

Abstract
Faithful chromosome segregation during mitosis is tightly regulated by opposing activities of Aurora B kinase and protein phosphatase-1 (PP1). PP1 function at kinetochores has been linked to SDS22, but the exact localization of SDS22 and how it affects PP1 are controversial. Here, we confirm that SDS22 is required for PP1 activity, but show that SDS22 does not normally localize to kinetochores. Instead, SDS22 is kept in solution by formation of a ternary complex with PP1 and inhibitor-3 (I3). Depletion of I3 does not affect the amount of PP1 at kinetochores but causes quantitative association of SDS22 with PP1 on KNL1 at the kinetochore. Such accumulation of SDS22 at kinetochores interferes with PP1 activity and inhibits Aurora B threonine-232 dephosphorylation, which leads to increased Aurora B activity in metaphase and persistence in anaphase accompanied with segregation defects. We propose a model in which I3 regulates an SDS22-mediated PP1 activation step in solution that precedes SDS22 dissociation and transfer of PP1 to kinetochores, and which is required for PP1 to efficiently antagonize Aurora B.
Keywords: Aurora B, chromosome segregation, kinetochore, mitosis, protein phosphatase-1
Introduction
To achieve faithful chromosome segregation during cell division, the kinetochores of replicated chromatid pairs need to attach to opposite poles of the mitotic spindle. Bipolar attachment of spindle microtubules to kinetochores is monitored by the spindle assembly checkpoint and dynamically regulated by phosphorylation to allow correction of improper attachment and stabilization of correct attachments (Cheeseman et al, 2002; Andrews et al, 2004; Lampson et al, 2004; Lan et al, 2004; Ciferri et al, 2008; Welburn et al, 2010). Key elements of this regulation are the mitotic kinase Aurora B and protein phosphatase-1 (PP1), whose opposing activities need to be tightly balanced (Pinsky et al, 2006; Emanuele et al, 2008; Kim et al, 2010; Liu et al, 2010).
Aurora B is localized to the centromere via its partner proteins in the chromosomal passenger complex and controlled by histone H3 and H2A phosphorylation (Ruchaud et al, 2007; Kelly et al, 2010; Yamagishi et al, 2010). PP1 has multiple binding partners that determine its localization, specificity and activity in many different cellular pathways in interphase and mitosis (Heroes et al, 2013). During prometaphase and metaphase, PP1 (in mammals isoforms α, β and γ) localizes to kinetochores (Trinkle-Mulcahy et al, 2003; Trinkle-Mulcahy & Lamond, 2006; Posch et al, 2010; Meadows et al, 2011; Rosenberg et al, 2011), mostly through binding to the RVXF motif of the outer kinetochore protein KNL1 (Liu et al, 2010).
SDS22 (also called PPP1R7 or Sds22p in yeast) is another PP1-interacting protein implicated in Aurora B regulation at the kinetochore (Heroes et al, 2013). In human cells, SDS22 was shown to positively regulate PP1 as evidenced by increased Aurora B autophosphorylation at threonine-232 (T232) in the activation loop and unbalanced Aurora B activity during establishment of bipolar attachment upon cellular depletion of SDS22 (Posch et al, 2010). This function was explained by data showing that SDS22 localized to kinetochores and regulated recruitment of PP1 (Posch et al, 2010). A second study confirmed the link between SDS22 and PP1 in human cells and showed a requirement for SDS22 in stabilizing kinetochore-spindle attachment during anaphase (Wurzenberger et al, 2012). However, whether SDS22 localizes to kinetochores was drawn into question (Liu et al, 2010).
