

Abstract
The small GTPase Arf1 plays critical roles in membrane traffic by initiating the recruitment of coat proteins and by modulating the activity of lipid-modifying enzymes. Here, we report an unexpected but evolutionarily conserved role for Arf1 and the ArfGEF GBF1 at mitochondria. Loss of function of ARF-1 or GBF-1 impaired mitochondrial morphology and activity in Caenorhabditis elegans. Similarly, mitochondrial defects were observed in mammalian and yeast cells. In Saccharomyces cerevisiae, aberrant clusters of the mitofusin Fzo1 accumulated in arf1-11 mutants and were resolved by overexpression of Cdc48, an AAA-ATPase involved in ER and mitochondria-associated degradation processes. Yeast Arf1 co-fractionated with ER and mitochondrial membranes and interacted genetically with the contact site component Gem1. Furthermore, similar mitochondrial abnormalities resulted from knockdown of either GBF-1 or contact site components in worms, suggesting that the role of Arf1 in mitochondrial functioning is linked to ER–mitochondrial contacts. Thus, Arf1 is involved in mitochondrial homeostasis and dynamics, independent of its role in vesicular traffic.
Keywords: Cdc48, ER–mitochondrial contact sites, mitochondria-associated degradation, mitophagy, stress response
See also: C Rabouille (November 2014)
Introduction
Intracellular trafficking is essential for the correct distribution of membrane-bound components throughout the cell. Generation of transport carriers along the secretory pathway is regulated by small GTPases of the Arf/Sar family. Small GTPases cycle between an inactive, cytosolic GDP-bound and an active, membrane-associated GTP-bound state. The exchange of GDP for GTP on Arf proteins is catalyzed by the SEC7 domain of guanine nucleotide exchange factors (GEF) (Jackson & Casanova, 2000; Donaldson & Jackson, 2011). Like Arfs, ArfGEFs exist in a dynamic equilibrium between membrane-bound and cytosolic pools making them a potent determinant of temporal and spatial Arf activation (Jackson & Casanova, 2000; Donaldson & Jackson, 2011). Mammalian BIG1/2 and GBF1 are the ArfGEFs acting at the Golgi apparatus. They are conserved throughout eukaryotes: the yeast homologues are Sec7 and Gea1/2, respectively, and in C. elegans AGEF-1 and GBF-1 (Sato et al, 2006; Balklava et al, 2007; Ackema et al, 2013).
Recently, the contact sites between organelles of the secretory pathway and mitochondria attracted much attention (Elbaz & Schuldiner, 2011; Rowland & Voeltz, 2012; Klecker et al, 2014). In particular, the ER–mitochondrial contact site was found to play major roles in physiology and disease, including neurodegenerative disorders. These contact sites are essential for the exchange of phospholipids and Ca2+ between the organelles. In yeast, the ERMES (ER–mitochondria encounter structure) complex is localized at these connections and coordinates phospholipid exchange and mitophagy (Kornmann et al, 2009; Bockler & Westermann, 2014; Klecker et al, 2014). Even though there are no obvious ERMES homologues in metazoa (Wideman et al, 2013), ER–mitochondria contact site fractions can be obtained in mammalian tissue (Vance, 1990). Proteins that have been suggested to play a role in the establishment of ER–mitochondrial contacts in mammals include the Ca2+-release channel inositol 1,4,5-trisphosphate receptor (IP3R) on the ER side and the voltage-dependent anion channel VDAC in the mitochondrial outer membrane as well as ER and mitochondria-localized mitofusin, Mfn2 (Rizzuto et al, 2012). The nature of the proteins establishing and maintaining the connections remains elusive.
Like the ER, mitochondria are highly dynamic structures that undergo fusion and fission (Hoppins et al, 2007; Westermann, 2010a). These dynamics reflect a balance between the antagonistic actions of the dynamin-like GTPases mediating fission (Dnm1 in yeast, DRP-1 in worms, DRP1 in mammals) and fusion (Fzo1 in yeast, FZO-1 in worms, mitofusins Mfn1 and Mfn2 in mammals) (Hales & Fuller, 1997; Smirnova et al, 1998; Hoppins et al, 2007; Westermann, 2010b). The finding that yeast cells lacking components of the secretory pathway frequently exhibit aberrant mitochondria points to an important role of the endomembrane system in mitochondrial dynamics (Altmann & Westermann, 2005). However, only little is known about the function of secretory pathway components in dynamics and physiology of mitochondria.
Here, we report that Arf1 and its ArfGEF GBF-1/Gea1/2 are important for mitochondrial dynamics and function. This conserved role for activated Arf1 is independent of its function in COPI vesicle generation. The yeast arf1-11Δarf2 mutation caused clustering/aggregation of the mitofusin Fzo1. Overexpression of the AAA-ATPase Cdc48, but not general activation of the unfolded protein response (UPR) pathway, partially rescued the mitochondrial defects in arf1-11Δarf2 through the reduction of Fzo1 protein levels. The clustering/aggregation of Fzo1 in arf1-11Δarf2 may be a consequence of the inability to efficiently recruit Cdc48 and/or of altered mitochondrial outer membrane homeostasis. Consistent with this notion, deletion of GEM1 in yeast or knockdown of VDAC-1 or MIRO-1 in C. elegans, proteins which can be found on ER–mitochondrial contact sites (Kornmann et al, 2011; Rowland & Voeltz, 2012; Murley et al, 2013), caused a phenotype similar to Arf1 or GBF-1/Gea1/2 depletion. Thus, Arf1 plays a role in Fzo1 and mitochondrial homeostasis.
Results
gbf-1(RNAi) but not agef-1(RNAi) affects mitochondrial morphology
We have previously shown that the ArfGEF GBF-1 is required for ER morphology, secretion, and endocytosis in C. elegans (Ackema et al, 2013). During these studies, we noticed in our electron micrographs that the morphology of mitochondria was strongly affected specifically in gbf-1(RNAi) maturing oocytes (Fig 1A and B). Compared to mock-treated worms, mitochondria of gbf-1(RNAi) oocytes appeared dramatically enlarged, sometimes interconnected by thin membrane tubes (Fig 1B). Intriguingly, this effect was not observed upon knockdown of another ArfGEF of the secretory pathway, AGEF-1, suggesting that the phenotype is independent of general secretion inhibition (Fig 1C).
Figure 1. GBF-1 affects mitochondrial morphology.
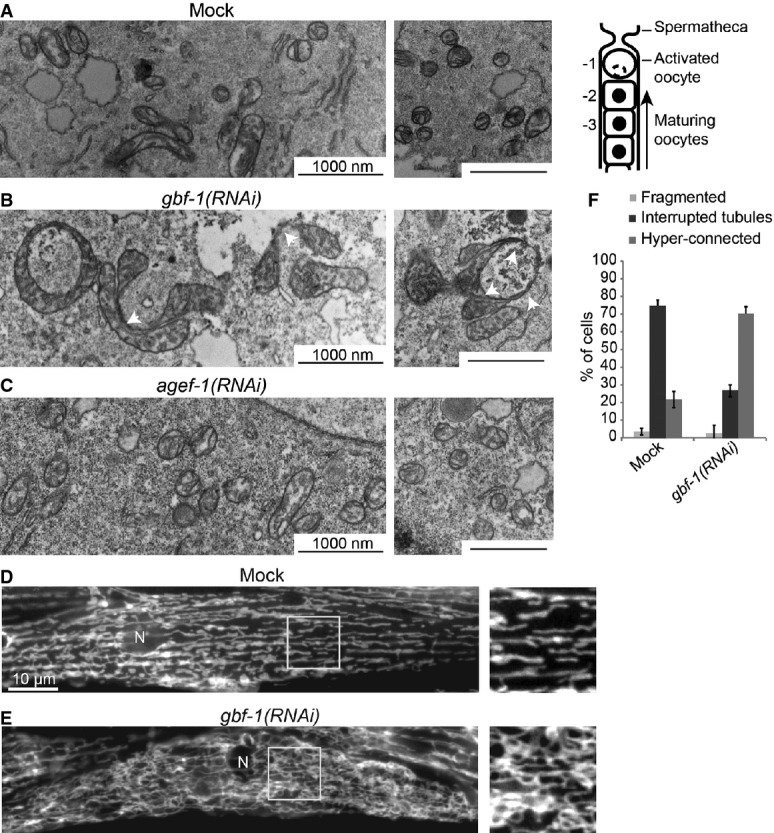
A–C Electron microscopy (EM) of mitochondria in Caenorhabditis elegans oocytes. Wild-type is shown in (A). On the right, a schematic view of the C. elegans proximal gonad is shown. Oocytes indicated with -2 were used for EM analysis. The depletion of GBF-1 (B) leads to the formation of enlarged mitochondria connected through thin membrane connections (indicated with white arrowheads). The mitochondria of agef-1(RNAi) worms (C) appear unaffected.
D, E Live imaging of TOM70::GFP in C. elegans muscle cells. In wild-type muscle cells (D), the mitochondrial network is organized in interrupted slightly branched tubules. gbf-1(RNAi) caused disorganization of the mitochondrial network and an increase of connections between mitochondria (E). N, nucleus.
F Quantification of (D) and (E) of three independent experiments, showing the percentage of muscle cells in a total of N = 392 (Mock) and N = 466 [gbf-1(RNAi)] cells, with fragmented, tubular or hyper-connected mitochondria. Error bars represent standard deviation.
To corroborate these results, we analyzed the mitochondrial morphology in adult C. elegans muscle cells by expressing the GFP-tagged mitochondrial outer membrane protein TOM-70::GFP (Labrousse et al, 1999). Like in most striated muscle, the mitochondrial network is organized in interrupted slightly branched tubules along the contractile apparatus in C. elegans body wall muscle cells (Fig 1D). Knockdown of GBF-1 caused disorganization of the mitochondrial network and an increase of connections between mitochondria in 70% of the cells (Figs 1E, F and 2A). These results indicate that GBF-1 has a role in maintaining mitochondrial morphology in at least two different tissues.
