

Abstract
Cyclic diadenosine monophosphate (c-di-AMP) is a second messenger that is essential for growth and homeostasis in bacteria. A recently discovered c-di-AMP-responsive riboswitch controls the expression of genes in a variety of bacteria, including important pathogens. To elucidate the molecular basis for specific binding of c-di-AMP by a gene-regulatory mRNA domain, we have determined the co-crystal structure of this riboswitch. Unexpectedly, the structure reveals an internally pseudo-symmetric RNA in which two similar three-helix-junction elements associate head-to-tail, creating a trough that cradles two c-di-AMP molecules making quasi-equivalent contacts with the riboswitch. The riboswitch selectively binds c-di-AMP and discriminates exquisitely against other cyclic dinucleotides, such as c-di-GMP and cyclic-AMP-GMP, via interactions with both the backbone and bases of its cognate second messenger. Small-angle X-ray scattering experiments indicate that global folding of the riboswitch is induced by the two bound cyclic dinucleotides, which bridge the two symmetric three-helix domains. This structural reorganization likely couples c-di-AMP binding to gene expression.
Keywords: cyclic di-AMP, riboregulation, riboswitch, small-angle X-ray scattering, X-ray crystallography
Introduction
Detection of pathogens by human cells is the critical first step in the innate immune response and is accomplished by the recognition of foreign molecules that are either unique to pathogens or characteristic of their infection. The bacterial pathogen Listeria monocytogenes releases cyclic dinucleotides, such as cyclic diadenosine monophosphate (c-di-AMP) (Woodward et al, 2010), which stimulates an immune response through a cytosolic surveillance pathway involving the stimulator of interferon genes (STING) protein (Burdette et al, 2011; Burdette & Vance, 2013). In human cells, the related cyclic dinucleotide cyclic-GMP-AMP, which also activates STING, is synthesized by the cytosolic DNA sensor cyclic-GMP-AMP synthase (reviewed in Cai et al, 2014).
C-di-AMP is a recently discovered bacterial second messenger that was first identified as a ligand for DisA, a bacterial diadenylate cyclase, during its crystallographic characterization (Witte et al, 2008). In bacteria, c-di-AMP levels are controlled by diadenylate cyclases that synthesize it from two molecules of ATP, and phosphodiesterases that degrade c-di-AMP to pApA (reviewed in Corrigan & Grundling, 2013). These are analogous to the enzymes that synthesize cyclic diguanosine monophosphate (c-di-GMP) from GTP, and the phosphodiesterases that break it down (reviewed in Schirmer & Jenal, 2009). C-di-GMP is an extensively characterized bacterial second messenger that regulates processes such as biofilm formation and virulence (reviewed in Boyd & O'Toole, 2012) and serves as a paradigm for elucidating the interactions of the newly discovered c-di-AMP. Mounting evidence suggests a widespread role of c-di-AMP in bacterial virulence and growth (Luo & Helmann, 2012; Bai et al, 2013, 2014; Mehne et al, 2013) akin to the better characterized functions of c-di-GMP. In some bacterial species, a variety of enzymes that control c-di-AMP levels are present, presumably to allow a nuanced response to cellular stimuli. Bacterial proteins that bind c-di-AMP are associated with potassium transport and cell wall homeostasis, which appears to be a primary regulatory function of c-di-AMP and explains why it is essential for growth (Corrigan et al, 2013). The central role that cyclic dinucleotides play in bacterial biofilm formation and growth may partly explain why human cells have evolved to detect them.
In addition to binding to receptor proteins, c-di-AMP has recently been shown to regulate gene expression in bacteria by binding to c-di-AMP-responsive riboswitches present in the 5′ untranslated regions of many mRNAs, including a virulence factor for Mycobacterium tuberculosis (Nelson et al, 2013). Riboswitches are highly structured mRNA domains that control gene expression in cis in response to direct binding by ions, small-molecule metabolites, or second messengers (reviewed in Peselis & Serganov, 2014; Zhang et al, 2014). The c-di-AMP riboswitch binds to its cognate ligand with an apparent dissociation constant (KD) of approximately 1 nM (Nelson et al, 2013). It is typified by sequences found in the Bacillus subtilis ydaO-yuaA operon. This RNA domain was first identified as a potential riboswitch in 2004 based on phylogenetic conservation (Barrick et al, 2004). Subsequent work demonstrated that it is phylogenetically widespread and suggested that it predominantly regulates genes important for cell wall homeostasis (Block et al, 2010). Bacteria employ two distinct classes of riboswitches to sense the structurally related second messenger c-di-GMP (Sudarsan et al, 2008; Lee et al, 2010). Crystallographic analyses of the c-di-GMP-I and c-di-GMP-II riboswitches have revealed the structural basis of their ligand specificity (Kulshina et al, 2009; Smith et al, 2009, 2011). The c-di-AMP riboswitch sequence is unrelated to those of either class of c-di-GMP riboswitches. Thus, the results of previous genetic and biochemical studies of the c-di-AMP riboswitch (Watson & Fedor, 2012; Nelson et al, 2013) could not be structurally rationalized. To elucidate the molecular basis for function of this important bacterial gene regulator, we have determined the co-crystal structure of the B. subtilis ydaO riboswitch in the presence of c-di-AMP. Our analysis reveals an RNA of unprecedented internally symmetric structure. Because the c-di-AMP riboswitch is widespread and regulates essential genes, targeting it with c-di-AMP mimics constitutes a promising strategy for the development of novel antibiotics (Deigan & Ferré-D'Amaré, 2011; Kalia et al, 2013). Our structure represents the starting point for the rational design of ligand analogs and inhibitors of this important bacterial regulatory RNA.
Results
Overall structure of the c-di-AMP riboswitch
The ydaO-yuaA c-di-AMP riboswitch from B. subtilis was engineered for crystallization by incorporating a binding site for the human spliceosomal protein U1A (Ferré-D'Amaré, 2010) in a region of the RNA with no phylogenetic conservation (Nelson et al, 2013). Isothermal titration calorimetry (ITC) analysis demonstrated that this did not adversely alter the affinity of the RNA for c-di-AMP (Supplementary Fig S1). The engineered RNA was co-crystallized with selenomethionyl U1A and c-di-AMP, and the structure solved by the single-wavelength anomalous dispersion (SAD) method. The electron density maps were of high quality and revealed the presence of two molecules of c-di-AMP bound to quasi-equivalent locations in the RNA (Supplementary Fig S2). The structure has been refined against two diffraction datasets extending to 3.1 and 3.2 Å resolution (Materials and Methods, Table 1). While this manuscript was under revision, independent structural determinations of c-di-AMP riboswitches from Thermoanaerobacter tengcongensis, T. pseudethanolicus, and Thermovirga lienii were published that are in general agreement with the results presented here (Gao & Serganov, 2014; Ren & Patel, 2014).
Table 1.
Summary of crystallographic statistics
| Dataset 1 | Dataset 2 | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 0.9793 | 0.9611 |
| Space group | I222 | I222 |
| Unit cell, a, b, c (Å) | 60.3, 83.1, 233.6 | 59.6, 84.7, 232.7 |
| Resolution | 49–3.2 (3.3–3.2) a | 47–3.1 (3.2–3.1) a |
| Unique reflections | 9,841 (909) | 9,322 (943) |
| Multiplicity | 12.3 (8.2) | 10.4 (10.3) |
| Completeness (%) | 98.5 (85.7) | 86.4 (83.8) |
| <I>/<σ(I)> | 14.3 (1.9) | 18.1 (1.9) |
| Rmerge | 0.156 (1.0) | 0.1708 (1.0) |
| Refinement | ||
| Rwork/Rfree | 0.252/0.289 | 0.234/0.286 |
| R.m.s. bonds (Å)/angles (°) | 0.003/0.73 | 0.003/0.79 |
| Non-hydrogen atoms | ||
| RNA | 2,033 | 2,146 |
| Protein | 658 | 639 |
| c-di-AMP | 88 | 88 |
| Ions | 10 | 5 |
| Mean B-factors (Å2) | ||
| RNA | 106.8 | 115.2 |
| Protein | 87.4 | 104.7 |
| c-di-AMP | 53.8 | 79.7 |
| Ions | 47.4 | 66.5 |
| Ramachandran, most favored/disallowed (%) | 98/0 | 97/0 |
| Mean coordinate precision (Å) | 0.41 | 0.47 |
| PDB code | 4W92 | 4W90 |
Values in parentheses correspond to highest resolution shell.