SDS22 interacts with PP1 through leucine-rich repeats (Ceulemans et al, 2002). Intriguingly, SDS22-PP1 forms a ternary complex with inhibitor-3 (I3, also called PPP1R11 or Ypi1 in yeast), which, like KNL1, binds PP1 via an RVXF motif (Garcia-Gimeno et al, 2003; Lesage et al, 2007; Pedelini et al, 2007). In vitro, binding of both SDS22 and I3 to PP1 inhibits PP1 phosphatase activity (Lesage et al, 2007). Consistently, overexpression of the orthologs Sds22p/SDS22 or Ypi1/I3 in yeast suppresses Ipl1/Aurora deficiency in Ipl1 mutants (Garcia-Gimeno et al, 2003; Pinsky et al, 2006; Pedelini et al, 2007), implying that they attenuate Glc7/PP1 activity. However, like in human cells, both factors are required for faithful chromosome segregation in yeast and positively regulate PP1 in order to antagonize Ipl1/Aurora (Peggie et al, 2002; Pedelini et al, 2007; Bharucha et al, 2008). Thus, despite the obvious importance of SDS22 (and potentially of I3) for chromosome segregation, their functional relationship to PP1, as well as their localization at the kinetochore, and regulation remain obscure.
In this study, we analyzed the mitotic role of I3 in an attempt to clarify the regulation and functional relationship of SDS22 to PP1 at the kinetochore. We establish that I3 is required for faithful bipolar chromosome attachment in human cells. However, I3 does not localize to kinetochores, nor does I3 or SDS22 control recruitment of PP1 to kinetochores. Instead, I3 limits association of SDS22 with KNL1-bound PP1 at the kinetochore that would otherwise inhibit PP1-mediated dephosphorylation of Aurora B. Because SDS22 inhibits PP1 while bound to PP1, yet SDS22 is also required for full PP1 activity, our data suggest that SDS22 in cooperation with I3 acts as a chaperone that activates PP1 in solution prior to recruitment to the kinetochore.
Results
Both PP1-interacting proteins, I3 and SDS22, are essential for proper chromosome alignment and timely progression through mitosis
To explore a possible role of I3 in bipolar spindle attachment in human cells and revisit the involvement of SDS22 in the process, we depleted both factors in HeLa cells using siRNA (Fig 1A). As a control, we depleted NIPP1 that, like I3, interacts with PP1 via an RVXF motif, but has no known mitotic functions (Nuytten et al, 2008). We first assessed chromosome alignment efficacy by light microscopy in unsynchronized cells that were fixed and DAPI-stained. Visual inspection revealed a significant increase of metaphase cells with misaligned chromosomes to almost 20% in SDS22 or I3 depleted cells compared to around 5% in control populations (Fig 1B). To ensure that this was not an off-target effect of the I3 siRNA, we generated a stable inducible HeLa cell line expressing an siRNA-resistant GFP-I3 variant. Indeed, expression of GFP-I3 significantly reduced misalignment caused by I3 siRNA treatment, while endogenous I3 was still efficiently depleted confirming that the effect was specific (Supplementary Fig S1A). We next monitored the timing of mitosis from nuclear envelope breakdown to anaphase onset by live-cell microscopy of HeLa cell populations expressing the markers histone H2B-RFP and importin-beta binding domain (IBB)-GFP (Supplementary Fig S1B). Consistent with the detected misalignment, we observed a significant delay of anaphase onset in fractions of I3- or SDS22-depleted cells compared to control-depleted cells, suggesting that the spindle assembly checkpoint was activated (Fig 1C and Supplementary Fig S1B). Again, NIPP1-depleted cells displayed no delay of anaphase onset.
Figure 1. Inhibitor-3 (I3) and SDS22 are required for proper chromosome alignment and progression through mitosis.

- Efficiency of siRNA-mediated depletion of indicated PP1-interacting proteins shown by Western blot. p97 was probed as a loading control. Luciferase siRNA (siLuc) was used as a negative control.
- Increased chromosome misalignment in SDS22- or I3-depleted HeLa cells. Cells were treated with indicated siRNAs, fixed and DAPI-stained. Depletion of the RVXF-motif protein NIPP1 served as a control. Representative confocal images. Scale bar, 5 μm. Percentage of metaphase cells with misaligned chromosomes was quantified. Error bars indicate s.d. of three independent experiments with 60 cells per condition. P-values were calculated using a Mann–Whitney U-test (***P < 0.001; **P ≤ 0.01; *P ≤ 0.05). See Supplementary Fig S1A for rescue experiments.