Figure 2. GBF-1 is required for mitochondrial morphology and function.
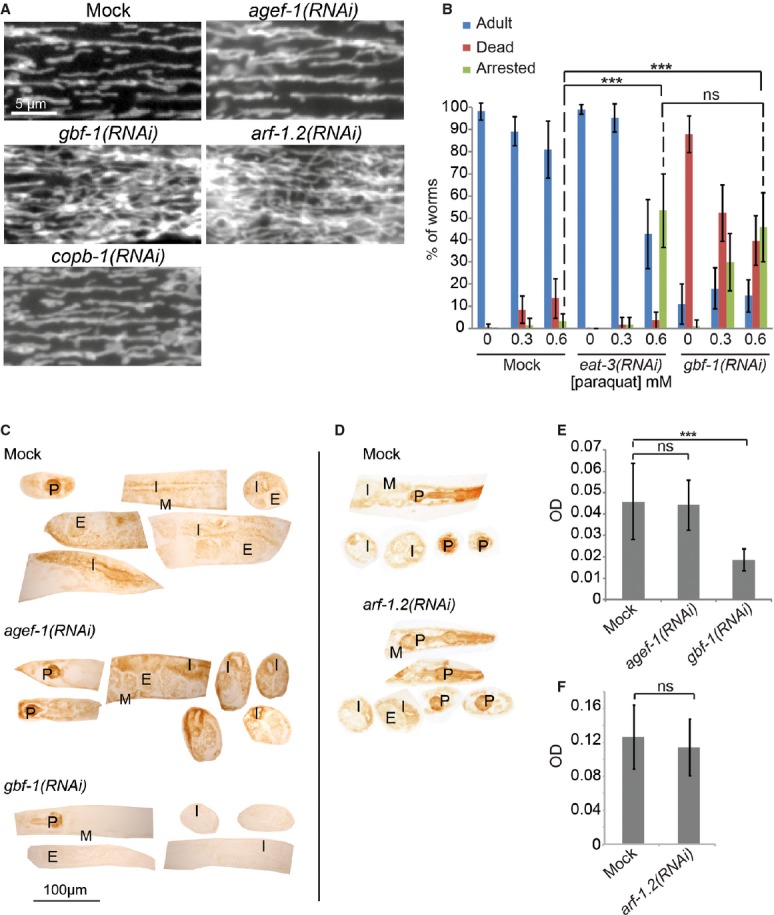
- Live imaging of TOM70::GFP in C. elegans muscle cells. In wild-type, agef-1(RNAi) and copb-1(RNAi) muscle cells, mitochondria show the typical tubular morphology along the length of the contractile apparatus, while mitochondria in gbf-1(RNAi) and arf-1.2(RNAi) muscle cells formed a hyper-connected network.
- gbf-1(RNAi) worms are ROS sensitive. eat-3(RNAi) served as a positive control. The experiment was performed four times in triplicates of 20 worms each. Standard deviation is given. For significance testing, one-way ANOVA was used, followed by a Tukey's test (arrested phenotype at 0.6 mM paraquat: mock versus eat-3(RNAi) P < 1 × 10−6; mock versus gbf-1(RNAi) P < 1 × 10−6; eat-3(RNAi) versus gbf-1(RNAi) P = 0.79).
- The enzymatic activity of cytochrome c oxidase is decreased in somatic tissues and the gonad of gbf-1(RNAi) worms compared to wild-type and agef-1(RNAi). Representative examples are shown.
- The enzymatic activity of cytochrome c oxidase was not decreased in arf-1(RNAi) compared to wild-type.
- Light microscopic quantification of the resulting optical density (OD) in the body wall muscles. Standard deviation is indicated by error bars. Mock: n = 20, agef-1(RNAi): n = 24, gbf-1(RNAi): n = 25. The statistically significant difference as indicated with asterisks between mock and gbf-1(RNAi) (P < 1 × 10−7) was determined by a t-test (two-tailed, unequal variance).
- Analysis as in (E). Mock: n = 13, arf-1(RNAi): n = 12. Mock and arf-1(RNAi) are statistically significantly different (P < 7 × 10−5). P, pharynx; I, intestine; G, gonad; E, embryos; M, muscle.
Mitochondrial activity is strongly reduced in gbf-1(RNAi) animals
gbf-1(RNAi) worms appeared less active on agar plates (not shown) and showed a strong reduction in muscle performance in a body bend assay (Supplementary Fig S1A, Supplementary Movies S1 and S2). This phenotype could be due to reduced mitochondrial performance or defects in the cytoskeleton. The actin organization was not altered in body wall muscle cells in gbf-1(RNAi) animals (Supplementary Fig S1B). To corroborate these results, we tested whether disruption of the cytoskeleton with low doses of latrunculin A (LatA) for actin or benomyl for microtubules causes a mitochondrial phenotype similar to gbf-1(RNAi) animals. Both treatments were efficient as judged by partial paralysis (not shown) and reduced body size (Supplementary Fig S1C). In neither benomyl nor LatA-treated worms did the mitochondria form the characteristic hyper-connected disorganized structure that we had observed for gbf-1(RNAi) (compare Supplementary Fig S1D with Fig 1E). We conclude that the gbf-1(RNAi)-dependent hyper-connected mitochondrial morphology is not caused by disruption of the actin or microtubule cytoskeleton.
Next, we assessed mitochondrial activity by determining the sensitivity to reactive oxygen species (ROS) using paraquat, an enhancer of ROS production (Kondo et al, 2005). Mitochondria with defects in oxidative phosphorylation complexes are more prone to free radical damage, and ROS-susceptible worms arrest in development when exposed to paraquat. EAT-3 is the C. elegans ortholog of yeast Mgm1 and mammalian OPA1 dynamin-related GTPases, crucial for mitochondrial inner membrane fusion. eat-3(RNAi) is known to cause paraquat sensitivity (Kanazawa et al, 2008). We determined the survival rate for adult worms and the number of arrested larvae 5 days after hatching on paraquat containing plates. To avoid “contamination” by progeny from unaffected adults, we used the sterility-conferring glp-4(bn2) ts-allele (Beanan & Strome, 1992). Adult gbf-1(RNAi) worms die due to a weak cuticle, which causes bursting. As a consequence, paraquat-induced developmental arrest increases the survival of gbf-1(RNAi) worms. gbf-1(RNAi) and eat-3(RNAi) worms were equally sensitive to paraquat (Fig 2B) (Kanazawa et al, 2008), supporting a role for GBF-1 in mitochondrial function.
To assess mitochondrial activity more directly, we histochemically examined the enzymatic activity of cytochrome c oxidase (COX), complex IV of the respiratory chain (Hench et al, 2011; Rolland et al, 2013). The intensity of the stain correlates with the enzymatic activity in the tissue (Jung et al, 2002; Hench et al, 2011). COX activity was severely reduced in all tissues in gbf-1(RNAi) worms compared to wild-type or agef-1(RNAi) animals (Fig 2C and E). Taken together, our results suggest that loss of GBF-1 impairs both mitochondrial morphology and function.
Mitochondrial morphology is dependent on ARF-1.2
GBF1 is a GEF for the small GTPase ARF1 (Peyroche et al, 1996; Donaldson & Jackson, 2011). We tested whether the C. elegans ARF1 homologue, ARF-1.2, also influences mitochondrial morphology. A hyper-connected network was observed in arf-1.2(RNAi) reminiscent of the gbf-1(RNAi) phenotype (Fig 2A). The efficiency of the knockdowns was confirmed by qPCR (Supplementary Fig S2). ARF-1.2 function on mitochondrial structure was independent from its role in vesicle formation at the Golgi because knockdown of the coatomer subunit COPB-1, which is essential for ARF-1-dependent retrograde transport within the Golgi and back to the ER, did not affect mitochondrial morphology (Fig 2A), while we observed embryonic lethality (unpublished observation).
The COX activity in arf-1.2(RNAi) tissues was indistinguishable from mock-treated worms (Fig 2D and F). This is in agreement with the overall phenotype of the arf-1.2(RNAi) worms, which were fertile and moved normally on the plate. This mild phenotype is likely due to the existence of another ARF class I/II protein in C. elegans, ARF-3. In mammalian cells, class I and class II Arfs have overlapping functions; for example, class I Arf1 and class II Arf4 and Arf5 can all generate COPI-coated vesicles in vitro (Popoff et al, 2011). Similarly, yeast Arf1 and Arf2 have redundant functions (Stearns et al, 1990). These results indicate that mitochondrial morphological abnormalities and dysfunction can be separated as indicated by the partial redundancy of the Arf family proteins.
GBF-1 and ARF-1 deficiencies do not suppress mitochondrial fission or fusion defects
The phenotypes of loss of function of ARF-1 and its GEF may reflect defects in mitochondrial dynamics. We first tested whether ARF-1 function was involved in the regulation of mitochondrial fission because partial knockdown of DRP-1 by RNAi (Labrousse et al, 1999) caused a phenotype very similar to gbf-1(RNAi) and arf-1.2(RNAi) (Fig 3A and C). However, neither concomitant knockdown of GBF-1 and DRP-1 nor arf-1.2(RNAi) in drp-1(tm1108) loss-of-function animals altered the described phenotype (Labrousse et al, 1999; Breckenridge et al, 2008) (Fig 3). This lack of phenotypic enhancement may be expected as the drp-1 mutants have already a very strong phenotype. Moreover, DRP-1 mitochondrial localization was independent of GBF-1 as overexpressed DRP-1 was still present on mitochondria in gbf-1(RNAi) muscle cells, without being able to rescue the hyper-connected phenotype (unpublished observation). Taken together, our data suggest that DRP-1 acts independent of ARF-1.2 and GBF-1.
Figure 3. Epistasis analysis.

A–C Live imaging of worms expressing TOM70::GFP in the muscle cells. fzo-1(tm1133) produces fragmented mitochondria (B). drp-1(tm1108) causes fusion and formation of blebs in the mitochondrial network (C).
