX Plus Molecular Imager (Bio-Rad) and the NIH ImageJ software.
Cardiac function
Intact cardiac function was assessed in conscious animals by echocardiography, as described (Zhang et al, 2012). Data were obtained and analyzed by operators blinded to genotypes. Electrocardiography (EKG) recordings were performed in conscious mice, as described (Sysa-Shah et al, 2012). A heat lamp was used to maintain the pups' body temperature during the recording. Surface probes were inserted subcutaneously, and EKG signal (standard lead II) was obtained for 30–60 s using a PowerLab data acquisition system (ML866) and Animal Bio Amp (ML136; AD Instruments, Colorado Springs, CO, USA). LabChart Pro 7.2 software (AD Instruments) was used for automated EKG tracing analysis. After clearing the tracing for noise, 20 s of lead II tracing was used for analysis. Data recorded at 2-s intervals were averaged by the software, and the data points obtained were averaged further to receive an averaged value for each pup. If there was no identifiable P wave in lead II, lead I was used for calculations. For isoproterenol treatment, (−)-isoproterenol diluted in 0.9% saline was administered via intraperitoneal injection to P7 control and Drp1KO mice and EKG was recorded immediately, as described (Sysa-Shah et al, 2012). Baseline EKG recordings performed prior to the isoproterenol injection were used as control.
Electron microscopy
Hearts were quickly removed from the chest after euthanasia. Retrograde perfusion was performed with Ca2+-free HEPES buffer containing 130 mM NaCl, 5.4 mM KCl, 0.33 mM NaH2PO4, 0.5 mM MgCl2, 22 mM glucose, and 20 mM HEPES (pH 7.4) at 1.0 ml/min for 3 min, followed by 0.15 M sodium cacodylate buffer containing 2% paraformaldehyde and 2.5% glutaraldehyde for 7 min. The heart was then post-fixed in the same fixative for 24 h. Small tissue blocks (1–2 mm3) were then excised from left ventricular lateral mid-walls, stained, dehydrated, and resin-embedded as descried previously (Hayashi et al, 2009). Thin sections (70–90 nm thickness) were observed at 5,000–15,000× magnification in a JEOL 1200 operated at 80 kV. For electron tomography, semi-thin sections (500 nm thickness) were imaged at 15,000× magnification and angular increments of 1° from −60° to +60° using an FEI Titan intermediate-voltage electron microscope operated at 300 kV. Double-tilt series of projected images were reconstructed into tomograms using a transform-based back projection algorithm, TxBR (Lawrence et al, 2006) or the IMOD (Boulder Laboratory for 3-Dimensional Electron Microscopy of Cells, University of Colorado) (Kremer et al, 1996). Tomograms were viewed, objects of interest were manually traced, and the triangular surface meshes were generated to determine, visualize, and quantify the 3-dimensional structure of objects in IMOD followed by the use of Amira (Mercury Computer Systems Inc.).
Immunohistochemistry
Hearts were fixed using retrograde perfusion as described above for electron microscopy, except that PBS containing 4% paraformaldehyde was used as the fixative. The heart was then post-fixed in the same fixative for 2 h at 4°C. Fixed hearts were frozen in the OCT compound (Tissue-Tek). Frozen sections were cut, washed in PBS, and blocked in 10% donkey serum. Sections were incubated with antibodies to p62 (Progen Biotechnik), ubiquitin (DAKO), pyruvate dehydrogenase (Mitosciences), Lamp1 (BD Biosciences), Hsp60 (Cell Signaling Technology), and Parkin (Abcam) followed by the appropriate secondary antibodies. Samples were viewed using a Zeiss LSM510-Meta laser scanning confocal microscope (Kageyama et al, 2012).
For immunofluorescence of cerebellar PNs, mice were fixed by perfusion with ice-cold 4% paraformaldehyde in PBS as described (Kageyama et al, 2012). The brain was dissected, further fixed in 4% paraformaldehyde in PBS for 2 h at 4°C, and frozen in the OCT compound (Tissue-Tek). Frozen sections were cut, washed in PBS, and blocked in 5–10% donkey serum. Sections were incubated with antibodies to Car8, Drp1 (BD Biosciences), ubiquitin (Invitrogen), and Tom20 (Santa Cruz Biotechnology) followed by the appropriate secondary antibodies.