- SDS22 or I3 depletion causes delay of anaphase onset. The timing between nuclear envelope breakdown (NEBD) and anaphase onset (AO) was determined in live cells expressing H2B-RFP and IBB-GFP after treatment with indicated siRNAs (see also Supplementary Fig S1B for image sequences of example movies). NEBD was monitored as a nuclear efflux of IBB-GFP, and AO was monitored as the start of separation of the sister chromatids using H2B-RFP. Percentages of cells committing anaphase in the indicated time windows are plotted. n ≥ 133 cells per condition, summed up from three independent experiments.
Source data are available online for this figure.
Depletion of SDS22 and I3 affect activity, but not amount of PP1 at the kinetochore
To understand the basis for the alignment defects, we first analyzed Aurora B at the centromeres, which is negatively regulated by PP1 during establishment of bipolar spindle attachment. We again depleted SDS22 or I3, or NIPP1 as a control, and processed cells for immunofluorescence to monitor Aurora B protein amounts and threonine-232 phosphorylation (pT232), which represents the activated state of Aurora B (Fig 2A). The signal intensities of confocal images were quantified and statistically evaluated (Fig 2B and C). In agreement with a previous report (Posch et al, 2010), SDS22 depletion led to an increase of Aurora B pT232 compared to control-depleted cells (Fig 2A and B), confirming that SDS22 is required for full PP1 activity at kinetochores. We also observed an increase of Aurora B protein. This is consistent with a positive feedback on Aurora B recruitment, because Aurora B also inhibits the PP1-Repo-Man complex, which in turn leads to increased phospho-threonine-3 of histone H3 that constitutes a binding site of Aurora B at the centromere (Kelly et al, 2010; Qian et al, 2011, 2013). Nevertheless, the increase in pT232 exceeded the increase of Aurora B protein (Fig 2C). Importantly, also I3 depletion led to an increase of Aurora B phosphorylation at the centromere to a comparable degree (Fig 2A–C). To clarify whether this increase had any functional impact, we analyzed the phosphorylation status of Dsn1, an Aurora B substrate at the kinetochore (Welburn et al, 2010), with phospho-specific antibodies. Ser109 phosphorylation was indeed elevated in SDS22 and I3, compared to NIPP1 or control-depleted cells (Fig 2D and E). Increased Aurora B activity is expected to destabilize microtubule–kinetochore attachments. Consistent with this, we found increased recruitment of the spindle assembly checkpoint protein BubR1 to kinetochores at metaphase plates of SDS22 and I3-depleted cells, while Aurora B-depleted cells had largely reduced BubR1 signals even on misaligned chromosomes (Fig 2F and G). These data suggest that I3, like SDS22, acts as a positive regulator of PP1 with respect to Aurora B dephosphorylation and therefore as an antagonist of Aurora B function during spindle attachment.