D Quantification of the mitochondrial phenotypes in gbf-1(RNAi) combined with either fzo-1(RNAi) or drp-1(RNAi). Standard deviation is given. Statistically significant differences of RNAis compared to Mock are indicated by colored asterisks. ns: P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001. For significance testing, one-way ANOVA was used, followed by a Tukey's test. The RNAi of GBF-1 did not significantly change the phenotype of drp-1(RNAi) or fzo-1(RNAi) (indicated with the black brackets). Muscle cells from at least three independent experiments were scored. Mock and gbf-1(RNAi) as shown in Fig 1, drp-1(RNAi): n = 427, drp-1(RNAi) + gbf-1(RNAi): n = 381, fzo-1(RNAi): n = 454, fzo-1(RNAi) + gbf-1(RNAi): n = 636, fzo-1(RNAi) + drp-1(RNAi): n = 499.
E arf-1.2(RNAi) did not change the drp-1(tm1108) phenotype. Three experiments were counted. drp-1(tm1108) + mock, n = 472; drp-1(tm1108) + arf-1.2(RNAi), n = 462;. Standard deviation is indicated by error bars.
Next, we tested whether gbf-1 or arf-1.2 would regulate mitochondrial morphology through genetic interaction with the mitofusin fzo-1. Animals with fzo-1 loss-of-function mutations or being treated with fzo-1(RNAi) have fragmented mitochondria due to lack of fusion activity (Breckenridge et al, 2008; Rolland et al, 2009) (Fig 3A and B). This phenotype was unaffected by additional gbf-1(RNAi) or arf-1.2(RNAi) (Fig 3A, B and D). These findings indicate that there is no genetic interaction between fzo-1 and arf-1.2 or gbf-1.
The defects in mitochondrial morphology caused by GBF-1 loss of function are conserved from yeast to man
To test whether the effect of ARF1 on mitochondria is evolutionarily conserved across the animal kingdom, we assessed mitochondrial morphology in HeLa cells. Because mammalian cells have a large repertoire of ARF proteins but only one GBF-1 homologue, we knocked down GBF1, which strongly affected mitochondrial structure (Fig 4A). We conclude that GBF-1-dependent activation of ARF-1 is important for maintaining mitochondrial morphology and function and is conserved up to man.
Figure 4. The role of Arf1 in mitochondrial morphology is conserved and independent of its role in secretion.

A Mitochondrial morphology is affected by siRNA-based knockdown of GBF1 in HeLa cells. Confocal microscopy of fixed cells. Mitochondria are visualized with MitoTracker, and the nucleus is stained with DAPI.
B arf1-11Δarf2 causes fragmentation of mitochondria. Live confocal microscopy of yeast strains ARF1Δarf2, arf1-11Δarf2 expressing mtGFP and shifted to 37°C for 1 h. Maximum projection of an overlay of the DIC and GFP channels.
C gea1-19Δgea2 gives a similar fragmentation phenotype than arf1-11Δarf2.
D–F Analysis of yeast ts-mutants of the (D) GBF-1 homologues GEA1/2, (E) the small GTPase ARF1 and (F) the coatomer subunit SEC27 carrying a mito-GFP marker at 23°C (permissive temperature) and 37°C (restrictive temperature) by epifluorescence microscopy. For each of three independent experiments, we scored around 100 cells per genotype. Standard deviation is given.
G Mutants in COPI and COPII do not affect mitochondrial morphology. sec21-3 (COPI) and sec23-1 (COPII) displayed a wild-type-like mitochondrial morphology at 37°C. gea1-6Δgea2 shows a similar globular phenotype as gea1-19Δgea2 at 37°C.
Next, we asked whether the function of active Arf1 on mitochondria is also conserved in the budding yeast Saccharomyces cerevisiae. GBF-1 and ARF-1.2 each have two functionally redundant homologues in yeast, Gea1 and Gea2, and Arf1 and Arf2, respectively (Stearns et al, 1990; Peyroche et al, 1996; Spang et al, 2001). Deletion of either pair is lethal. The function of Arf and the ArfGEF can be assessed using conditional mutants. Strikingly, the temperature-sensitive (ts) alleles gea1-6Δgea2, gea1-19Δgea2, and arf1-11Δarf2 formed dense globular mitochondrial structures at the restrictive temperature (Fig 4B–E and G). In addition, arf1-11Δarf2 mutants had impaired mitochondrial function as indicated by poor growth on a non-fermentable carbon source (Supplementary Fig S3B and D). This phenotype was not due to impaired actin-dependent mitochondrial inheritance (Supplementary Fig S3A). Like in C. elegans, the yeast ts-alleles of the coatomer subunits SEC21 and SEC27 established that the Arf1-dependent mitochondrial phenotype is independent of COPI transport (Fig 4F and G). Moreover, the arf1 mutant alleles arf1-17Δarf2 and arf1-18Δarf2 did not share the mitochondrial phenotype (Supplementary Fig S3C and D, and unpublished observations). The arf1-11 allele mainly affects ER export and cause UPR activation, while arf1-17 and arf1-18 attenuate transport at the cis- and trans-Golgi, respectively (Yahara et al, 2001; Kilchert et al, 2010). Why arf1-11Δarf2 has such a strong ER phenotype in comparison to arf1-17Δarf2 remains unclear, as a direct Arf1 function at the ER is still elusive. Our data suggest a role for Gea1/2 and Arf1 in mitochondria function also in yeast. Thus, yeast can be employed as model system to further study Arf1 function at mitochondria.
DNM1 shows a weak genetic interaction with ARF1 and GEA1/2
The mitochondrial morphological changes upon loss of Arf1 function were not identical in yeast and C. elegans muscle cells. Therefore, we repeated and extended the genetic interaction analysis of Arf1 and its GEF with the mitochondrial fission and fusion mediators, Dnm1 and Fzo1, respectively. When we deleted the DRP-1 homologue DNM1 in arf1-11Δarf2, we observed a mixed phenotype in which the mitochondria formed interconnected globular structures that looked like beads-on-a-string (Fig 5A and B). Likewise, Dnm1 overexpression caused a slight rescue of the mitochondrial phenotype (compare Figs 4E and 5E, F) in arf1-11Δarf2 cells. As reported previously (Sesaki & Jensen, 1999), overexpression of DNM1 caused fragmentation of mitochondria, which we also observed. Finally, similarly to the results in C. elegans, Dnm1-GFP was still localized to mitochondria when Arf1 function was impaired (Supplementary Fig S4). As a control, we deleted FIS1, which encodes a protein that recruits Dnm1 to mitochondria (Mozdy et al, 2000). In Δfis cells, Dnm1 no longer localized to the mitochondria, but formed large accumulations in the cytoplasm (Supplementary Fig S4). Taken together, we observe a weak genetic interaction between DNM1 and ARF1 or GEA1/2. Yet, Arf1 does not appear to directly regulate Dnm1 function.
Figure 5. ARF1 interacts genetically with DNM1.
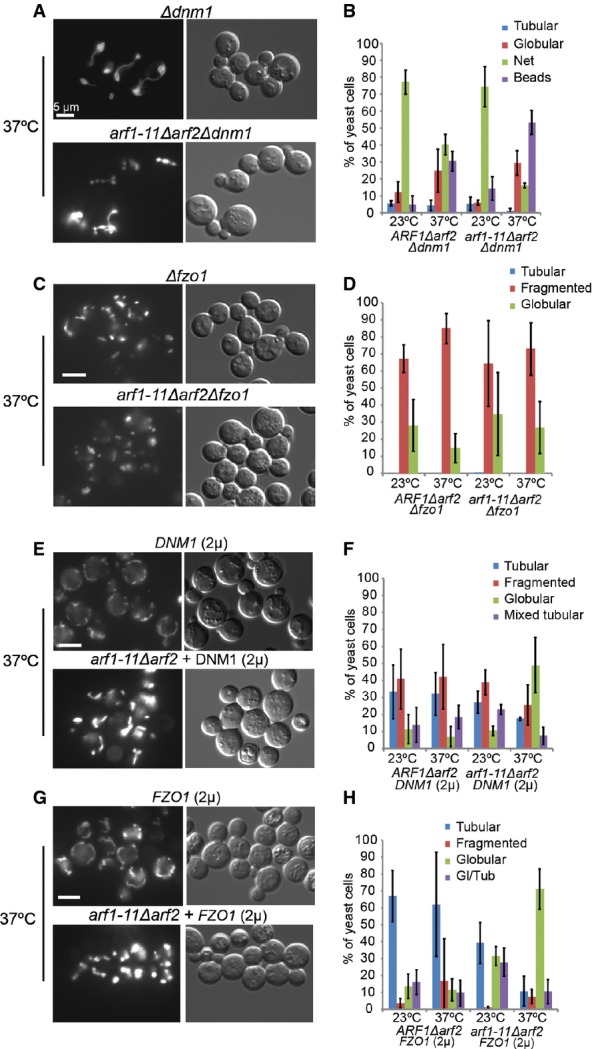
A–H Live imaging of yeast strains expressing mt-GFP at 23 and 37°C. Epistasis experiments of arf1-11Δarf2 the isogenic wild-type together with: (A, B) Δdnm1, (C, D) Δfzo1, (E, F) overexpression of DNM1 or (G, H) overexpression of FZO1. arf1-11Δarf2 (A) did not enhance the Δdnm1-induced network, but caused a mixed phenotype. arf1-11Δarf2 (C) did not rescue the Δfzo1-induced fragmentation. The globular morphology of arf1-11Δarf2 (E) was slightly dominant over DNM1 (2μ)-induced fragmentation. No effect of FZO1 overexpression on the arf1-11Δarf2 mitochondrial phenotype was observed (G). Quantification of the phenotypes presented in (A), (C), (E), and (G) are shown in (B), (D), (F), and (H), respectively. For each of the three experiments, we scored about 100 cells per genotype. Standard deviation is given.