The c-di-AMP riboswitch adopts a strikingly symmetrical structure composed of six helical elements (paired regions P1 through P6), in which the first three helices adopt an arrangement that is repeated by the latter three (Fig 1). Thus, P1 connects at an acutely angled three-way junction with the coaxially stacked P2 and P3, and P4 connects with the coaxially stacked P5 and P6. In three dimensions, the P1-P3 and P4-P6 substructures associate head-to-tail, forming a trough-like intramolecular interface that traverses the molecule diagonally and envelops two molecules of c-di-AMP (Figs 1 and 2), burying over 80% of solvent-accessible surface area of the second messengers. These bridge the minor grooves of P3 and P4 (c-di-AMP molecule 1, hereafter A1) and of P1 and P6 (c-di-AMP molecule 2, hereafter A2). Although the overall three-dimensional structure of the c-di-AMP riboswitch can be characterized by a twofold rotation axis passing through the center of the structure (Fig 1C), the connectivity of the RNA chain is such that the P4-P6 substructure is inserted into the distal end of P3. In addition to breaking symmetry, this relationship obscures the secondary structural self-similarity, explaining why it was not detected in previous structural and biochemical characterization (Supplementary Fig S3).
Figure 1. Overall structure of the c-di-AMP riboswitch.
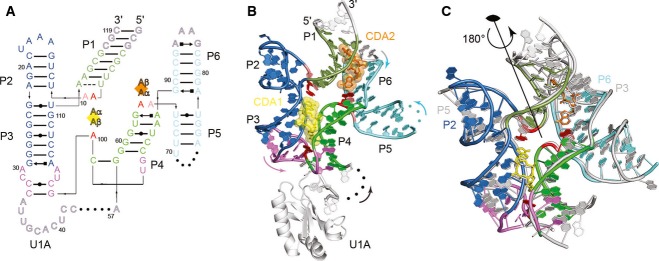
- Secondary structure of the B. subtilis c-di-AMP riboswitch. Non-canonical base pairs are depicted with Leontis and Westhof ( 2001) symbols. Nucleotides unresolved in the crystal structures are indicated by dots. Bound c-di-AMP molecules A1 (yellow) and A2 (orange) are denoted by squares with individual c-di-AMP bases indicated as Aα and Aβ. RNA colored as follows: P1 helix in dark green, P2 and P3 helices in blue, P4 helix in light green, P5 and P6 helices in light blue, and linking region in purple. Adenosine residues stacking on c-di-AMP ligands are shown in red, with adjacent stacked residues in pink. Nucleotides modified for crystallization are shown in outlined gray letters.
- Structure of the c-di-AMP riboswitch. The coloring scheme is the same as A. Arrows denote 5′ to 3′ chain direction. Spheres denote breaks in the crystallographic model.
- Internal twofold symmetry of the c-di-AMP riboswitch. The structure of the c-di-AMP riboswitch is shown colored as in (B), superimposed on the same molecule rotated by 180° (gray). Asymmetric elements (e.g. U1A protein) are removed for clarity. Approximate position of the twofold rotational symmetry axis is indicated.
Figure 2. Binding of c-di-AMP in an intramolecular RNA trough.
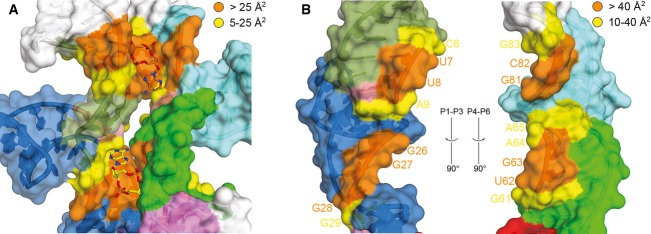
- Surface representation of the two c-di-AMP binding sites. The coloring scheme is the same as in Fig 1 except for residues contacting c-di-AMP molecules, which are colored orange (> 25 Å2 solvent-accessible interaction surface) and yellow (5–25 Å2 interaction surface). Binding to the riboswitch buries 650 Å2 of solvent-accessible surface for c-di-AMP A1 and 610 Å2 for c-di-AMP A2, corresponding to 85 and 80% of the total accessible surface area for each ligand, respectively. RNA–RNA interactions between P1–P3 and P4–P6 bury an additional 350 Å2 per interface.
- Open-book view of the P1–P3 (left) and P4–P6 helices (right). The coloring scheme is the same as in Fig 1 except for residues involved in RNA–RNA interactions, which are labeled and colored orange (> 40 Å2 interaction surface) and yellow (10–40 Å2 interaction surface).
Three-way junctions resemble tetraloop–receptor interactions
The two three-way junctions in the c-di-AMP riboswitch structure are A-minor junctions (Lescoute & Westhof, 2006; Geary et al, 2011), in which the RNA is globally similar to a GAAA tetraloop interacting with a tetraloop–receptor helix (Fig 3). In the canonical tertiary interaction, exemplified by the P4–P6 domain of the Tetrahymena group I intron (Cate et al, 1996), the three adenosines of the tetraloop from L5b stack on each other and make A-minor interactions (Nissen et al, 2001) with the minor groove of the receptor helix J6a/6b. In addition, the first of the three adenine bases of the tetraloop stacks on an adenine from a homopurine platform in the receptor (A151 and A226 in the Tetrahymena group I intron, respectively, Fig 3A).
Figure 3. The three-way junctions of the c-di-AMP riboswitch resemble a tetraloop–receptor interface.
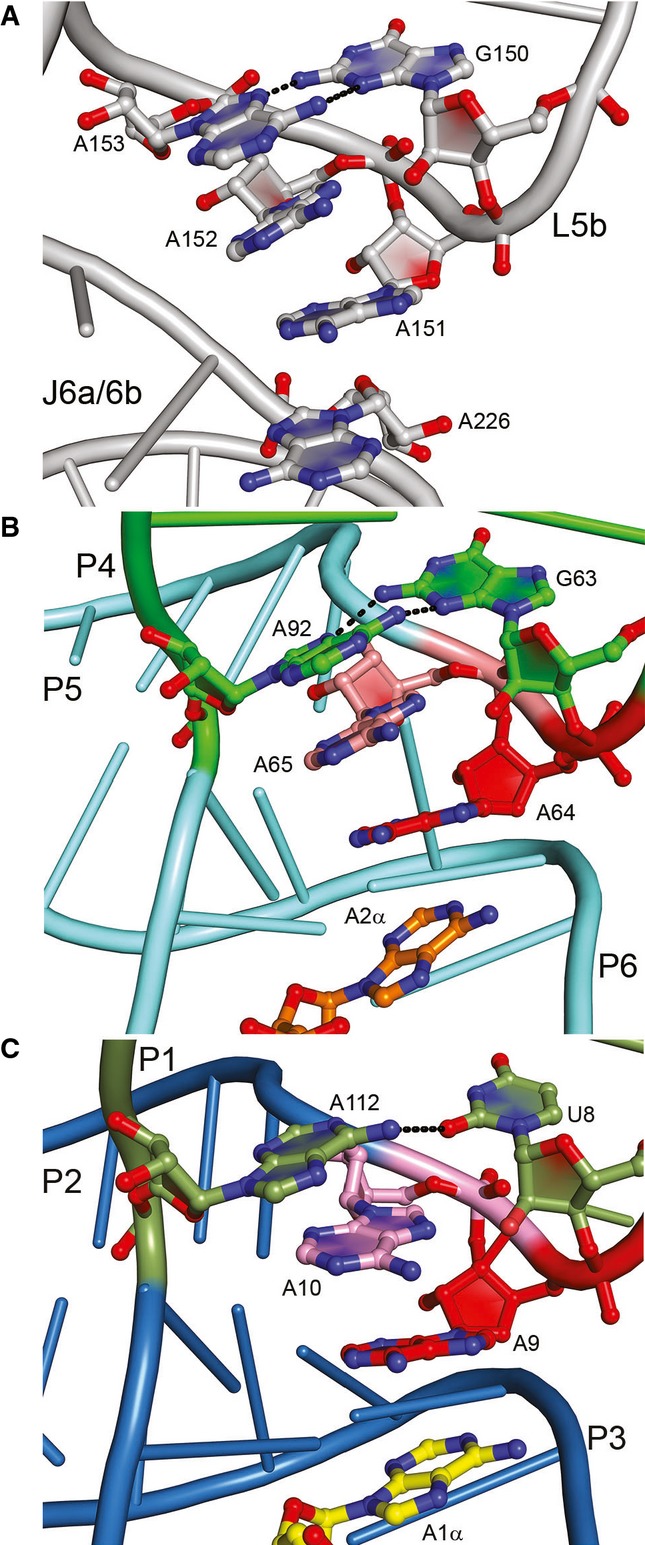
- The GAAA tetraloop of the Tetrahymena thermophila group I intron (Cate et al, 1996). The GAAA tetraloop motif (nts 150–153) of loop L5b dock onto helix J6a/6b. A226 is part of a homopurine platform motif.