Figure 2. Depletion of SDS22 and I3 affect PP1 activity at the kinetochore.
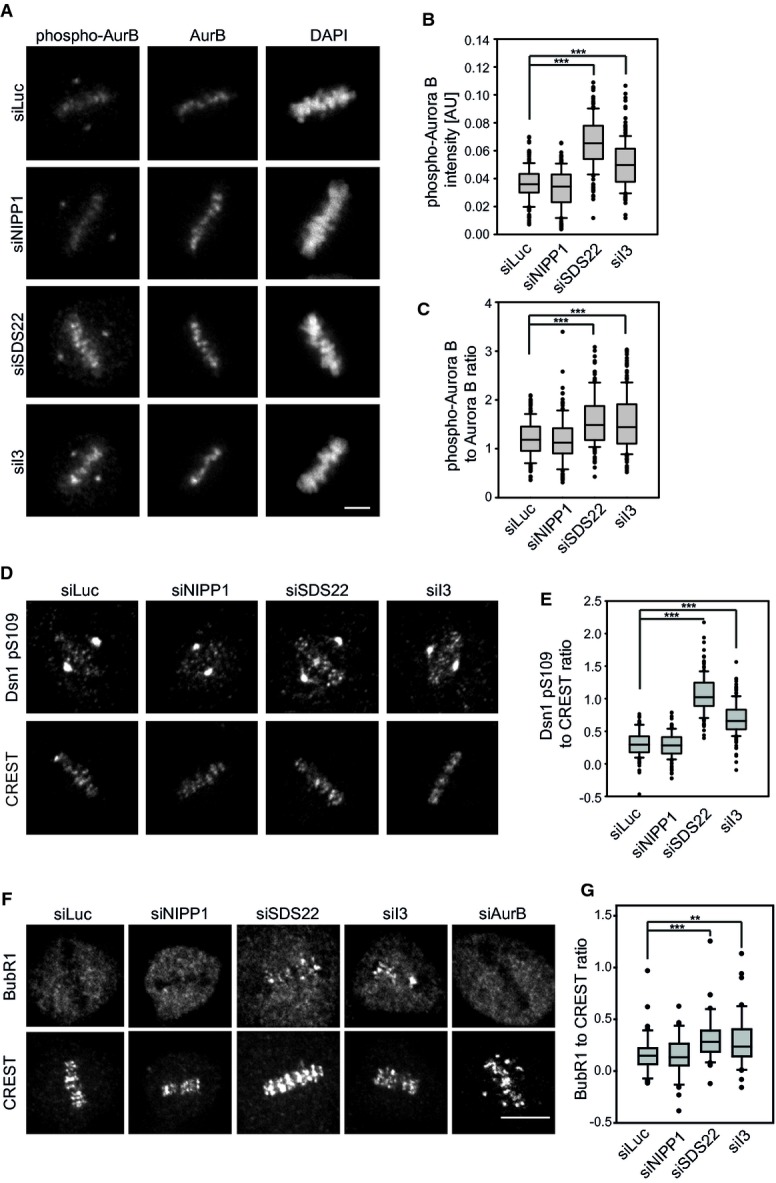
A Increased Aurora B autophosphorylation upon SDS22 or I3 depletion. Representative images of HeLa cells in metaphase depleted with indicated siRNAs. Cells were stained with Aurora B-specific (AurB), phospho-T232 (phospho-AurB) antibodies and DAPI. Note non-specific staining of the centrosome with pT232 antibodies. Scale bar, 5 μm.
B, C Quantification of phospho-AurB (B) or the ratio of phospho-AurB versus total AurB (C) signal intensity on chromatin. Box blots show median, lower and upper quartiles (line and box), 10th and 90th percentiles (whiskers) and outliers (•). ***P < 0.001. Data from three independent experiments with 50 cells per condition.
D Increased Dsn1 phosphorylation at S109 upon SDS22 or I3 depletion. Representative images of HeLa cells in metaphase treated with indicated siRNAs and stained with Dsn1 phospho-S109-specific antibodies and CREST serum. Note non-specific staining of the centrosome with S109 antibodies. Scale bar, 5 μm.
E Quantification of the ratio of Dsn1 pS109 versus CREST signal intensity at kinetochores. Box blots show median, lower and upper quartiles (line and box), 10th and 90th percentiles (whiskers) and outliers (•). ***P < 0.001. Data from three independent experiments with 50 cells per condition.
F Increased BubR1 recruitment at metaphase kinetochores upon SDS22 or I3 depletion. Representative images of HeLa cells in metaphase treated with indicated siRNAs and stained with BubRI and CREST antibodies. Also note for comparison the absence of BubR1 signal in AurB-depleted cells in spite of severe misalignment. Scale bar, 10 μm.
G Quantification of the ratio of BubR1 versus CREST signal intensity on kinetochores of aligned metaphase plates. Box blots show median, lower and upper quartiles (line and box), 10th and 90th percentiles (whiskers) and outliers (•). **P < 0.01, ***P < 0.001. Data from three independent experiments with a total of 42 cells per condition. AurB-depleted cells were not quantified due to absence of metaphase plates.
We next asked whether possible changes in PP1 amounts at the kinetochore may explain the reduced PP1 activity as evidenced by increased Aurora B phosphorylation upon SDS22 or I3 depletion. To visualize PP1, we analyzed previously described HeLa cells that express a GFP fusion of PP1γ at near endogenous levels [(Trinkle-Mulcahy et al, 2003) and see below]. However, unlike reported before (Posch et al, 2010), we did not find evidence for a loss of GFP-PP1γ localization from kinetochores by either SDS22 or I3 depletions compared to control or NIPP1 depletions (Fig 3A and B). In SDS22-depleted cells, we did observe a slight increase of PP1γ diffusely associated with the periphery of the metaphase plate, but not at the core region at or near the kinetochores.