The arf1-11 and gea1/2 mutant phenotypes were somewhat reminiscent of a fusion defect. Combining Δfzo1 with arf1-11Δarf2 did neither rescue nor enhance the mitochondrial fragmentation (Fig 5C and D). While strong overexpression of FZO1 leads to aggregated mitochondria (Fritz et al, 2003), mild overexpression of FZO1 did not affect wild-type mitochondria morphology but did also not rescue the arf1-11Δarf2 phenotype (Fig 5G and H). Thus, we did not detect a genetic interaction between ARF1 and FZO1.
Arf1 co-fractionates with ER and mitochondria
Arf1 is localized to and has established functions at the Golgi apparatus but, to our knowledge, has never been localized to mitochondria. We explored the possibility that a sub-fraction of Arf1 is localized on mitochondria and analyzed highly enriched yeast mitochondria fractions (Meisinger et al, 2000, 2006) (Supplementary Fig S5A). As observed previously, although most of the ER was eliminated from the preparations, the fraction with the purified mitochondria still contained ER due to ER–mitochondria contact sites, which cannot be resolved (Supplementary Fig S5B). Most importantly, Arf1 was present in both ER and mitochondrial fractions, with higher enrichment in the mitochondrial fraction (Supplementary Fig S5B). Thus, a proportion of Arf1 is localized on mitochondria and/or ER–mitochondria contacts, supporting a distinct role in mitochondrial dynamics and function.
Arf1 promotes mitophagy
Recent data suggest that autophagy is initiated at ER–mitochondria contact sites (Hailey et al, 2010; Hamasaki et al, 2013). In addition, the ERMES complex has been shown to be important for turnover of mitochondria (mitophagy) through its involvement in isolation membrane formation (Bockler & Westermann, 2014). Furthermore, Arf1 and Gea1/2 were reported to play a role in the expansion of autophagosomal membranes during macrophagy (van der Vaart et al, 2010). Therefore, we asked whether Arf1 is required for mitophagy. The reporter protein mt-Rosella consists of a mitochondrial targeting sequence, a pH-sensitive GFP and RFP (Rosado et al, 2008). In mitochondria, the GFP and RFP signals overlap. Upon mitophagy, the mitochondrial GFP signal disappears due to the pH drop upon arrival of mitochondrial fragments in the vacuole and only the red fluorescence persists. Using this assay, we tested two different alleles of ARF1: one with (arf1-11Δarf2) and one without (arf1-18Δarf2) aberrant mitochondria (Supplementary Fig S3C and D). Both arf1 mutants were strongly impaired in mitophagy (Fig 6A and B). The difference between the two arf1 alleles is that arf1-11 is defective at the ER, whereas arf1-18 impairs Arf1 function at the trans-Golgi (Yahara et al, 2001). Although it is conceivable that the different alleles block distinct stages during mitophagy, Arf1 might also be essential for autophagy in general. Indeed, both arf1 alleles were defective in autophagy as monitored by GFP-Atg8 arrival in the vacuole (Klionsky et al, 2012) (Fig 6C). Taken together, our data suggest that Arf1 impacts mitochondria by two distinct functions: It affects turnover of mitochondria through its role in autophagy and has an additional, independent role in mitochondrial dynamics. As arf1-18Δarf2 mutants display normal mitochondrial morphology but are defective in mitophagy, the role of Arf1 in mitochondrial dynamics appears to be independent of its role in mitophagy.
Figure 6. Arf1 is involved in mitophagy and autophagy.
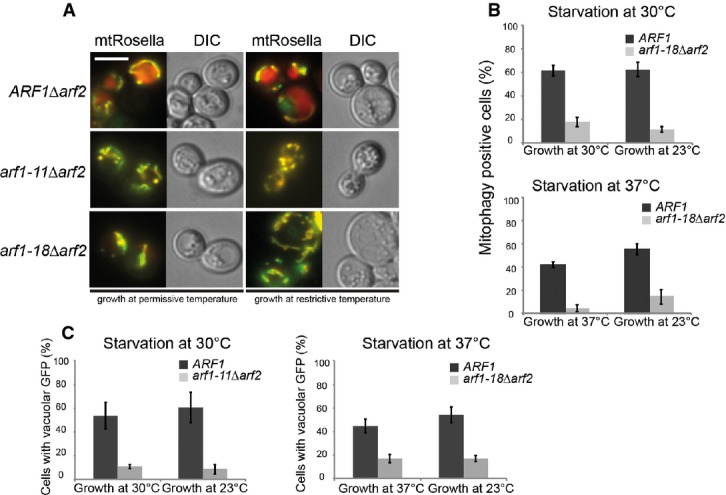
- arf1 mutants are defective in mitophagy. Strains expressing mt-Rosella were grown at permissive temperature overnight, diluted, grown to logarithmic phase at the indicated temperatures, then shifted to starvation in SD-N for 14 h at indicated temperatures and imaged thereafter. Scale bar, 5 μm. Half of the culture was shifted to restrictive temperature for 1 h and imaged thereafter.
- Quantification of data in (A) from three independent experiments in which at least 100 cells were scored. Standard deviation is given.
- arf1 mutants are defective in autophagy. Strains expressing GFP-Atg8 from its endogenous promoter were cultured as in (A) and starved in SD-N for 5 h. The arrival of GFP-Atg8 in the vacuole as a readout for autophagy was scored in arf1 mutant cells. Data from three independent experiments in which at least 100 cells were scored are shown. Standard deviation is given.
CDC48 overexpression partially restores mitochondrial morphology in arf1-11Δarf2
To better understand how Arf1 acts on mitochondria, we aimed to identify Arf1 interacting proteins from enriched mitochondrial membranes using differential affinity chromatography with recombinant Arf1. The basis of the approach is that the active and inactive forms of Arf1 have distinct conformations, which enables us to isolate proteins that specifically bind to either conformation. We have used this approach successfully in the past (Trautwein et al, 2004, 2006). The analysis revealed that the AAA-ATPase Cdc48 was strongly enriched on the Arf1-GTP column (Supplementary Table S1). Cdc48 has been described to be part of the unfolded protein response, the ER- and the mitochondria-associated degradation pathways (Rabinovich et al, 2002; Caruso et al, 2008; Nakatsukasa et al, 2008; Heo et al, 2010). We tested whether overexpression of CDC48 would rescue the arf1-11Δarf2 induced mitochondrial phenotype. Overexpression of CDC48 caused a slight fragmentation of mitochondria in wild-type cells (Fig 7A), while it partially restored mitochondrial morphology in arf1-11Δarf2 (Fig 7B). The slight mitochondrial fragmentation observed in the wild-type might be explained by reduced Fzo1 levels under these conditions (see below). In spite of the partial morphological restoration, CDC48 overexpression did not rescue mitochondrial function in arf1-11Δarf2 (Supplementary Fig S6A).
Figure 7. Overexpression of CDC48 alleviates the mitochondrial defect in arf1-11Δarf2 cells and causes Fzo1 degradation.

- Overexpression of CDC48 causes slight mitochondrial fragmentation in ARF1Δarf2 cells. Quantification of the phenotype from cells expressing mt-GFP from at least three independent experiments, scoring about 100 cells/experiment.
- Mild overexpression of CDC48 in arf1-11Δarf rescues the tubular phenotype at the restrictive temperature. For arf1-11Δarf2, four experiments with 1,670 cells were counted; and for arf1-11Δarf2 CDC48(OE), five experiments (1,520 cells). The statistically significant difference (P < 0.006) was determined by a t-test (two-tailed, unequal variance).
- Fzo1 levels are reduced upon CDC48 overexpression. Fzo1-FLAG was detected by immunoblotting in lysates from Arf1 wild-type and mutant cells with or without CDC48 overexpression. Pgk1 serves as a loading control.
- Fzo1-GFP forms bright foci in arf1-11Δarf2 at the restrictive temperature that are resolved by overexpression of CDC48. Mutant and wild-type cells chromosomally expressing Fzo1-GFP and overexpressing Cdc48 were analyzed by epifluorescence microscopy at the restrictive temperature.
- The Cdc48-dependent mitochondrial rescue is independent of UPR. Expressing the constitutively active Hac1 in arf1-11Δarf2 did not rescue the mitochondrial phenotype.
Source data are available online for this figure.
Next, we asked whether cdc48 mutants would display a mitochondrial defect themselves. However, the mutants cdc48-3 and cdc48-6 did not show any strong alteration in mitochondrial morphology when shifted to the restrictive temperature (Supplementary Fig S6B and C). Thus, under normal growth conditions, the level and function of Cdc48 is dispensable for mitochondrial morphology.
Usually, Cdc48 interacts with a co-factor, which provides specificity for a particular Cdc48 function. Vms1 was proposed to constitute such a co-factor in mitochondria-associated degradation (MAD) under certain stresses (Heo et al, 2010, 2013; Esaki & Ogura, 2012). MAD is supposed to act similar to ERAD in removing misfolded proteins from the organelle for subsequent degradation by the proteasome. We tested whether the rescue of the arf1-11Δarf2 phenotype by CDC48 overexpression would require Vms1. Deleting VMS1 did not interfere with the rescue capability of CDC48 overexpression (Supplementary Fig S6D). However, we noticed that in Δvms1 cells, the CDC48 overexpression-induced slight mitochondrial fragmentation phenotype was rescued, consistent with a role of a Cdc48–Vms1 complex at mitochondria (compare Supplementary Fig S6D and Fig 7A). Yet, Vms1 is unlikely to cooperate with Cdc48 in the rescue of the arf1-11Δarf2 mutant.
Fzo1 is mislocalized in abnormal clusters on mitochondria in arf1-11Δarf2 cells
In mammalian cells, mitofusins are substrates of MAD through ubiquitylation in a Cdc48/p97 and Parkin-dependent manner (Youle & van der Bliek, 2012; Escobar-Henriques & Langer, 2014). Therefore, we decided to determine Fzo1-GFP localization in dependence of Cdc48 expression levels. In wild-type cells, Fzo1-GFP was present throughout the mitochondrial membrane system. Surprisingly, Fzo1-GFP localization was drastically altered in arf1-11Δarf2 cells where Fzo1 clustered into bright foci (Fig 7D). Thus, the mitochondrial defect might be at least in part due to the mislocalization of Fzo1 into aberrant clusters that could lead to altered mitochondrial dynamics and thus function. These clusters were resolved by overexpression of CDC48, suggesting that the rescuing ability of Cdc48 could be connected to degradation of Fzo1 clusters.