- The GAAA tetraloop–receptor-like fold of the P4-P5-P6 three-helix junction is shown with the nucleotides G63, A64, A65, and distant residue A92 in the interaction with c-di-AMP A2α.
- The UAAA tetraloop–receptor-like fold of the P1-P2-P3 three-helix junction is shown with the three consecutive nucleotides U8, A9, and A10 along with A112 interacting with c-di-AMP A1α.
The junction where P4 of the c-di-AMP riboswitch connects with the coaxially stacked P5 and P6 helices most closely resembles the tetraloop–receptor arrangement (Fig 3B). G63 of the riboswitch corresponds to the guanine of a GAAA tetraloop. As in such loops, the backbone flips immediately 3′, and A64 forms the first of three stacked adenosines that pack against the minor groove of P5 of the riboswitch. P5 and P6, which stack coaxially, are therefore analogous to a canonical receptor helix. Moreover, A64 makes type I A-minor interactions (Nissen et al, 2001) with the G79:C90 pair (Supplementary Fig S4A). As in a conventional tetraloop, G63 closes the tetraloop by pairing through its sugar edge with the Hoogsteen edge of the last adenosine. The crucial difference with the canonical interaction is that this last adenosine (A92 in the c-di-AMP riboswitch) comes from a separate chain, thereby leading to the strand swap that makes this a three-way junction, rather than a tetraloop–receptor tertiary interaction.
The P1–P3 junction (Fig 3C) is similar, but has the sequence UAA(A) (the last purine is A112 from a separate strand) in the B. subtilis c-di-AMP riboswitch. As a result, the closing pair of the “tetraloop,” rather than being a sheared G•A pair, is a single-hydrogen bond U•A pair in which the purine is in the syn conformation. As with A64, the P1–P3 junction also contains type I A-minor interactions between A9 and the C24:G110 pair (Supplementary Fig S4B). Completion of a tetraloop–receptor-like motif by an external nucleotide was first observed in the structure of an AMP aptamer, in which the AMP ligand acts as the fourth base of the pseudo-tetraloop (Jiang et al, 1996).
Structurally and functionally furthering the similarity between the A-minor junctions of the riboswitch and the canonical tetraloop–receptor interaction, one of the two adenine bases of each of the two bound c-di-AMP molecules stacks on the first adenosine of each of the two NAA(A) tetraloops of the RNA (N is either G63 or U8 in this case). These c-di-AMP nucleobases (hereafter A1α and A2α, Fig 3B and C) are therefore analogous to the homopurine platform nucleobase of the tetraloop receptor (A226, Fig 3A). In summary, the c-di-AMP riboswitch contains two NAA(A) A-minor three-helix junctions that not only organize the overall structure of the riboswitch, but also contribute to forming the binding site for the two bound second messenger molecules.
Riboswitch recognition of c-di-AMP
As expected from the overall pseudo-symmetry of the riboswitch, the two c-di-AMP ligands make structurally similar interactions with the RNA (Fig 4). In the co-crystal structure, the adenine bases of the two c-di-AMPs stack with unpaired riboswitch adenosines, except for A2β, whose stacking partner was omitted from our crystallization construct but is likely present in the wild-type B. subtilis riboswitch (Supplementary Fig S5A). In addition to stacking, the c-di-AMP adenine bases make hydrogen bonds with the minor groove faces of the RNA through their Hoogsteen, Watson–Crick and, sugar edges (Fig 4). The riboswitch recognizes all the 2′-OH atoms of the bound c-di-AMP molecules through extensive ribose-zipper-type (Cate et al, 1996) hydrogen bonding interactions. The A1α 2′-OH contacts the O2 of C109, while the A1β 2′-OH contacts the O2 of U95. The A2α makes corresponding contacts with the O2 of C89, while the A2β 2′-OH contacts the O2 of C115. This dense network of interactions would break down if c-di-AMP were replaced with its deoxynucleotide analog, thus explaining why the c-di-AMP riboswitch discriminates by at least 100-fold against c-di-dAMP (Nelson et al, 2013). Moreover, the majority of these hydrogen bonds would be absent for ATP, which was previously proposed to be the cognate ligand (Watson & Fedor, 2012) but binds the riboswitch with at least 106-fold weaker affinity than c-di-AMP (Nelson et al, 2013).
Figure 4. Structure of the c-di-AMP binding sites of the riboswitch.
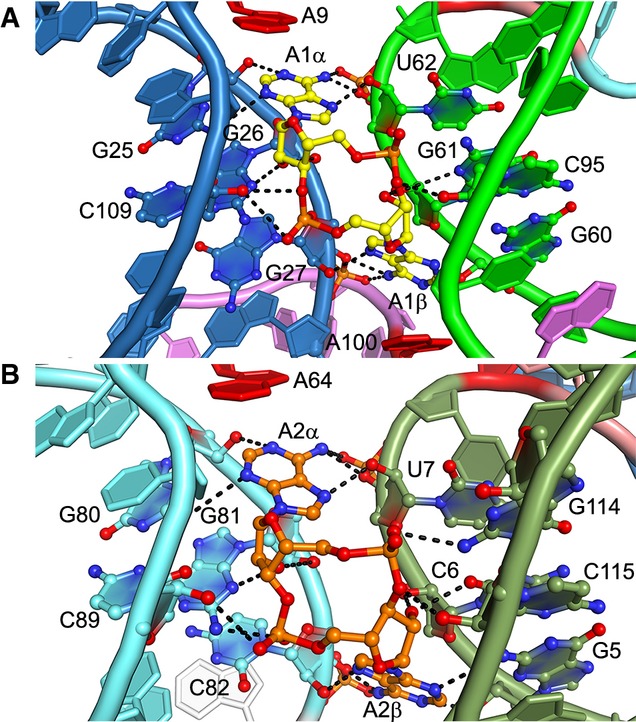
- Binding site of c-di-AMP A1 highlighting interacting nucleotides. Black dashes represent putative hydrogen bonds (the mean precision of the current atomic coordinates is 0.41 Å, Table 1).
- Binding site of c-di-AMP A2.
The c-di-AMP riboswitch also discriminates strongly against the closely related bacterial second messenger c-di-GMP (Nelson et al, 2013). Our structure suggests that this discrimination arises primarily by recognition of the N6 exocyclic amine of c-di-AMP and partly by steric exclusion of c-di-GMP (Fig 5). The four c-di-AMP exocyclic amines form hydrogen bonds with the 3′ oxygen atoms of nearby U62, G27, C82, and U7, respectively, as well as the bridging phosphate oxygen of the subsequent riboswitch residues. These interactions would be absent in the case of c-di-GMP as O6 cannot serve as a hydrogen bond donor. Additionally, the Watson–Crick faces of each c-di-AMP nucleobase are closely apposed to the sugar edges of nearby riboswitch residue bases G25, G60, G5, and G80, respectively (Fig 5). If the second messenger were comprised of guanines, the exocyclic N2 amines of the nucleobases would clash with the nearby 2′-OH and N4 of the riboswitch guanine bases. The presence of exocyclic N2 substituents in guanines at these locations might potentially result in favorable hydrogen bonding but would necessitate that the small-molecule ligands be pushed away from the riboswitch. This is consistent with the molecular selectivity of the c-di-AMP riboswitch, which binds cyclic-AMP-GMP at least five orders of magnitude more weakly than c-di-AMP (Nelson et al, 2013). Indeed, the RNA crystallized here did not bind either cyclic-AMP-GMP or cyclic diinosine phosphate detectably when examined using ITC (data not shown).
Figure 5. Recognition of c-di-AMP nucleobases by neighboring G residues and riboses.
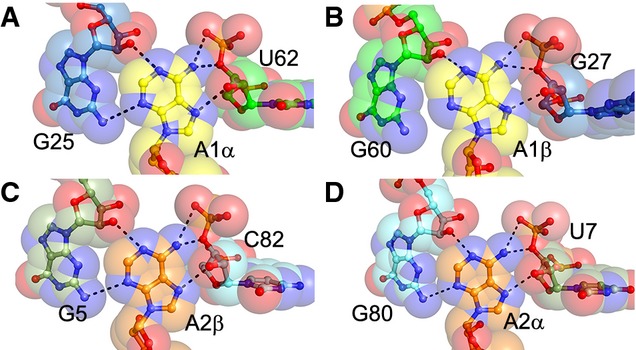
- U62 ribose of helix P4 and G25 of helix P3 in proximity to c-di-AMP A1α.
- Binding site of A1β adjacent to sugar edge of G60 of helix P4 and G27 ribose of helix P3.
- C82 ribose of helix P6 and G5 of helix P1 in proximity to c-di-AMP A2β.