Culture of neonatal cardiomyocytes
Primary cultures of neonatal mouse cardiac myocytes were prepared as described previously (Lader et al, 1998). Hearts were harvested from pups within 24 h after birth. Dissected hearts were immediately placed in a Ca2+- and Mg2+-free Hank's balanced salt solution and washed twice. Hearts were minced thoroughly and subjected to trypsin digestion at 4°C for 17 h. Trypsin digestion was stopped by trypsin inhibitor, and further digestion of connective tissue was conducted by collagenase at 37°C for 40 min. Cell clumps were removed using a cell strainer (70 μm mesh size). Cells were centrifuged at 80 × g for 5 min at room temperature, and cell pellets were resuspended in Ham's F-10 medium with l-glutamine (Corning Cellgro) containing 5% bovine serum (Sigma) and 10% horse serum (Sigma). Cells were seeded onto 35-mm glass coverslip bottom culture dish and incubated at 37°C with 5% CO2. For immunofluorescence, cardiomyocytes were fixed using PBS containing 4% paraformaldehyde. Fixed cells were washed in PBS, permeabilized with 0.1% Triton X-100/PBS, and blocked in 0.5% BSA/PBS. The cells were incubated with antibodies to PDH (Abcam), Tom20 (Santa Cruz Biotechnology), and HNEJ-2 (Abcam), followed by the appropriate secondary antibodies.
Mitochondrial OCR in isolated cardiomyocytes was analyzed in an XF24 Extracellular Flux Analyzer (Seahorse Bioscience), as described previously (Cooper et al, 2012). Approximately 40,000 cardiomyocytes were plated per well in XF24 cell culture microplates pre-coated with laminin (Sigma). The cultures were incubated for 4 days in Ham's F-10 medium with l-glutamine containing 5% bovine serum and 10% horse serum. For OCR recording, the culture medium was replaced with Seahorse XF medium supplemented with 20 mM glucose (Sigma), 2 mM sodium pyruvate (Sigma), and 4 mM l-glutamine (Gibco). OCR was measured at 37°C with a 2-min mix, 2-min wait, and 2-min measurement protocol. 1 μg/ml oligomycin was sequentially injected into each well to access basal respiration. OCRs were normalized relative to the amount of protein in each well, and the data are presented as % change relative to control.
Histochemical measurements of electron transport chain complex activity
The activity of NADH dehydrogenase, succinate dehydrogenase, and cytochrome c oxidase was measured as described previously (De Paepe et al, 2009; Kageyama et al, 2012). Hearts were dissected out and immersed in OCT compound. Samples were quick-frozen in isopentane pre-cooled with liquid nitrogen for 10 s. Then they were wrapped in aluminum foil and placed into liquid nitrogen for at least 30 min. Frozen hearts were sectioned (7 μm thickness) and mounted onto glass slides. To measure NADH dehydrogenase activity, sections were incubated with 0.625 mg/ml NADH and 1.25 mg/ml nitro blue tetrazolium in phosphate/saline buffer at room temperature for 30 min. For succinate dehydrogenase activity, sections were incubated with 13.5 μg/ml sodium succinate and 250 μg/ml nitro blue tetrazolium in phosphate/K-EGTA buffer at 37°C for 2 h. For cytochrome c oxidase, sections were incubated with 0.5 mg/ml diaminobenzidine tetrahydrochloride, 2 μg/ml catalase, and 1 mg/ml cytochrome c in phosphate buffer adjusted at pH 7.4 at 37°C for 2 h. The enzymatic reaction was terminated by rinsing with distilled water. Samples for NADH and succinate dehydrogenase were mounted with aqueous medium. Samples for cytochrome c oxidase were dehydrated, mounted with non-aqueous mounting medium. Samples were viewed using an Olympus BX51 microscope equipped with a DP-70 color camera.