Cdc48 removes abnormal Fzo1 clusters through MAD
To test this hypothesis, we appended Fzo1 with a FLAG-tag and monitored its levels in different strains by immunoblot. Unexpectedly, CDC48 overexpression caused Fzo1 degradation even in ARF1 wild-type cells (Fig 7C), suggesting that Cdc48 is rate limiting for Fzo1 degradation already under normal circumstances. In arf1-11Δarf2 cells, Fzo1 concentration was slightly reduced (Fig 7C). This reduction in Fzo1 levels was drastically enhanced when CDC48 was overexpressed. Our data suggest that the arf1 mutant-dependent Fzo1 clusters are degraded through Cdc48-dependent MAD.
Given that Cdc48 functions together with the ubiquitin proteasome system (UPS) in both UPR and MAD, it is conceivable that activation of UPR could also trigger MAD. Yet, UPR is activated in arf1-11Δarf2 mutant cells at the restrictive temperature (Kilchert et al, 2010) and this activation did not prevent the mitochondrial phenotype. A possible explanation could be that since UPR activation occurs upon shift to the non-permissive temperature, the delay to mount the response on the protein level or the amplitude of the UPR may be insufficient to prevent mitochondrial defects. To exclude these possibilities, we transformed a plasmid expressing spliced HAC1 mRNA (Ng et al, 2000) and shifted the cells to the non-permissive temperature. Hac1 is a transcriptional activator of UPR response genes, and is itself converted into an active form by a cytoplasmic splicing event of its mRNA. Thus, expression of spliced HAC1 causes a constitutively active UPR. Under these conditions, we still observed the same mitochondrial phenotype in arf1-11Δarf2 cells, without having any noticeable effect on wild-type cells (Fig 7E). Therefore, the function of Cdc48 at arf1-11Δarf2 mitochondria is independent of UPR activation, and activated UPR is not the cause for the mitochondrial phenotype in arf1-11Δarf2. Taken together, our results suggest that Fzo1 is partially aggregated or non-functional in arf1-11Δarf2 cells and is a substrate of Cdc48 and MAD.
Tethering of ER and mitochondria appears unaltered in arf1-11Δarf2 cells
An explanation for the clustering of Fzo1 could be a change in the lipid composition in arf1-11Δarf2. Mitochondrial lipids are imported from the ER and some are further modified in the mitochondrial inner membrane. The transfer of lipids is thought to occur at ER–mitochondrial contact sites. Thus, we wondered whether changes in mitochondria morphology could be caused by loss of ER–mitochondrial contact sites. To investigate this possibility, we employed the yeast artificial ER–mitochondrial tether chiMERA, which has been shown to be able to restore mitochondrial morphology when contact sites are compromized (Kornmann et al, 2009). This artificial tether consists of one transmembrane domain anchored in the ER and one in the outer membrane of mitochondria, joined by a GFP (Kornmann et al, 2009). As previously described (Yahara et al, 2001), the ER morphology in arf1-11Δarf2 is strongly affected, which is most likely due to the accumulation of cargo in the ER because of the transport block in the ER–Golgi shuttle (Fig 8A) (Nakano & Muramatsu, 1989; Yahara et al, 2001; Spang, 2009). Given the elaborate ER, most of the mitochondrial fragments were in close proximity to ER membranes in arf1-11Δarf2 (Fig 8C). Expression of chiMERA in arf1-11Δarf2 cells did not significantly restore mitochondrial morphology and did not increase the percentage of tubular mitochondria (Supplementary Fig S7A). To analyze the presence of the contact sites more directly, we determined the localization of the ERMES component Mdm34 (Kornmann et al, 2009). Again, we did not observe any difference in distribution or number of spots per cell in the mutant compared to wild-type (Supplementary Fig S7B). Thus, it appears that tethers between ER and mitochondria are present in arf1-11Δarf2 cells. Nevertheless, the possibility remains that these contact sites may have lost part of their function.
Figure 8. Analysis of ER and ER–mitochondria contact sites.

A–C The ER is affected in arf1 mutant cells. Confocal images of live yeast cells tagged with a Pho88-mCherry ER marker (A–C) and mtGFP (B, C).
D ARF1 and GEM1 interact genetically. The arf1-11Δarf2 or gea1-19Δgea2 globular phenotype is strongly enhanced by the deletion of GEM1 at 37°C.
E Quantification of the arf1-11Δarf2Δgem1 and ARF1Δarf2Δgem1 phenotypes at 37°C. For each of the three independent experiments, we scored around 100 cells per genotype. Standard deviation is given.
F, G Single confocal planes of MitoTracker and the ER marker 3ER-YFP in the muscle cells of live worms. MitoTracker is shown in red, ER–YFP in green. Two confocal planes are displayed for each strain. The first plane (indicated with “1”) corresponds to a layer flanking the sarcomeres at which the ER is organized in stripes; the second plane (indicated with “2”) is from the level of the nucleus at which the ER is organized in the characteristic reticulate structure. The positions of the confocal planes are indicated in the schematic drawing of a muscle cell on the right in (F).
H Knockdown of MIRO-1, VDAC-1, and ARF-1.2 results in a similar phenotype.
I Co-feeding with RNAi against GBF-1 and MIRO-1 or VDAC-1 did not enhance the fusion phenotype. Standard deviation is given. Mock and gbf-1(RNAi) as shown in Fig 1, miro-1(RNAi): n = 479, miro-1(RNAi) + gbf-1(RNAi): n = 398. VDAC-1(RNAi) induces a hyper-connected phenotype in 50% of the muscle cells. VDAC-1(RNAi): n = 429. Standard deviation is indicated.
To explore the involvement of contact sites further, we deleted the conserved Rho-like GTPase Gem1 (Miro1 in mammals; MIRO-1 in C. elegans). Gem1 is part of the ERMES complex and negatively regulates connections between ER and mitochondria in yeast (Kornmann et al, 2011; Murley et al, 2013). Intriguingly, Δgem1 showed a phenotype very reminiscent of the one observed in arf1-11Δarf2 and gea1-19Δgea2 cells at the restrictive temperature (compare Figs 4 and 8D, (Frederick et al, 2004, 2008). Strikingly, combining Δgem1 with the arf1-11Δarf2 or gea1-19Δgea2 mutation enhanced the phenotype dramatically, causing the residual tubules in Δgem1 to completely collapse into globular structures (Fig 8D and E). These data suggest that Arf1 and Gea1/2 may have a function at ER–mitochondrial contact sites and could be involved in maintaining the functionality of these sites.
ER and mitochondria remain in close proximity in arf-1.2 (RNAi) and gbf-1(RNAi) animals
We went back to C. elegans to confirm some of our findings from the yeast system. C. elegans has two isoforms of Cdc48, CDC-48.1 and CDC-48.2. Consistent with the results obtained in yeast, simultaneous knockdown of CDC-48.1 and CDC-48.2 did not affect mitochondria in body wall muscle cells. Overexpression of CDC-48.1 or CDC-48.2 resulted in strong phenotypes precluding an analysis in arf-1.2(RNAi) and gbf-1(RNAi) worms.
Next, we examined ER–mitochondrial connections in C. elegans muscle cells. The ERMES complex is not conserved in metazoa and therefore we concentrated on the localization of the ER with respect to mitochondria. We noticed that the ER organization in C. elegans muscle cells displays two major patterns: Right underneath the sarcomeres, the ER is organized in stripes (slice 1), while the ER proximal to the nucleus is more reticulate (slice 2) (Fig 8F). In contrast, mitochondrial organization seems to be location independent. We decided to assess ER and mitochondrial organizations at both locations. In wild-type muscle cells, both organelles were in close proximity, with mitochondrial tubules mainly positioned at the gaps of the ER network and vice versa (Fig 8F). As described above, in arf-1.2(RNAi) muscle cells, mitochondria became hyper-connected and the stripe-like organization was mostly lost (Fig 8G), whereas the ER morphology remained unaffected. The effect on the ER of arf1.2(RNAi) or gbf-1(RNAi) appears to be tissue dependent, because we have found previously that knockdown of either protein affected ER morphology in oocytes (Ackema et al, 2013). Despite the change in mitochondrial morphology, ER and mitochondria were still in close contact, and thus, physical contact between the organelles may persist.
Knockdown of MIRO and VDAC-1 leads to hyper-connected mitochondria in C. elegans
Even though the ERMES core components are not conserved in metazoans, the mammalian homologue of Gem1, Miro1, was found to localize to ER–mitochondrial contact sites in COS-7 cells (Kornmann et al, 2011). Therefore, we decided to knockdown MIRO-1 in C. elegans. miro-1(RNAi) resulted in a strong hyper-connected mitochondrial phenotype, which resembled the gbf-1(RNAi) phenotype (Fig 8H and I). A double knockdown of MIRO-1 and GBF-1 did not enhance the phenotype further (Fig 8I). We consider it possible that the maximal penetrance of the phenotype already occurred with the single knockdowns and that the difference in phenotypes in yeast and C. elegans is likely a reflection of sensitivity and penetrance of the individual phenotypes. In addition, at least in Drosophila and mammalian cells, Miro1 connects mitochondria to Kif5 motor proteins (Guo et al, 2005; Glater et al, 2006; Macaskill et al, 2009), indicating that MIRO-1 has also additional functions in C. elegans. On the other hand, the miro-1(RNAi) phenotype in the body wall muscle cells resembled the gbf-1 and arf-1.2 knockdown and not the one after benomyl treatment. We sought another way to interfere with ER–mitochondrial connections. Unlike in yeast, ER–mitochondrial contact sites are also sites of Ca2+ exchange in metazoa. The voltage-dependent anion channel VDAC-1 is located in the mitochondrial outer membrane and has been shown to be physically linked to the ER Ca2+-release channel inositol 1,4,5-trisphosphate receptor (IP3R) through Grp75 (Szabadkai et al, 2006). vdac-1(RNAi) caused hyper-connected mitochondria to a similar extent as the knockdowns of miro-1, arf-1.2, and gbf-1 (Fig 8H and I). We conclude that our data are consistent with a conserved role of Arf1 and its GEF at ER–mitochondria contact sites.