- Sugar edge of G80 of helix P6 and U7 ribose of helix P1 form the binding pocket for c-di-AMP A2α.
Data information: In this figure, translucent spheres depict van der Waals surfaces.
Ligand binding induces folding of the c-di-AMP riboswitch
To probe the moieties of the c-di-AMP riboswitch necessary for ligand binding, we examined RNA sequence variants using ITC (Table 2). In the presence of 10 mM MgCl2, the wild-type RNA (Supplementary Fig S5A) bound c-di-AMP with a dissociation constant (KD) of 53 nM, about fivefold weaker than the apparent KD derived from in-line probing experiments under similar solution conditions (Nelson et al, 2013). To examine whether the proposed pseudoknot contributes to c-di-AMP binding, residues 132–139 were removed and residues 88–99 were replaced with a GAAA tetraloop (Supplementary Fig S5B). This riboswitch variant (ΔPS) bound c-di-AMP with a KD (˜61 nM) similar to that of the wild-type, indicating that either the pseudoknot is not required for c-di-AMP binding under these conditions or that the pseudoknot has not formed. This construct also proved to be more conformationally homogeneous as judged by size-exclusion chromatography (Supplementary Fig S6A). Therefore, it formed the basis for further mutagenesis. A U1A-binding loop was placed in the bulge at 41–48 (Supplementary Fig S5C), a segment that is variable among c-di-AMP riboswitch sequences (Nelson et al, 2013). This construct (U1A) bound with ˜threefold tighter KD to c-di-AMP (˜18 nM), indicating that modification of this region of the RNA is well tolerated and possibly even stabilizing. This construct also displayed excellent conformational homogeneity, as judged by SEC (Supplementary Fig S6B). The 5′ and 3′ ends were mutagenized to assess whether the stability of P1 has an effect on c-di-AMP binding, as this helix interacts with both ligands. Removing the two A-U pairs (ΔPS Δ5′/3′) weakened binding by about 15-fold (KD ˜908 nM), and replacing the two A-U pairs with three G-C pairs—the construct that crystallized—had no effect on binding (KD ˜18 nM). These results suggest that the stability of P1 modulates the affinity of the riboswitch for c-di-AMP, consistent with the participation of this helix in ligand binding (e.g. G5, U7, and A9) revealed by our co-crystal structure, and implicate P1 in transducing ligand binding energy into the ultimate genetic control decision.
Table 2.
ITC binding measurements of c-di-AMP to riboswitch variants
| RNA variant | KD (nM) |
|---|---|
| Wild-type | 53.1 ± 30 |
| Wild-type U1A | 37.2 ± 8.1 |
| ΔPS a | 60.7 ± 3.1 |
| ΔPS U1A | 18.4 ± 6.0 |
| ΔPS Δ5′/3′ | 908 ± 280 |
| ΔPS U1A 5′ GCG 3′ CGC (crystal construct) | 18.1 ± 9.8 |
| A9G b | N.B. |
| A9U | 140 ± 19 |
| A100G | N.B. |
| A100U | N.B. |
| A64G | N.B. |
| A64U | 896 ± 170 |
| A10U | N.B. |
| A112U | 108 ± 5.8 |
| T. tengcongensis ydaO | 156 ± 88 |
Pseudoknot deletion based upon secondary structure of the previously reported B. subtilis sequence (Nelson et al, 2013). The sequence and secondary structures of DPS and other constructs are shown in Supplementary Fig S3.
Point mutations were made to the crystal construct. N.B., no binding.
In the ITC experiments summarized above, all isotherms displayed a 1:1 ligand:RNA binding stoichiometry, despite the observation of two pseudo-symmetrical binding sites in the crystal structure. In contrast to these B. subtilis-derived sequences, T. tengcongensis ydaO RNA displayed 2:1 binding (Supplementary Fig S7A) but was heterogeneous as judged by SEC (Supplementary Fig S7B). Thus, to ascertain the role of each ligand binding site as revealed by the crystal structure of the B. subtilis riboswitch, we mutated the crystal construct at key residues in proximity to each c-di-AMP ligand. Each of the adenosine residues (A9, A64, and A100) that directly interact with the c-di-AMP ligands were mutated to either U or G. Interacting directly with A2α, A64 is required for high-affinity c-di-AMP binding, as no binding was observed for the A64G mutant and ˜50-fold weaker binding was observed for the A64U mutant (Table 2, KD = 896 nM). Both the A100U and A100G point mutants displayed no binding to c-di-AMP, suggesting that the A100-A1β interaction is also essential for c-di-AMP binding. In contrast, while A9G failed to bind c-di-AMP, the A9U mutant showed only approximately eightfold reduced binding (KD = 140 nM). Interestingly, this mutant displayed a 2:1 c-di-AMP:RNA binding stoichiometry (Supplementary Fig S7C), suggesting that the A9U mutation stabilizes the binding of a second c-di-AMP ligand, either directly or indirectly. A9 stacks with A1α and forms a Hoogsteen A(GC) triplet with the G110:C24 pair (Supplementary Fig S4B). Mutation of nearby residue A10U, which stacks on A9 and participates in the tetraloop-like motif (Fig 3C), was necessary for binding (Table 2). In contrast, A112U, which stacks on A10 and also participates in the tetraloop-like motif, still bound c-di-AMP, although with a sixfold weaker affinity (KD = 108 nM).
We examined the c-di-AMP riboswitch in solution in the presence and absence of c-di-AMP using small-angle X-ray scattering (SAXS) with the conformationally homogeneous crystallization construct that binds the second messenger with high affinity (Fig 6, Table 2, and Materials and Methods). From the SAXS data, we calculated the radius of gyration (Rg), maximum end-to-end distance (Dmax), and Porod volume (VP) (Table 3). The riboswitch compacts substantially upon ligand binding, as indicated by a decrease in Rg (Rg = 33.7 and 28.6 Å, free and bound, respectively). The modest change in VP (59,300 Å3 and 60,900 Å3 for free and bound, respectively) suggests that this is not due to a change in the oligomerization state of the RNA, nor were concentration-dependent effects (i.e. to Rg) observed over the measured RNA concentration range. Kratky plots, which qualitatively describe the extent of folding of macromolecules (Putnam et al, 2007), indicate that the c-di-AMP riboswitch is partially unfolded in the absence of c-di-AMP. The Kratky plot of the free RNA exhibits two maxima at q = 0.07 Å and 0.11 Å (Fig 6A), suggestive of two distinct domains connected by a flexible linker, possibly the two three-helix domains. Upon addition of c-di-AMP, the Kratky plot changes markedly, exhibits a single maximum, and is consistent with a well-folded, compact RNA. This pronounced ligand-binding-induced change is also apparent in the pair-distance distribution functions [P(r)] (Fig 6B). The P(r) for the ligand-bound riboswitch, characterized by a Dmax of 99.5 Å, is indicative of a more compact RNA than that for the ligand-free riboswitch (Table 3, Dmax = 116 Å), consistent with our co-crystal structure in which the two bound c-di-AMP molecules bridge the two halves of the pseudo-symmetric RNA. Taken together, our SAXS analyses indicate that cognate ligand binding to the minimal functional riboswitch domain (as defined by our mutagenesis, calorimetry, and co-crystal structure) induces a global folding transition, a phenomenon observed previously in other riboswitches, including the c-di-GMP-I riboswitch (Kulshina et al, 2009; Baird & Ferré-D'Amaré, 2010; Baird et al, 2010; Wood et al, 2012; Zhang et al, 2014).
Figure 6. SAXS analysis of c-di-AMP riboswitch.
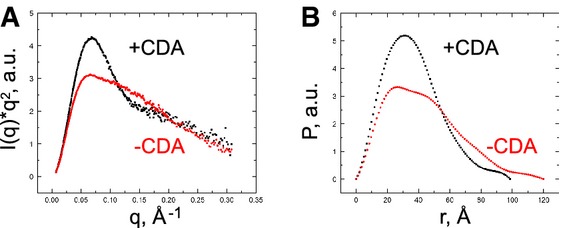
- Kratky plot of c-di-AMP riboswitch in the absence (red) and presence (black) of c-di-AMP. a.u., arbitrary units.
- Pair-distance distribution in the absence (red) and presence (black) of c-di-AMP.
Table 3.