Mouse embryonic fibroblasts
Mouse embryonic fibroblasts were isolated from WT, Drp1−/−, Drp1flox/flox, and Drp1flox/floxParkin−/− embryos and immortalized spontaneously by serial passage (over 30 times) as described (Wakabayashi et al, 2009). MEFs were cultured in IMDM supplemented with 10% FBS (Wakabayashi et al, 2009).
Mitochondrial OCR in WT, Drp1KO, ParkinKO, and ParkinDrp1KO MEFs was analyzed in a XF96 Extracellular Flux Analyzer (Seahorse Bioscience) as described previously (Nadanaciva et al, 2012). Approximately 10,000 MEFs were plated per well in XF 96-well cell culture microplate for 17–22 h. For OCR recording, the culture medium was replaced with Seahorse XF medium supplemented with 25 mM glucose (Sigma) and 1 mM sodium pyruvate (Sigma). Multiple wells without cells were set as background controls. OCR analysis was initiated with a 20-min equilibration followed by three time cycles of 2-min mix, 2-min wait, and 4-min measurement. All measurement cycles were performed at 37°C. Baseline OCR was recorded three times, and then, 1 μg/ml oligomycin, 1 μM FCCP, and 0.1 μM rotenone plus 2 μM antimycin A were sequentially injected into each well. The mix–wait–measurement cycles were repeated three times after each drug injection. OCRs were normalized relative to the amount of protein in each well, and the data are presented as % change relative to control.
Lentiviruses
Lentiviruses were generated as described previously (Kim et al, 2007; Kageyama et al, 2012). Briefly, pHR-SIN-CSGW expressing mt-Keima (Katayama et al, 2011) was cotransduced into HEK293T cells with two other constructs, pHR-CMV8.2ΔR and pCMV-VSVG, using Lipofectamine 2000 (Invitrogen). Two days after transfection, the supernatant of transduced cells containing released viruses was collected. The viruses were quick-frozen in liquid nitrogen and stored at −80°C.
Acknowledgments
We thank Dan Stevens for technical support, Atsushi Miyawaki for the mt-Keima plasmid, and Todd Waldman for the lentivirus constructs. EM imaging and tomography used resources provided by the National Center for Microscopic and Imaging Research at UCSD. This work was supported by NIH grants GM084015 (MI), GM089853 (HS), NS084154 (HS), HL107153 (DAK), HL59480 (DAK), HL077180 (DAK) and NS38377 (SA, VLD, TMD), an AHA grant 0840013N (MH) and 12GRANT1173006 (MH). The authors (SA, VLD, TMD) acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through their direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation's Parkinson's Disease Program. T.M.D. is the Leonard and Madlyn Abramson Professor in neurodegenerative diseases.
Author contributions
YK, MI, and HS designed the study. YK performed the experiments with contribution from MH (electron microscopy), KS (immunoblotting, histology, and immunofluorescence of hearts), DB and DAK (echocardiography), PS-S and KG (electrocardiography), SAA, VLD, and TMD (oxygen consumption rate measurements in cardiomyocytes), and WC and AH (oxygen consumption rate measurements in MEFs). HS, MI, and YK wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
Supplementary Figures
Supplementary Tables
Review Process File
References
- Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraud N, Pelloux S, Usson Y, Kuznetsov AV, Ronot X, Tourneur Y, Saks V. Mitochondrial dynamics in heart cells: very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J Bioenerg Biomembr. 2009;41:195–214. doi: 10.1007/s10863-009-9214-x. [DOI] [PubMed] [Google Scholar]
- Chang CR, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann N Y Acad Sci. 2010;1201:34–39. doi: 10.1111/j.1749-6632.2010.05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, Hargus G, Deleidi M, Lawson T, Bogetofte H, Perez-Torres E, Clark L, Moskowitz C, Mazzulli J, Chen L, Volpicelli-Daley L, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson's disease. Sci Transl Med. 2012;4:141ra190. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paepe B, De Bleecker JL, Van Coster R. Histochemical methods for the diagnosis of mitochondrial diseases. Curr Protoc Hum Genet. 2009;63: 19.2:19.2.1–19.2.19. doi: 10.1002/0471142905.hg1902s63. [DOI] [PubMed] [Google Scholar]