Discussion
In this study, we have revealed previously unanticipated and evolutionarily conserved roles of the small GTPase Arf1 and its GEF GBF-1/Gea1/2 in mitochondrial morphology and function. These tasks are independent of the previously established responsibilities in vesicular transport and Golgi structure, pointing to a novel function of ARF-1.2/Arf1 and its activator GBF-1/Gea1/2.
Activated Arf1 may play multiple roles at mitochondria (Fig 9). First, Arf1 plays are role in mitochondrial dynamics, influencing the connectivity of the network. Second, Arf1 is required for mitophagy, presumably through its role in macroautophagy (van der Vaart et al, 2010; Orlichenko et al, 2012). Isolation membrane formation during autophagy was proposed to occur at ER–mitochondrial contact sites (Hailey et al, 2010; Hamasaki et al, 2013). However, this function is independent of the one in mitochondrial dynamics.
Figure 9. Model for Arf1 function at ER–mitochondrial contact sites.
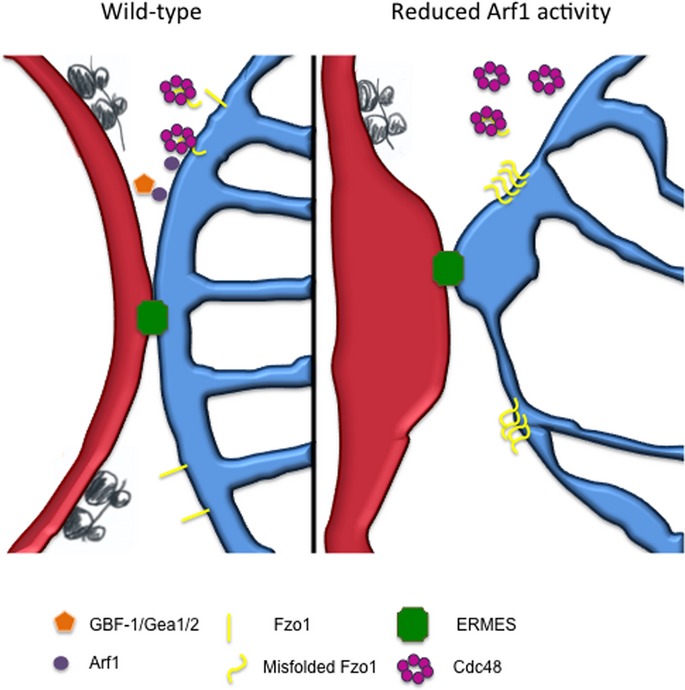
Yeast components are pictured. For explanations, see text.
In addition, the arf1-11Δarf2 mutation results in aggregated Fzo1, which is removed by overexpression of Cdc48. We identified Cdc48 as an interactor of Arf1-GTP. Thus, a third function could be the recruitment of the AAA-ATPase Cdc48 to mitochondria to remove Fzo1 and potentially other mitochondrial outer membrane proteins. In the wild-type situation, Arf1 would promote recruitment of Cdc48 to mitochondria to maintain proper Fzo1 homeostasis, whereas in the mutant situation aberrant Fzo1 would accumulate because Cdc48 is recruited less efficiently. Cdc48 has a known role in the quality control of mitochondrial outer membrane proteins in yeast and worms and promotes degradation of Fzo1 (Heo et al, 2010). Consistently, the mammalian Cdc48 homologue p97 promotes ubiquitylation and proteasome-dependent degradation of the mitofusins Mfn1 and Mfn2 (Tanaka et al, 2010). Our data are in agreement with the recently reported role of Cdc48/p97/VCP in the retro-translocation of mitochondrial outer membrane proteins like Mcl1 and Mfn1 (Xu et al, 2011; Kim et al, 2013).
In an alternative, mutually non-exclusive scenario, Arf1 could have a function at ER–mitochondrial contact sites without directly contacting Cdc48. Our data suggest that contact sites are still present when Arf1 function is compromised. We envision that lipid transfer could be impaired at the contact sites resulting in altered lipid composition of mitochondria, which would be consistent with the abnormal morphology of both the mitochondrial outer and inner membranes as revealed by EM in C. elegans and the loss of the functionality of mitochondria in both yeast and worms. Moreover, at least in C. elegans, knockdown of proteins with a reported function at ER–mitochondria contact sites display a very similar phenotype. In mammalian cells, VDAC1 is physically linked to the ER Ca2+-release channel inositol 1,4,5-trisphosphate receptor (IP3R) (Szabadkai et al, 2006). Knockdown of VDAC-1 in C. elegans caused a similar phenotype as gbf-1(RNAi) and arf-1.2(RNAi). Similarly, reducing the Ca2+-dependent GTPase MIRO-1 also caused hyper-connection of mitochondria. The yeast homologue, Gem1, is part of ER–mitochondria contact sites without being a structural component (Kornmann et al, 2011; Stroud et al, 2011; Nguyen et al, 2012; Murley et al, 2013). Impaired lipid transfer could cause a change in the lipid composition of mitochondrial membranes and thereby promote aggregation of Fzo1.
It is also conceivable that the role of Arf1 in mitochondrial morphology and function is indirect through controlling ER morphology (Yahara et al, 2001; Poteryaev et al, 2005) and function. We do not favor this possibility because we did not observe any defects in ER morphology in C. elegans muscle cells after arf-1.2 or gbf-1 knockdown, while mitochondria were severely affected under the same conditions. Unlike in C. elegans oocytes (Poteryaev et al, 2005; Ackema et al, 2013), the ER did not show any gross abnormalities in muscle cells upon arf-1.2 (RNAi) or gbf-1(RNAi), yet mitochondrial function was disturbed in both tissues. Consistently, knockdown of the mammalian GBF1 left the ER structure intact (Citterio et al, 2008). In yeast, however, ER morphology was clearly perturbed in arf1-11Δarf2 mutant cells, most likely due to a transport block in the ER–Golgi shuttle or a defect in lipid homeostasis. Thus, there is no direct correlation between ER and mitochondrial morphology defects. A reason for the different ER phenotypes in various tissues upon loss of Arf1 function could be the difference in the amounts of cargo that has to be transported through the secretory pathway in these tissues. High cargo load would cause ER defects, while low cargo load would not grossly affect ER structure. Moreover, in tissues with high secretory capacity, the lipid demand would be higher and hence the mitochondria-derived phosphatidylethanolamine, which is an important substrate for synthesis of phosphatidylcholine in the ER (Osman et al, 2011; Tatsuta et al, 2014), may become limiting and cause structural changes in the ER membrane.
The yeast and C. elegans mitochondrial phenotypes are not identical when Arf1 function is impaired. In C. elegans body wall muscle cells, we observed a hyper-connected mitochondrial network, while in yeast, the mitochondria were compact and globular. While we cannot exclude the possibility that Arf1 plays different roles in C. elegans muscle cells and yeast, we note that removing the function of MIRO-1/Gem1 gave us phenotypes very similar to those seen upon loss of Arf1 or GBF-1/Gea1/2. In all cases, the mitochondria became hyper-connected in worms and globular in yeast. It is conceivable that the fusion and fission dynamics and the rate of mitophagy are very different between muscle and yeast cells and likely correlate with the amount of ATP that needs to be produced by the different cell types. Therefore, we propose that the role of Arf1 on mitochondria is conserved from yeast to C. elegans and man.
Given that ubiquitin-dependent degradation or regulation of mitochondrial proteins is vital for the maintenance of mitochondrial function as well as mitochondrial membrane remodeling and communication with other organelles (Yonashiro et al, 2006; Karbowski et al, 2007; Neutzner et al, 2008; Escobar-Henriques & Langer, 2014), it is tempting to speculate that activation and function of Arf1 are essential for the maintenance of mitochondrial homeostasis. This phenomenon might have been overlooked because secretion is an equally essential cellular process, and Arf1's role as a master regulator of vesicle formation has been established over 20 years ago (Stearns et al, 1990; Balch et al, 1992; Traub et al, 1993).
Materials and Methods
C. elegans strains and plasmids
Caenorhabditis elegans was cultured and maintained as described previously (Brenner, 1974) at 20°C. Bristol N2 was used as wild-type control. For the paraquat assay SS104, glp-4(bn2)I was used (Beanan & Strome, 1992). drp-1(tm11.8),fzo-1(tm1133) (Breckenridge et al, 2008; Rolland et al, 2009), and MD2913 (bcEx847) [Pmyo-3::ER::YPF, rol-6(su1006)] were obtained from B. Conradt. The pmyo-3::TOM70::GFP plasmid (Labrousse et al, 1999) was co-injected with pRF4 [rol-6(su1006)] (Mello et al, 1991).
RNAi experiments
RNAi was performed as described (Kamath & Ahringer, 2003). For RNAi feeding, plasmid L4440, containing agef-1 (Y6B3A.1), gbf-1 (C24H11.7), arf1.2 (B0336.2), sar-1 (ZK180.4), copbp-1 (Y25C1A.5), drp-1 (T12E12.4), or eat-3 (D2013.5) sequences, was retrieved from the Ahringer library and sequenced to confirm their identity (Kamath & Ahringer, 2003). An RNAi construct for miro-1 (K08F11.5) was generated by amplification of cDNA using primer sequences: ceMiro1-3 fw 5′-CGTGCTGGGAATACACTCGAC-3′ and ceMiro1-3 re 5′-CAGCTAGAGCTACTATGGCAG-3′. The PCR was first cloned using the TOPO® TA Cloning® kit (Invitrogen) and then cloned into L4440 using XhoI/HindIII sites. An RNAi construct for vdac-1 (R05G6.7) was generated by cDNA amplification using primer sequences: vdac-1 fw 5′-ATGGCCCCACCAACCTTCGC-3′ and vdac-1 re 5′-GAAGATCTAGTTGGATGGATC-3′. The PCR was sub-cloned using the TOPO® TA Cloning® (Invitrogen) and cloned into L4440 using EcoRI. The RNAi construct pBC567(fzo-1; ZK1248.14) was described earlier (Rolland et al, 2009).