SAXS analysis of the c-di-AMP riboswitch
| Rg (Å) | Dmax (Å) | VP (Å3) | |
|---|---|---|---|
| Riboswitch | 33.7 ± 0.61 | 116 ± 3.2 | 59,300 ± 2,800 |
| Riboswitch + c-di-AMP | 28.6 ± 0.16 | 99.5 ± 0.94 | 60,900 ± 890 |
Discussion
In this study, we define the functional core of the c-di-AMP riboswitch and lay bare the molecular basis for its exquisite specificity for c-di-AMP over other cyclic dinucleotides. Our findings are fully consistent with the pattern of phylogenetic sequence conservation and the published in-line probing data for this riboswitch (Nelson et al, 2013). The nucleotides most highly conserved across members of this riboswitch class (Supplementary Fig S8A) line the intramolecular trough that accommodates the two c-di-AMP molecules (Fig 2A). Highly conserved nucleotides include most of P4 as well as the apices of the NAA(A) tetraloop-like motifs that directly interact with the bound second messengers, reminiscent of but distinct from the three-helix junctions present in purine riboswitches (Lescoute & Westhof, 2005). RNA–RNA interfacial interactions between the two halves of the riboswitch bury 350 Å2 of solvent-accessible surface, compared to the approximately 625 Å2 buried by the two bound c-di-AMP molecules (Fig 2). Thus, the ligands contribute the majority of the binding interactions that bridge the two domains. Our structure also agrees with the pattern of reduction of in-line cleavage resulting from ligand binding (Supplementary Fig S8B). The residues that exhibit the largest reductions in scission are in P4, which binds A1 directly through G60 and C95 (Fig 4A) and also plays a role in positioning A65 for stacking on A2α (Fig 4B). Not surprisingly, peripheral nucleotides, such as those in the loops that cap the distal ends of P2 and P5, do not exhibit a change in in-line cleavage susceptibility in response to c-di-AMP binding (Supplementary Fig S8B).
As expected, mutation of residues in contact with c-di-AMP generally results in a loss of binding. Despite observing 1:1 binding in all B. subtilis-derived RNAs except for A9U, the two binding sites appear to be linked as mutation to either site in most cases eliminates binding under our conditions. This may be partly explained by the extensive stacking interactions and domain–domain interactions connecting the two binding sites. For example, A100 is located at the hinge connecting the P1–P3 riboswitch subdomain to the P4–P6 subdomain and is conserved across riboswitch species. Both A9 and A64 participate in tetraloop-like interactions at the centers of three-helix junctions and partly tolerate U at this position. Mutations to the RNA for the purposes of crystallization may have disrupted site A2, which lacks a stacking nucleotide provided by the pseudoknot. However, we hypothesize that the weaker site was still occupied in the crystal due to the high concentrations (400 μM) of c-di-AMP present in crystallization conditions. Both the wild-type B. subtilis and T. tengcongensis RNAs were poorly folded as judged by SEC (Supplementary Figs S6 and S7), which may be due to the lack of proper pseudoknot folding in vitro. Although the pseudoknot was not essential for ligand binding, the base pairing of this sequence may dictate the preference for pairing of a downstream region containing a previously identified intrinsic termination hairpin (Nelson et al, 2013). During transcription, pseudoknot formation occurs last, as it bridges the 5′ and 3′ ends of the riboswitch. Thus, c-di-AMP binding may facilitate pseudoknot formation during transcription and actively determine the genetic output via this structural folding pathway.
Riboswitch recognition of cyclic dinucleotides
The recognition mechanism employed by the c-di-AMP riboswitch is distinctly different from those of the class I and II c-di-GMP riboswitches (Kulshina et al, 2009; Smith et al, 2009, 2011). Both riboswitch-bound c-di-AMP molecules adopt an extended conformation in which the Watson–Crick faces of the nucleobases point in opposite directions. In contrast, when bound to either of its cognate riboswitches, c-di-GMP is in a C-shaped conformation in which the two nucleobases point in the same direction and sandwich an interdigitated adenine nucleobase of the riboswitch (A47 and A70, respectively, for c-di-GMP-I and c-di-GMP-II). Moreover, in both c-di-GMP riboswitches, the bound nucleotide and interdigitated adenine form part of a continuous helical stack (Fig 7A and B). In the case of the c-di-AMP riboswitch, both of the purine bases of the bound cyclic dinucleotide stack on purine nucleobases contributed by the RNA (A9 and A100 for c-di-AMP A1, Fig 7C), but because of its extended conformation, the bound ligand is not an integral part of a single continuous riboswitch helical stack.
Figure 7. Comparison of RNAs that recognize cyclic dinucleotides.
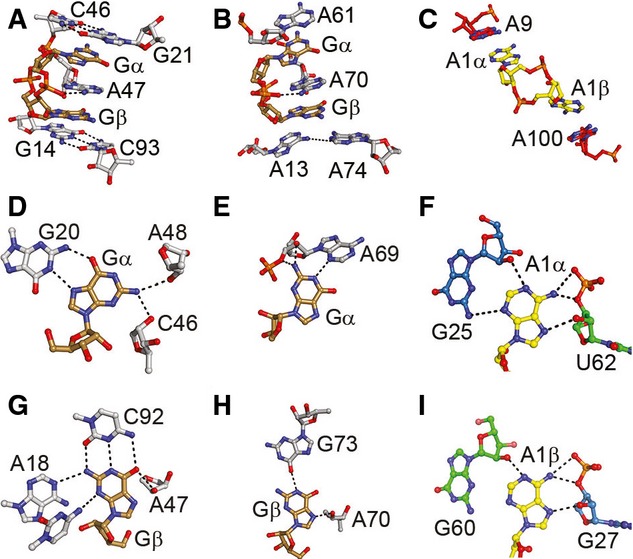
- Binding site of the class I c-di-GMP riboswitch (Smith et al, 2009). the c-di-GMP ligand (brown) surrounds the interdigitated A46 and is sandwiched between the G21–C46 and G14–C93 pairs.
- Binding site of the class II c-di-GMP riboswitch (Smith et al, 2011). c-di-GMP (brown) is surrounded by A61 and the A13–A70 base pair with A70 pairing between Gα and Gβ.
- C-di-AMP riboswitch binding site presented herein. c-di-AMP A1 (yellow) stacks with A9 and A100 via A1α and A1β, respectively.
- Hydrogen bonds between Gα and nearby residues in the class I c-di-GMP riboswitch.
- Interactions between Gα and A69 in the class II C-di-GMP riboswitch.
- C-di-AMP riboswitch A1α interactions with the G25 sugar edge of helix P3 and U62 bridging phosphate, 3′-OH, and 2′-OH helix P4.
- Hydrogen bonds between Gβ and surrounding residues in the class I c-di-GMP riboswitch.
- Two hydrogen bonds, one each from G73 and sugar A70, interact with Gβ in the c-di-GMP class II riboswitch.
- C-di-AMP riboswitch A1β interactions with the G60 sugar edge of helix P4 and G27 U62 bridging phosphate, 3′-OH, and 2′-OH helix P3.
The c-di-GMP-I riboswitch relies on canonical and non-canonical base pairing for specificity (Kulshina et al, 2009; Smith et al, 2009). Thus, Gβ makes a conventional base pair with C92, while Gα is recognized through a Hoogsteen pair with G20 (Fig 7D and G). The c-di-GMP-II riboswitch is less reliant on base pairing for specificity (Fig 7E and H) (Smith & Strobel, 2011). In addition to extensive interactions with the sugar–phosphate backbone of the cyclic dinucleotides (Fig 4), the c-di-AMP riboswitch employs the minor groove edge helical elements to contact the bases of its cognate ligand. Thus, A1α makes A-minor-type interactions with G25, and A1β makes A-minor-type interactions with G60 (Fig 7F and I). These interactions do not strictly specify A over G, as the 2′-OH of G25/G60 can act as either a hydrogen bond donor or acceptor to A or G, respectively. Thus, the specificity of the c-di-AMP riboswitch is likely to arise at least in part from steric exclusion of the latter (Fig 5). Interestingly, the other source of specificity for A arises from interaction between N3 of each A with a nearby 3′-OH and bridging phosphate oxygen, interactions which would be absent in the case of G. The A-minor interactions of both c-di-AMP molecules are mirrored by A-minor interactions between the symmetry-related A residues A9 and A64. These residues are the focal points of the communication between the three-helix junctions and the ligands as they stack upon A1α and A2α, respectively, while interacting with helices P2 and P5, respectively (Fig 3).
Cyclic dinucleotide recognition by proteins and RNA
As a result of the intrinsic twofold rotational symmetry of cyclic dinucleotides, the local trajectory of the dinucleotide phosphodiester backbone is inverted around their two constituent purine residues. To accommodate this local inversion, c-di-GMP-I and c-di-GMP-II riboswitch structures incorporate a corresponding local inversion in the direction of their RNA chains such that a single nucleotide of each riboswitch has a backbone trajectory that is opposite that of the nucleotides that precede and follow it in the polynucleotide chain, but becomes antiparallel to one of the two residues of the bound c-di-GMP (Kulshina et al, 2009; Smith et al, 2009, 2011). Instead of resorting to such local chain inversions, the c-di-AMP riboswitch accommodates the twofold symmetry of its cognate ligand by being itself globally twofold symmetric (Fig 1). By rotating an entire three-helical subdomain 180° relative to the other, the c-di-AMP riboswitch can employ residues oriented in opposed directions to recognize each of the two oppositely oriented nucleobases of a bound cyclic dinucleotide.