- Gaussin V, Van de Putte T, Mishina Y, Hanks MC, Zwijsen A, Huylebroeck D, Behringer RR, Schneider MD. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc Natl Acad Sci USA. 2002;99:2878–2883. doi: 10.1073/pnas.042390499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Martone ME, Yu Z, Thor A, Doi M, Holst MJ, Ellisman MH, Hoshijima M. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci. 2009;122:1005–1013. doi: 10.1242/jcs.028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Sun L, Ji S, Zhao T, Zhang W, Xu J, Zhang J, Wang Y, Wang X, Franzini-Armstrong C, Zheng M, Cheng H. Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc Natl Acad Sci USA. 2013;110:2846–2851. doi: 10.1073/pnas.1300741110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013;23:64–71. doi: 10.1016/j.tcb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH, Iijima M, Sesaki H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol. 2012;197:535–551. doi: 10.1083/jcb.201110034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y, Zhang Z, Sesaki H. Mitochondrial division: molecular machinery and physiological functions. Curr Opin Cell Biol. 2011;23:427–434. doi: 10.1016/j.ceb.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol. 2007;27:662–677. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Lader AS, Xiao YF, Ishikawa Y, Cui Y, Vatner DE, Vatner SF, Homcy CJ, Cantiello HF. Cardiac Gsalpha overexpression enhances L-type calcium channels through an adenylyl cyclase independent pathway. Proc Natl Acad Sci USA. 1998;95:9669–9674. doi: 10.1073/pnas.95.16.9669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence A, Bouwer JC, Perkins G, Ellisman MH. Transform-based backprojection for volume reconstruction of large format electron microscope tilt series. J Struct Biol. 2006;154:144–167. doi: 10.1016/j.jsb.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Nadanaciva S, Rana P, Beeson GC, Chen D, Ferrick DA, Beeson CC, Will Y. Assessment of drug-induced mitochondrial dysfunction via altered cellular respiration and acidification measured in a 96-well platform. J Bioenerg Biomembr. 2012;44:421–437. doi: 10.1007/s10863-012-9446-z. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- Ong SB, Hall AR, Hausenloy DJ. Mitochondrial dynamics in cardiovascular health and disease. Antioxid Redox Signal. 2013;19:400–414. doi: 10.1089/ars.2012.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A, Ventura-Clapier R, Joubert F. Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front Physiol. 2013;4:102. doi: 10.3389/fphys.2013.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki H, Adachi Y, Kageyama Y, Itoh K, Iijima M. In vivo functions of Drp1: lessons learned from yeast genetics and mouse knockouts. Biochim Biophys Acta. 2013;1842:1179–1185. doi: 10.1016/j.bbadis.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- Sysa-Shah P, Xu Y, Guo X, Belmonte F, Kang B, Bedja D, Pin S, Tsuchiya N, Gabrielson K. Cardiac-specific over-expression of epidermal growth factor receptor 2 (ErbB2) induces pro-survival pathways and hypertrophic cardiomyopathy in mice. PLoS One. 2012;7:e42805. doi: 10.1371/journal.pone.0042805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Itoh K, Sesaki H. SnapShot: mitochondrial dynamics. Cell. 2011;145:1158. doi: 10.1016/j.cell.2011.06.018. 1158.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci USA. 2004;101:10744–10749. doi: 10.1073/pnas.0401297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, Zhou J, Chen Q. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem. 2011;286:11649–11658. doi: 10.1074/jbc.M110.144238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial dynamics in model organisms: what yeasts, worms and flies have taught us about fusion and fission of mitochondria. Semin Cell Dev Biol. 2010;21:542–549. doi: 10.1016/j.semcdb.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Takimoto E, Lee DI, Santos CX, Nakamura T, Hsu S, Jiang A, Nagayama T, Bedja D, Yuan Y, Eaton P, Shah AM, Kass DA. Pathological cardiac hypertrophy alters intracellular targeting of phosphodiesterase type 5 from nitric oxide synthase-3 to natriuretic peptide signaling. Circulation. 2012;126:942–951. doi: 10.1161/CIRCULATIONAHA.112.090977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Tables
Review Process File