NGM plates containing 1.5 mM IPTG and 25 μg/ml carbenicillin were inoculated with RNAi bacteria and induced for approximately 12 h at room temperature. Eggs or larvae were cultured at 20 or 25°C until adulthood. For muscle cells, RNAi strains were fed for 2 days at 20°C starting from L2/L3 larvae. For double RNAi experiments, TOM70::GFP L4 larvae were placed on RNAi plates against miro-1, fzo-1, or drp-1 and incubated at 25°C. The offspring was picked at L2 larval stage and transferred to double RNAi plates containing the original RNA together with gbf-1(RNAi) or L4440 as control. Plates were incubated for another 2 days at 25°C.
Yeast methods
Standard genetic techniques were employed (Sherman, 1991). Transformations and deletions were performed as described (Schiestl & Gietz, 1989; Knop et al, 1999). For drop assays, cells were grown to log phase in liquid YP media with 2% glucose (YPD). Cells were diluted to OD600 0.1, followed by 10× serial dilutions. Aliquots were spotted onto YPAD or YPAG plates and incubated at various temperatures. Yeast strains are specified in Supplementary Table S2. For microscopy, cultures were grown in HC + adenine O/N at 23°C, diluted to early log phase, and grown at 23°C for 4 h in YPAD. Half of each culture was shifted to 37°C for 1 h and imaged thereafter. For quantification, approximately 100 cells were counted in each of at least three individual experiments. The starvation experiments were performed as described (Bockler & Westermann, 2014). For quantification of autophagy, the arrival of GFP-Atg8 in the vacuole was quantified after 5 h of starvation.
Enzyme histochemical analysis
Worms were synchronized by bleaching as described (Bianchi & Driscoll, 2006). Approximately 600 young adults were embedded. Worms were washed off the NGM plates using M9 buffer and embedded in carbowax cryo-embedding media (TissueTek®, Sakura Fintek, Japan). The worm blocks were frozen in pre-cooled isopentane (−20°C) and then stored at −20°C. Seven micrometer sections were cut with a cryo-microtome, and samples from three experiments were combined on the same glass slide (“Super Frost”, Menzel, Germany) for co-staining. Enzymatic histochemical reactions for COX were performed and quantified as described, using the existing macros in ImageJ (Hench et al, 2011). Graphs and statistics were processed in OpenOffice.
Imaging
For live imaging, adult hermaphrodites were mounted in M9 without anesthetics between the slide and coverslip using Vaseline® at the edges to function as a spacer. Fixed samples were mounted on uncoated slides in anti-fade reagent CitiFluor (Citifluor Ltd., UK) with a small rim of Vaseline® on the side.
Yeast cells expressing Dnm1-GFP/mtRFP or Pho88-mCherry/mtGFP were imaged on an Andor Revolution spinning disk confocal system (Andor Technologies, Northern Ireland) mounted onto an IX-81 inverted microscope (Olympus, Center Valley, PA), equipped with iXonEM+ EMCCD camera (Andor Technologies). MitoTracker/Tom70::GFP and yeast mtGFP experiments were imaged on a Leica SP5-II-Matrix confocal microscope with a HCX PLAN APO lambda blue 63× oil objective (Leica Microsystems, Germany) using the high resonance scanner at 8,000 Hz, 2.5× zoom and 16× line average (worms) or with normal scanner settings at 1,000 Hz, 15× zoom and 8× line average (yeast). LAS AF version 2.6.0.7266 software was used. Images for quantification of yeast and worm phenotypes, muscle actin staining and Benomyl/LatA experiments were taken on a Zeiss Axioplan 2 microscope equipped with a Zeiss Axio Cam MRm camera (Carl Zeiss, Germany) and a Plan Apochromat 63×/NA1.40 oil objective. Images of HeLa cells were taken with a Zeiss LSM700 confocal microscope with 10 × 63 magnification and 2× zoom. ImageJ was used for post-processing and analysis of all images.
Statistical analysis
All multiple comparisons were tested for statistical significance with a one-way ANOVA followed by pairwise comparisons with a Tukey's test using OriginPro8. In case of percentages, we transformed the data into arcsin values to obtain a normal distribution of freedom. For the Tukey's test, a maximum of seven decimal digits was used to describe the adjusted P-value. Pairwise single comparisons were done by using a two-tailed unequal variance t-test.
Acknowledgments
We thank A. Nakano, B. Grant, C.L. Jackson, R. Deshaies, S. Munro, A. van der Bliek, A. Audhya, J. Shaw, D.T. Ng, F. Madeo, S. Rolland, B. Conradt and the Caenorhabditis Genetic Center for plasmids, antibodies and strains, and S. Ipsen for technical support. We thank S. Rolland and J. Shaw for the helpful discussions and I.G. Macara and M. Dekkers for critical reading of the manuscript. The Biozentrum Imaging and Proteome Core Facilities are acknowledged for instrumental and technical support. This research was supported by the Swiss National Science Foundation (31003A_141207) and the University of Basel.
Author contributions
KA and AS conceived the project; KA, US, JH, HM, SCW, SB and AS performed the experiments; KA, JH, SF, FB, SB and BW and AS analyzed and discussed the data; KA and AS wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Ackema KB, Sauder U, Solinger JA, Spang A. The ArfGEF GBF-1 is required for ER structure, secretion and endocytic transport in. PLoS ONE. 2013;8:e67076. doi: 10.1371/journal.pone.0067076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann K, Westermann B. Role of essential genes in mitochondrial morphogenesis in Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:5410–5417. doi: 10.1091/mbc.E05-07-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Kahn RA, Schwaninger R. ADP-ribosylation factor is required for vesicular trafficking between the endoplasmic reticulum and the cis-Golgi compartment. J Biol Chem. 1992;267:13053–13061. [PubMed] [Google Scholar]
- Balklava Z, Pant S, Fares H, Grant BD. Genome-wide analysis identifies a general requirement for polarity proteins in endocytic traffic. Nat Cell Biol. 2007;9:1066–1073. doi: 10.1038/ncb1627. [DOI] [PubMed] [Google Scholar]
- Beanan MJ, Strome S. Characterization of a germ-line proliferation mutation in C. elegans. Development. 1992;116:755–766. doi: 10.1242/dev.116.3.755. [DOI] [PubMed] [Google Scholar]
- Bianchi L, Driscoll M. Culture of embryonic C. elegans cells for electrophysiological and pharmacological analyses. The C. elegans Research Community. 2006:1551–8507. doi: 10.1895/wormbook.1.122.1. WormBook, September 30, 2006 http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockler S, Westermann B. Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev Cell. 2014;28:450–458. doi: 10.1016/j.devcel.2014.01.012. [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Kang BH, Kokel D, Mitani S, Staehelin LA, Xue D. Caenorhabditis elegans drp-1 and fis-2 regulate distinct cell-death execution pathways downstream of ced-3 and independent of ced-9. Mol Cell. 2008;31:586–597. doi: 10.1016/j.molcel.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso ME, Jenna S, Bouchecareilh M, Baillie DL, Boismenu D, Halawani D, Latterich M, Chevet E. GTPase-mediated regulation of the unfolded protein response in Caenorhabditis elegans is dependent on the AAA+ ATPase CDC-48. Mol Cell Biol. 2008;28:4261–4274. doi: 10.1128/MCB.02252-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citterio C, Vichi A, Pacheco-Rodriguez G, Aponte AM, Moss J, Vaughan M. Unfolded protein response and cell death after depletion of brefeldin A-inhibited guanine nucleotide-exchange protein GBF1. Proc Natl Acad Sci USA. 2008;105:2877–2882. doi: 10.1073/pnas.0712224105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Jackson CL. ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat Rev Mol Cell Biol. 2011;12:362–375. doi: 10.1038/nrm3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz Y, Schuldiner M. Staying in touch: the molecular era of organelle contact sites. Trends Biochem Sci. 2011;36:616–623. doi: 10.1016/j.tibs.2011.08.004. [DOI] [PubMed] [Google Scholar]
- Esaki M, Ogura T. Cdc48p/p97-mediated regulation of mitochondrial morphology is Vms1p-independent. J Struct Biol. 2012;179:112–120. doi: 10.1016/j.jsb.2012.04.017. [DOI] [PubMed] [Google Scholar]
- Escobar-Henriques M, Langer T. Dynamic survey of mitochondria by ubiquitin. EMBO Rep. 2014;15:231–243. doi: 10.1002/embr.201338225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick RL, McCaffery JM, Cunningham KW, Okamoto K, Shaw JM. Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J Cell Biol. 2004;167:87–98. doi: 10.1083/jcb.200405100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick RL, Okamoto K, Shaw JM. Multiple pathways influence mitochondrial inheritance in budding yeast. Genetics. 2008;178:825–837. doi: 10.1534/genetics.107.083055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz S, Weinbach N, Westermann B. Mdm30 is an F-box protein required for maintenance of fusion-competent mitochondria in yeast. Mol Biol Cell. 2003;14:2303–2313. doi: 10.1091/mbc.E02-12-0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales KG, Fuller MT. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997;90:121–129. doi: 10.1016/s0092-8674(00)80319-0. [DOI] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- Hench J, Bratic Hench I, Pujol C, Ipsen S, Brodesser S, Mourier A, Tolnay M, Frank S, Trifunovic A. A tissue-specific approach to the analysis of metabolic changes in Caenorhabditis elegans. PLoS ONE. 2011;6:e28417. doi: 10.1371/journal.pone.0028417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JM, Livnat-Levanon N, Taylor EB, Jones KT, Dephoure N, Ring J, Xie J, Brodsky JL, Madeo F, Gygi SP, Ashrafi K, Glickman MH, Rutter J. A stress-responsive system for mitochondrial protein degradation. Mol Cell. 2010;40:465–480. doi: 10.1016/j.molcel.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JM, Nielson JR, Dephoure N, Gygi SP, Rutter J. Intramolecular interactions control Vms1 translocation to damaged mitochondria. Mol Biol Cell. 2013;24:1263–1273. doi: 10.1091/mbc.E13-02-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- Jackson CL, Casanova JE. Turning on ARF: the Sec7 family of guanine-nucleotide-exchange factors. Trends Cell Biol. 2000;10:60–67. doi: 10.1016/s0962-8924(99)01699-2. [DOI] [PubMed] [Google Scholar]