Although the c-di-AMP riboswitch is the first known example of a twofold symmetric RNA recognizing a twofold symmetric ligand, this is a common structural solution among proteins that bind, synthesize, or hydrolyze cyclic dinucleotides, which achieve such symmetry by virtue of dimeric or higher-order quaternary structures (Fig 8A) (Witte et al, 2008; Schirmer & Jenal, 2009). The co-crystal structures of the STING protein bound to c-di-GMP (Shang et al, 2012; Shu et al, 2012; Yin et al, 2012) reveal an extended dinucleotide conformation in which each nucleobase stacks on a tyrosine residue contributed by either subunit of the dimeric protein (Fig 8A). The tyrosines are functionally equivalent to the adenosines A9, A64, and A100 of the c-di-AMP riboswitch that stack on the bases of its cognate second messenger. A second example of an extended cyclic nucleotide conformation is provided by c-di-GMP phosphodiesterases (Barends et al, 2009; Sundriyal et al, 2014). As seen in the co-crystal structure of the phosphodiesterase YahA (Sundriyal et al, 2014), a monomeric enzyme which hydrolyzes c-di-GMP, an extended c-di-GMP ligand is bound asymmetrically via a tyrosine stacking with one of the guanine nucleobases (Fig 8B).
Figure 8. Comparison with proteins that bind cyclic dinucleotides.
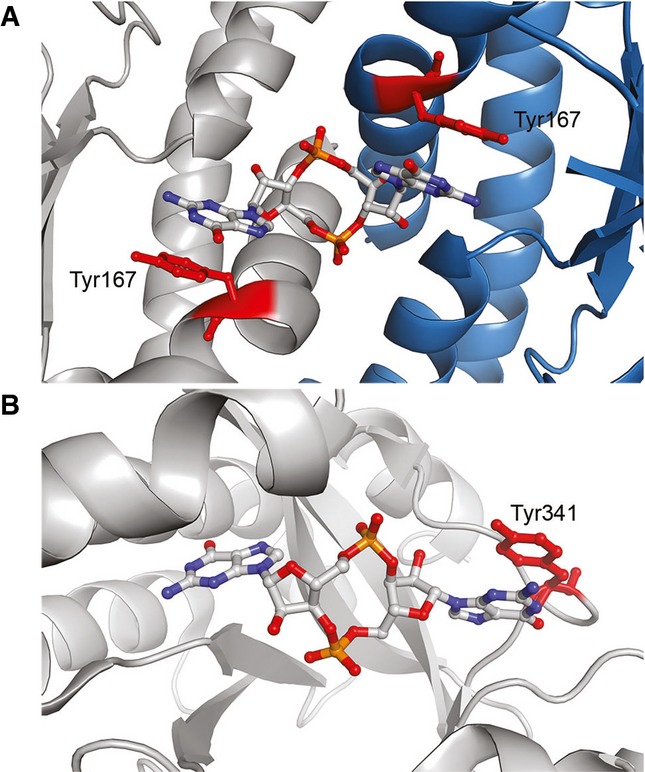
- Human STING binds c-di-GMP (Shu et al, 2012) flanking the cyclic nucleotide the Tyr167 residues (red) from each protomer of the dimeric protein (the two protomers are in tray and blue, respectively).
- The Escherichia coli YahA c-di-GMP phosphodiesterase (Sundriyal et al, 2014) binds its ligand in an extended dinucleotide conformation, in which one nucleobase contacts Tyr341 (red).
Internal symmetry and pseudo-quaternary RNA structure
Unlike proteins, which commonly associate into oligomeric structures with diverse quaternary symmetry (reviewed in Brändén & Tooze, 2009), very few biological RNA molecules are known to function as higher-order oligomers (Guo et al, 1998; Mujeeb et al, 1998). Rather than exhibiting true quaternary structure, the c-di-AMP riboswitch folds thorough symmetric association two self-similar domains that reside in a single polynucleotide chain. Nonetheless, this RNA is functionally a symmetric dimer whose ability to recognize c-di-AMP requires specific association of its two three-helix-junction domains, each of which constitutes one half of the ligand-binding trough (Fig 2). Although our co-crystal structure provides the first example of a globally twofold symmetric RNA recognizing a twofold symmetric small molecule, previous structural analyses of the glycine and FMN riboswitches have documented recognition of asymmetric ligands by RNAs with pseudo-quaternary structures. The glycine riboswitch is comprised of two domains of closely related structure that are connected through a structured linker (Mandal et al, 2004; Huang et al, 2010; Butler et al, 2011; Baird & Ferré-D'Amaré, 2013). Each of the modules contains a functional glycine binding site, but their association through a specific inter-domain interface is required for efficient glycine binding and genetic control. Rather than being a functional dimer that binds to two equivalents of its cognate ligand, like the c-di-AMP or glycine riboswitches, the FMN riboswitch is structurally pseudo-symmetric but recognizes a single molecule of FMN through asymmetric interactions (Serganov et al, 2009). Proteins exploit the symmetry of quaternary association for recognizing symmetric ligands (Fig 8), but also to achieve sophisticated biochemical functions, such as allostery and cooperativity (reviewed in Perutz, 1990). Our SAXS analyses indicate that in the case of the c-di-AMP riboswitch, folding and ligand recognition are tightly coupled. Since the stability of the ligand-bound aptamer domain determines the outcome of its genetic control decision (Nelson et al, 2013), the symmetric structure of the c-di-AMP riboswitch exemplifies how RNA quaternary structure formation gives rise to an adaptive biological response.
Materials and Methods
RNA and protein constructs
All RNAs were prepared by in vitro transcription (Xiao et al, 2008), from templates generated by PCR from plasmids encoding the B. subtilis QB928 ydaO-yuaA cDNA (synthesized by Integrated DNA Technologies). The wild-type B. subtilis plasmid pydaO used herein is identical to GenBank CP003783.1, nts 486,110–486,233, except for the C47G and G53C (numbering hereafter as in Fig 1A) mutations that reverse a C-G pair (Supplementary Fig S4A). The T. tengcongensis sequence is identical to GenBank NC_003869.1, nts 127,258–127,392. Additionally, all plasmids encoded a hammerhead ribozyme at the 5′ end of the riboswitch sequence to yield homogeneous 5′ RNA termini upon transcription with T7 RNA polymerase (Ferré-D'Amaré & Doudna, 1996). To reduce 3′ heterogeneity, 3′ PCR primers incorporated two 2′-O-methyl nucleotides at their 5′ end (Kao et al, 1999) complementary to the 3′ end of the template strand. RNAs were purified by denaturing polyacrylamide gel electrophoresis. Site-directed mutagenesis was performed using the QuikChange kit (Agilent). Selenomethionyl U1A protein RNA binding domain (Y31H, Q36R; SeMet-U1A) was prepared as previously described (Ferré-D'Amaré & Doudna, 2000). Prior to use, RNA was brought up in a buffer containing 50 mM HEPES–KOH pH 7.4, 150 mM KCl, heated to 95°C for 2 min, MgCl2 was added to 10 mM final concentration, and the solution immediately placed on ice and incubated for at least 30 min. C-di-AMP, cyclic-AMP-GMP, and cyclic diinosine monophosphate were purchased from Invivogen or Biolog and used without further purification.
Crystallization and diffraction data collection
For crystallization by the vapor diffusion method, 1 μl of RNA complexed with SeMet-U1A and c-di-AMP (200 μM RNA, 50 mM HEPES–KOH pH 7.4, 150 mM KCl, 400 μM c-di-AMP, 300 μM SeMet-U1A) was mixed with an equal volume of reservoir solution [25% (w/v) PEG 3350, 5% (v/v) ethylene glycol, 0.1 M BisTris–HCl pH 5.2] and equilibrated with 500 μl of the reservoir at 21°C. Plate-shaped co-crystals appeared within 1 week and grew to maximum dimensions of 400 × 200 × 30 μm3 in a month. Prior to harvesting, 1 μl of a solution containing 25% (w/v) PEG 3350, 5% (v/v) ethylene glycol, 0.1 M BisTris–HCl pH 5.2, 10 mM MgCl2, 150 mM KCl, 400 μM c-di-AMP was added directly to the drop. Crystals were mounted in nylon loops and flash-cooled by plunging into liquid nitrogen. SAD data were collected using the inverse beam method from a single crystal at the selenium K edge at 100 K at beamline 5.0.2 of the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory. Data were integrated and scaled with the HKL package (Otwinowski & Minor, 1997). Of the approximately 1,000 crystals grown under these conditions that were examined by oscillation photography and synchrotron X-radiation, approximately two dozen diffracted to a resolution higher than 4 Å, and of those, only a few diffracted beyond 3.5 Å.