- Jung C, Higgins CM, Xu Z. A quantitative histochemical assay for activities of mitochondrial electron transport chain complexes in mouse spinal cord sections. J Neurosci Methods. 2002;114:165–172. doi: 10.1016/s0165-0270(01)00524-6. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Kanazawa T, Zappaterra MD, Hasegawa A, Wright AP, Newman-Smith ED, Buttle KF, McDonald K, Mannella CA, van der Bliek AM. The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. 2008;4:e1000022. doi: 10.1371/journal.pgen.1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007;178:71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilchert C, Weidner J, Prescianotto-Baschong C, Spang A. Defects in the secretory pathway and high Ca2+ induce multiple P-bodies. Mol Biol Cell. 2010;21:2624–2638. doi: 10.1091/mbc.E10-02-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, Winborn BJ, Moore J, Lee JY, Yao TP, Pallanck L, Kundu M, Taylor JP. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron. 2013;78:65–80. doi: 10.1016/j.neuron.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klecker T, Bockler S, Westermann B. Making connections: interorganelle contacts orchestrate mitochondrial behavior. Trends Cell Biol. 2014;24:537–545. doi: 10.1016/j.tcb.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Siegers K, Pereira G, Zachariae W, Winsor B, Nasmyth K, Schiebel E. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;15:963–972. doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Kondo M, Senoo-Matsuda N, Yanase S, Ishii T, Hartman PS, Ishii N. Effect of oxidative stress on translocation of DAF-16 in oxygen-sensitive mutants, mev-1 and gas-1 of Caenorhabditis elegans. Mech Ageing Dev. 2005;126:637–641. doi: 10.1016/j.mad.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Osman C, Walter P. The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc Natl Acad Sci USA. 2011;108:14151–14156. doi: 10.1073/pnas.1111314108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. Caenorhabditis elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell. 1999;4:815–826. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisinger C, Sommer T, Pfanner N. Purification of Saccharomcyes cerevisiae mitochondria devoid of microsomal and cytosolic contaminations. Anal Biochem. 2000;287:339–342. doi: 10.1006/abio.2000.4868. [DOI] [PubMed] [Google Scholar]
- Meisinger C, Pfanner N, Truscott KN. Isolation of yeast mitochondria. Methods Mol Biol. 2006;313:33–39. doi: 10.1385/1-59259-958-3:033. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley A, Lackner LL, Osman C, West M, Voeltz GK, Walter P, Nunnari J. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. Elife. 2013;2:e00422. doi: 10.7554/eLife.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano A, Muramatsu M. A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol. 1989;109:2677–2691. doi: 10.1083/jcb.109.6.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutzner A, Benard G, Youle RJ, Karbowski M. Role of the ubiquitin conjugation system in the maintenance of mitochondrial homeostasis. Ann N Y Acad Sci. 2008;1147:242–253. doi: 10.1196/annals.1427.012. [DOI] [PubMed] [Google Scholar]
- Ng DT, Spear ED, Walter P. The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J Cell Biol. 2000;150:77–88. doi: 10.1083/jcb.150.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TT, Lewandowska A, Choi JY, Markgraf DF, Junker M, Bilgin M, Ejsing CS, Voelker DR, Rapoport TA, Shaw JM. Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic. 2012;13:880–890. doi: 10.1111/j.1600-0854.2012.01352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlichenko L, Stolz DB, Noel P, Behari J, Liu S, Singh VP. ADP-ribosylation factor 1 protein regulates trypsinogen activation via organellar trafficking of procathepsin B protein and autophagic maturation in acute pancreatitis. J Biol Chem. 2012;287:24284–24293. doi: 10.1074/jbc.M111.328815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyroche A, Paris S, Jackson CL. Nucleotide exchange on ARF mediated by yeast Gea1 protein. Nature. 1996;384:479–481. doi: 10.1038/384479a0. [DOI] [PubMed] [Google Scholar]
- Popoff V, Langer JD, Reckmann I, Hellwig A, Kahn RA, Brugger B, Wieland FT. Several ADP-ribosylation factor (Arf) isoforms support COPI vesicle formation. J Biol Chem. 2011;286:35634–35642. doi: 10.1074/jbc.M111.261800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteryaev D, Squirrell JM, Campbell JM, White JG, Spang A. Involvement of the actin cytoskeleton and homotypic membrane fusion in ER dynamics in Caenorhabditis elegans. Mol Biol Cell. 2005;16:2139–2153. doi: 10.1091/mbc.E04-08-0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- Rolland SG, Lu Y, David CN, Conradt B. The BCL-2-like protein CED-9 of C. elegans promotes FZO-1/Mfn1,2- and EAT-3/Opa1-dependent mitochondrial fusion. J Cell Biol. 2009;186:525–540. doi: 10.1083/jcb.200905070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland SG, Motori E, Memar N, Hench J, Frank S, Winklhofer KF, Conradt B. Impaired complex IV activity in response to loss of LRPPRC function can be compensated by mitochondrial hyperfusion. Proc Natl Acad Sci USA. 2013;110:E2967–E2976. doi: 10.1073/pnas.1303872110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado CJ, Mijaljica D, Hatzinisiriou I, Prescott M, Devenish RJ. Rosella: a fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy. 2008;4:205–213. doi: 10.4161/auto.5331. [DOI] [PubMed] [Google Scholar]
- Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13:607–625. doi: 10.1038/nrm3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Sato M, Audhya A, Oegema K, Schweinsberg P, Grant BD. Dynamic regulation of caveolin-1 trafficking in the germ line and embryo of Caenorhabditis elegans. Mol Biol Cell. 2006;17:3085–3094. doi: 10.1091/mbc.E06-03-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl RH, Gietz RD. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- Sesaki H, Jensen RE. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol. 1999;147:699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM. A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol. 1998;143:351–358. doi: 10.1083/jcb.143.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A, Herrmann JM, Hamamoto S, Schekman R. The ADP ribosylation factor-nucleotide exchange factors Gea1p and Gea2p have overlapping, but not redundant functions in retrograde transport from the Golgi to the endoplasmic reticulum. Mol Biol Cell. 2001;12:1035–1045. doi: 10.1091/mbc.12.4.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A. On vesicle formation and tethering in the ER-Golgi shuttle. Curr Opin Cell Biol. 2009;21:531–536. doi: 10.1016/j.ceb.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Stearns T, Willingham MC, Botstein D, Kahn RA. ADP-ribosylation factor is functionally and physically associated with the Golgi complex. Proc Natl Acad Sci USA. 1990;87:1238–1242. doi: 10.1073/pnas.87.3.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud DA, Oeljeklaus S, Wiese S, Bohnert M, Lewandrowski U, Sickmann A, Guiard B, van der Laan M, Warscheid B, Wiedemann N. Composition and topology of the endoplasmic reticulum-mitochondria encounter structure. J Mol Biol. 2011;413:743–750. doi: 10.1016/j.jmb.2011.09.012. [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T, Scharwey M, Langer T. Mitochondrial lipid trafficking. Trends Cell Biol. 2014;24:44–52. doi: 10.1016/j.tcb.2013.07.011. [DOI] [PubMed] [Google Scholar]
- Traub LM, Ostrom JA, Kornfeld S. Biochemical dissection of AP-1 recruitment onto Golgi membranes. J Cell Biol. 1993;123:561–573. doi: 10.1083/jcb.123.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein M, Dengjel J, Schirle M, Spang A. Arf1p provides an unexpected link between COPI vesicles and mRNA in Saccharomyces cerevisiae. Mol Biol Cell. 2004;15:5021–5037. doi: 10.1091/mbc.E04-05-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein M, Schindler C, Gauss R, Dengjel J, Hartmann E, Spang A. Arf1p, Chs5p and the ChAPs are required for export of specialized cargo from the Golgi. EMBO J. 2006;25:943–954. doi: 10.1038/sj.emboj.7601007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vaart A, Griffith J, Reggiori F. Exit from the Golgi is required for the expansion of the autophagosomal phagophore in yeast Saccharomyces cerevisiae. Mol Biol Cell. 2010;21:2270–2284. doi: 10.1091/mbc.E09-04-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- Westermann B. Mitochondrial dynamics in model organisms: what yeasts, worms and flies have taught us about fusion and fission of mitochondria. Semin Cell Dev Biol. 2010a;21:542–549. doi: 10.1016/j.semcdb.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010b;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Wideman JG, Gawryluk RM, Gray MW, Dacks JB. The ancient and widespread nature of the ER-mitochondria encounter structure. Mol Biol Evol. 2013;30:2044–2049. doi: 10.1093/molbev/mst120. [DOI] [PubMed] [Google Scholar]
- Xu S, Peng G, Wang Y, Fang S, Karbowski M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol Biol Cell. 2011;22:291–300. doi: 10.1091/mbc.E10-09-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahara N, Ueda T, Sato K, Nakano A. Multiple roles of Arf1 GTPase in the yeast exocytic and endocytic pathways. Mol Biol Cell. 2001;12:221–238. doi: 10.1091/mbc.12.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, Ohmura-Hoshino M, Sada K, Hotta H, Yamamura H, Inatome R, Yanagi S. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006;25:3618–3626. doi: 10.1038/sj.emboj.7601249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.