Structure determination and refinement
Three of four possible selenium sites were identified using HySS (Grosse-Kunstleve & Adams, 2003) in Phenix (Adams et al, 2010) and SHELX (Schneider & Sheldrick, 2002), using Dataset 1 (Table 1). Experimental phases were calculated using PHASER (McCoy et al, 2007) and density modified using RESOLVE (Terwilliger, 2000). Model building commenced by placement of the known structure of U1A bound to an RNA hairpin (Oubridge et al, 1994), a GAAA tetraloop, and two 5-base pair duplexes using PHASER. Rounds of manual model building in Coot (Emsley & Cowtan, 2004) interspersed with rigid body, restrained individual atomic B-factor, simulated annealing and TLS refinement in Phenix using experimental phase restraints yielded the current Dataset 1 crystallographic model. C-di-AMP molecules were not placed into the |Fo|-|Fc| residual electron density until the model had an Rfree-factor of 0.347. A version of that model, lacking ligands and ions, was placed into Dataset 2 amplitudes, which provided better maps for ligand placement (Supplementary Fig S2) and allowed refinement to an Rfree-factor of 0.286. The current models have cross-validated σA coordinate precisions of 0.50 Å. Structure figures were prepared using PyMOL (Schrödinger, LLC). Solvent-accessible surface area calculations were performed using StrucTools provided by Helix Systems at the National Institutes of Health, Bethesda, MD (http://helix.nih.gov).
Isothermal titration calorimetry
All ITC measurements were recorded with a MicroCal iTC200 microcalorimeter (GE). Typically, 5–10 μM RNA in the cell was titrated with 50–100 μM c-di-AMP in the syringe. Concentrations as high as 60 μM RNA in the cell and 1 mM c-di-AMP in the syringe were used for weaker binding constructs. For all experiments, RNA was folded as described above, and the cell and syringe contained matching buffers (50 mM HEPES–KOH pH 7.4, 150 mM KCl, and 10 mM MgCl2). Data were analyzed and fit using NITPIC and SEDPHAT (Schuck, 2000; Keller et al, 2012). For T. tengcongensis ydaO, the RNA concentration was normalized using the relative population of RNA corresponding the monomeric peak as judged by SEC with identical samples in the same buffer (Supplementary Fig S6B).
Small-angle X-ray scattering
For SAXS experiments, approximately 200 μg RNA samples (crystal construct) were folded as described above and purified by size-exclusion chromatography using a Superdex 200 column (GE). The column was equilibrated with a buffer identical to the ITC buffer above, and fractions containing the monomeric RNA peak were concentrated in Amicon centrifugal concentrators and stored at 4°C. Analysis of the purified RNA by native gel electrophoresis revealed a single oligomeric state (not shown). Analytical SEC, as shown in Supplementary Fig S6, was performed similarly, except with injections of 100 μl of 17.5 μM RNA. Samples (˜25 μM RNA) and matching SEC buffers were shipped overnight at 4°C to beamline 12-ID-C of the Advanced Photon Source (APS), Argonne National Laboratory, where SAXS data were collected as described (Baird & Ferré-D'Amaré, 2013) for RNA samples in the presence and absence of 100 μM c-di-AMP. Scattering curves were buffer-subtracted, truncated to 0.3 Å q, and averaged for three RNA concentrations (˜1, 0.5, and 0.25 g/l). Data were analyzed in PRIMUS (Konarev et al, 2003) to calculate Kratky and Guinier plots and estimate radius of gyration (Rg) and Porod volume (VP).
Accession codes
Atomic coordinates and structure factor amplitudes for the c-di-AMP riboswitch bound to c-di-AMP have been deposited with the Protein Data Bank under accession codes 4W92 and 4W90.
Acknowledgments
We thank the staff at beamlines 5.0.1, 5.0.2, and 8.2.1 of the ALS for crystallographic data collection, G. Piszczek (National Heart, Lung and Blood Institute, NHLBI) for ITC support, Y.-X. Wang and X. Fang (National Cancer Institute) and the staff of APS beamline 12-ID-C for SAXS support, N. Baird, K. Collins, M. Lau, A. Roll-Mecak, K. Warner, and J. Zhang for discussions. This work was partly conducted at the ALS on the BCSB beamlines which are supported by the National Institutes of Health. Use of ALS and APS and was supported by the U.S. Department of Energy. This work was supported in part by the intramural program of the NHLBI, NIH.
Author contributions
CPJ and ARF designed experiments, CPJ carried out all biochemistry, crystallography, and biophysics, and both authors wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Yang J, Eisele LE, Underwood AJ, Koestler BJ, Waters CM, Metzger DW, Bai G. Two DHH subfamily 1 proteins in Streptococcus pneumoniae possess cyclic di-AMP phosphodiesterase activity and affect bacterial growth and virulence. J Bacteriol. 2013;195:5123–5132. doi: 10.1128/JB.00769-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Yang J, Zarrella TM, Zhang Y, Metzger DW, Bai G. Cyclic di-AMP impairs potassium uptake mediated by a cyclic di-AMP binding protein in Streptococcus pneumoniae. J Bacteriol. 2014;196:614–623. doi: 10.1128/JB.01041-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NJ, Ferré-D'Amaré AR. Idiosyncratically tuned switching behavior of riboswitch aptamer domains revealed by comparative small-angle X-ray scattering analysis. RNA. 2010;16:598–609. doi: 10.1261/rna.1852310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NJ, Kulshina N, Ferré-D'Amaré AR. Riboswitch function: flipping the switch or tuning the dimmer? RNA Biol. 2010;7:328–332. doi: 10.4161/rna.7.3.11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NJ, Ferré-D'Amaré AR. Modulation of quaternary structure and enhancement of ligand binding by the K-turn of tandem glycine riboswitches. RNA. 2013;19:167–176. doi: 10.1261/rna.036269.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barends TR, Hartmann E, Griese JJ, Beitlich T, Kirienko NV, Ryjenkov DA, Reinstein J, Shoeman RL, Gomelsky M, Schlichting I. Structure and mechanism of a bacterial light-regulated cyclic nucleotide phosphodiesterase. Nature. 2009;459:1015–1018. doi: 10.1038/nature07966. [DOI] [PubMed] [Google Scholar]
- Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, Collins J, Lee M, Roth A, Sudarsan N, Jona I, Wickiser JK, Breaker RR. New RNA motifs suggest an expanded scope for riboswitches in bacterial genetic control. Proc Natl Acad Sci USA. 2004;101:6421–6426. doi: 10.1073/pnas.0308014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block KF, Hammond MC, Breaker RR. Evidence for widespread gene control function by the ydaO riboswitch candidate. J Bacteriol. 2010;192:3983–3989. doi: 10.1128/JB.00450-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd CD, O'Toole GA. Second messenger regulation of biofilm formation: breakthroughs in understanding c-di-GMP effector systems. Annu Rev Cell Devel Biol. 2012;28:439–462. doi: 10.1146/annurev-cellbio-101011-155705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brändén C-I, Tooze J. Introduction to Protein Structure. 2nd edn. New York, NY: Garland Publishers; 2009. [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nature Immunol. 2013;14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- Butler EB, Xiong Y, Wang J, Strobel SA. Structural basis of cooperative ligand binding by the glycine riboswitch. Chem Biol. 2011;18:293–298. doi: 10.1016/j.chembiol.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. 2014;54:289–296. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Kundrot CE, Cech TR, Doudna JA. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- Corrigan RM, Campeotto I, Jeganathan T, Roelofs KG, Lee VT, Grundling A. Systematic identification of conserved bacterial c-di-AMP receptor proteins. Proc Natl Acad Sci USA. 2013;110:9084–9089. doi: 10.1073/pnas.1300595110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan RM, Grundling A. Cyclic di-AMP: another second messenger enters the fray. Nat Rev Microbiol. 2013;11:513–524. doi: 10.1038/nrmicro3069. [DOI] [PubMed] [Google Scholar]
- Deigan KE, Ferré-D'Amaré AR. Riboswitches: discovery of drugs that target bacterial gene-regulatory RNAs. Acc Chem Res. 2011;44:1329–1338. doi: 10.1021/ar200039b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crstallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ferré-D'Amaré AR, Doudna JA. Use of cis- and trans-ribozymes to remove 5′ and 3′ heterogeneities from milligrams of in vitro transcribed RNA. Nucleic Acid Res. 1996;24:977–978. doi: 10.1093/nar/24.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré-D'Amaré AR, Doudna JA. Crystallization and structure determination of a hepatitis delta virus ribozyme: use of the RNA-binding protein U1A as a crystallization module. J Mol Biol. 2000;295:541–556. doi: 10.1006/jmbi.1999.3398. [DOI] [PubMed] [Google Scholar]
- Ferré-D'Amaré AR. Use of the spliceosomal protein U1A to facilitate crystallization and structure determination of complex RNAs. Methods. 2010;52:159–167. doi: 10.1016/j.ymeth.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao A, Serganov A. Structural insights into recognition of c-di-AMP by the ydaO riboswitch. Nat Chem Biol. 2014;10:787–792. doi: 10.1038/nchembio.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary C, Chworos A, Jaeger L. Promoting RNA helical stacking via A-minor junctions. Nucleic Acids Res. 2011;39:1066–1080. doi: 10.1093/nar/gkq748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse-Kunstleve RW, Adams PD. Substructure search procedures for macromolecular structures. Acta Crystallogr D. 2003;59:1966–1973. doi: 10.1107/s0907444903018043. [DOI] [PubMed] [Google Scholar]
- Guo P, Zhang C, Chen C, Garver K, Trottier M. Inter-RNA interaction of phage phi29 pRNA to form a hexameric complex for viral DNA transportation. Mol Cell. 1998;2:149–155. doi: 10.1016/s1097-2765(00)80124-0. [DOI] [PubMed] [Google Scholar]
- Huang L, Serganov A, Patel DJ. Structural insights into ligand recognition by a sensing domain of the cooperative glycine riboswitch. Mol Cell. 2010;40:774–786. doi: 10.1016/j.molcel.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Kumar RA, Jones RA, Patel DJ. Structural basis of RNA folding and recognition in an AMP-RNA aptamer complex. Nature. 1996;382:183–186. doi: 10.1038/382183a0. [DOI] [PubMed] [Google Scholar]
- Kalia D, Merey G, Nakayama S, Zheng Y, Zhou J, Luo Y, Guo M, Roembke BT, Sintim HO. Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP, (p)ppGpp signaling in bacteria and implications in pathogenesis. Chem Soc Rev. 2013;42:305–341. doi: 10.1039/c2cs35206k. [DOI] [PubMed] [Google Scholar]
- Kao C, Zheng M, Rudisser S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. RNA. 1999;5:1268–1272. doi: 10.1017/s1355838299991033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S, Vargas C, Zhao H, Piszczek G, Brautigam CA, Schuck P. High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal Chem. 2012;84:5066–5073. doi: 10.1021/ac3007522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a windows PC-based system for small-angle scattering data analysis. J Appl Cryst. 2003;36:1277–1282. [Google Scholar]
- Kulshina N, Baird NJ, Ferré-D'Amaré AR. Recognition of the bacterial second messenger cyclic diguanylate by its cognate riboswitch. Nat Struct Mol Biol. 2009;16:1212–1217. doi: 10.1038/nsmb.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ER, Baker JL, Weinberg Z, Sudarsan N, Breaker RR. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science. 2010;329:845–848. doi: 10.1126/science.1190713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontis NB, Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA. 2001;7:499–512. doi: 10.1017/s1355838201002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescoute A, Westhof E. Riboswitch structures: purine ligands replace tertiary contacts. Chem Biol. 2005;12:10–13. doi: 10.1016/j.chembiol.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Lescoute A, Westhof E. Topology of three-way junctions in folded RNAs. RNA. 2006;12:83–93. doi: 10.1261/rna.2208106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Helmann JD. Analysis of the role of Bacillus subtilis sigma(M) in beta-lactam resistance reveals an essential role for c-di-AMP in peptidoglycan homeostasis. Mol Microbiol. 2012;83:623–639. doi: 10.1111/j.1365-2958.2011.07953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, Breaker RR. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–279. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehne FM, Gunka K, Eilers H, Herzberg C, Kaever V, Stulke J. Cyclic di-AMP homeostasis in Bacillus subtilis: both lack and high level accumulation of the nucleotide are detrimental for cell growth. J Biol Chem. 2013;288:2004–2017. doi: 10.1074/jbc.M112.395491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujeeb A, Clever JL, Billeci TM, James TL, Parslow TG. Structure of the dimer initiation complex of HIV-1 genomic RNA. Nat Struct Biol. 1998;5:432–436. doi: 10.1038/nsb0698-432. [DOI] [PubMed] [Google Scholar]
- Nelson JW, Sudarsan N, Furukawa K, Weinberg Z, Wang JX, Breaker RR. Riboswitches in eubacteria sense the second messenger c-di-AMP. Nat Chem Biol. 2013;9:834–839. doi: 10.1038/nchembio.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc Natl Acad Sci USA. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Crystal structure at 1.92 Å resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- Perutz MF. Mechanisms of Cooperativity and Allosteric Regulation in Proteins. Cambridge UK, New York: Cambridge University Press; 1990. [DOI] [PubMed] [Google Scholar]
- Peselis A, Serganov A. Themes and variations in riboswitch structure and function. Biochim Biophys Acta. 2014;1839:908–918. doi: 10.1016/j.bbagrm.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Ren A, Patel DJ. c-di-AMP binds the ydaO riboswitch in two pseudo-symmetry-related pockets. Nat Chem Biol. 2014;10:780–786. doi: 10.1038/nchembio.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer T, Jenal U. Structural and mechanistic determinants of c-di-GMP signalling. Nat Rev Microbiol. 2009;7:724–735. doi: 10.1038/nrmicro2203. [DOI] [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganov A, Huang L, Patel DJ. Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature. 2009;458:233–237. doi: 10.1038/nature07642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang G, Zhu D, Li N, Zhang J, Zhu C, Lu D, Liu C, Yu Q, Zhao Y, Xu S, Gu L. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat Struct Mol Biol. 2012;19:725–727. doi: 10.1038/nsmb.2332. [DOI] [PubMed] [Google Scholar]
- Shu C, Yi G, Watts T, Kao CC, Li P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat Struct Mol Biol. 2012;19:722–724. doi: 10.1038/nsmb.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KD, Lipchock SV, Ames TD, Wang J, Breaker RR, Strobel SA. Structural basis of ligand binding by a c-di-GMP riboswitch. Nat Struct Mol Biol. 2009;16:1218–1223. doi: 10.1038/nsmb.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KD, Shanahan CA, Moore EL, Simon AC, Strobel SA. Structural basis of differential ligand recognition by two classes of bis-(3′-5′)-cyclic dimeric guanosine monophosphate-binding riboswitches. Proc Natl Acad Sci USA. 2011;108:7757–7762. doi: 10.1073/pnas.1018857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KD, Strobel SA. Interactions of the c-di-GMP riboswitch with its second messenger ligand. Biochem Soc Trans. 2011;39:647–651. doi: 10.1042/BST0390647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, Breaker RR. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science. 2008;321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundriyal A, Massa C, Samoray D, Zehender F, Sharpe T, Jenal U, Schirmer T. Inherent regulation of EAL domain-catalyzed hydrolysis of second messenger cyclic di-GMP. J Biol Chem. 2014;289:6978–6990. doi: 10.1074/jbc.M113.516195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PY, Fedor MJ. The ydaO motif is an ATP-sensing riboswitch in Bacillus subtilis. Nat Chem Biol. 2012;8:963–965. doi: 10.1038/nchembio.1095. [DOI] [PubMed] [Google Scholar]
- Witte G, Hartung S, Buttner K, Hopfner KP. Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol Cell. 2008;30:167–178. doi: 10.1016/j.molcel.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Wood S, Ferré-D'Amaré AR, Rueda D. Allosteric tertiary interactions preorganize the c-di-GMP riboswitch and accelerate ligand binding. ACS Chem Biol. 2012;7:920–927. doi: 10.1021/cb300014u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Edwards TE, Ferré-D'Amaré AR. Structural basis for specific, high-affinity tetracycline binding by an in vitro evolved aptamer and artificial riboswitch. Chem Biol. 2008;15:1125–1137. doi: 10.1016/j.chembiol.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q, Tian Y, Kabaleeswaran V, Jiang X, Tu D, Eck MJ, Chen ZJ, Wu H. Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol Cell. 2012;46:735–745. doi: 10.1016/j.molcel.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Jones CP, Ferré-D'Amaré AR. Global analysis of riboswitches by small-angle X-ray scattering and calorimetry. Biochim Biophys Acta. 2014;1839:1020–1029. doi: 10.1016/j.bbagrm.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.