Figure 3. Depletion of SDS22 and I3 does not affect the amount of PP1γ at the kinetochore.
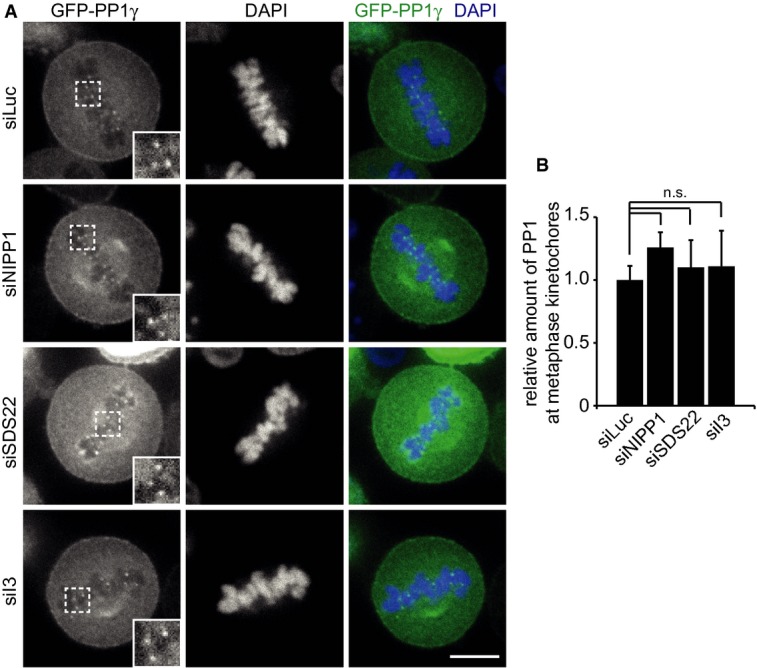
- PP1γ localization is not affected by SDS22 or I3 depletion. Stable GFP-PP1γ-expressing HeLa cells were depleted of indicated proteins, fixed and permeabilized, and DAPI-stained. Representative confocal images. Scale bar, 10 μm.
- Quantification of GFP-PP1γ intensity at kinetochores normalized to cytoplasmic signal intensity. The graph shows the mean and standard deviation of three independent experiments, in which the intensities of at least 3 kinetochores/cell in at least 10 metaphase cells per condition were measured. n.s., P > 0.05.
I3 limits association of SDS22 with the kinetochore, without itself localizing to kinetochores
Because PP1 localization to kinetochores was not affected by SDS22 or I3 depletions, we next asked whether the localization of SDS22 or I3 could explain the observed effect on PP1 activity. To monitor I3 localization, we generated stable HeLa cells that express a GFP-I3 fusion protein. Expression of GFP-I3 was above endogenous levels of I3 as assessed by Western blot analysis (Fig 4B). While GFP-I3 localized to nucleoli in interphase as previously reported (Huang et al, 2005) (Supplementary Fig S2), we could not detect any enrichment at kinetochores or centromeres at any stage of mitosis using confocal microscopy of live or fixed cells (Fig 4A, Supplementary Fig S2). To monitor SDS22 localization, we used a HeLa cell line stably transformed with a bacterial artificial chromosome coding for the whole SDS22 gene with an in-frame fusion to GFP (Poser et al, 2008). Expression levels of SDS22-GFP and endogenous SDS22 together corresponded to the endogenous level in parental cells (Fig 4B) and therefore allowed careful assessment of the distribution of SDS22. Unlike previously reported (Posch et al, 2010), we could not detect SDS22-GFP at kinetochores in live cells or after fixation using diverse methods in these cells (Fig 4A, data not shown). This suggests that SDS22-GFP does not quantitatively localize to kinetochores under control conditions when expressed at near endogenous levels.
Figure 4. Depletion of I3 induces re-localization of SDS22 to kinetochores.
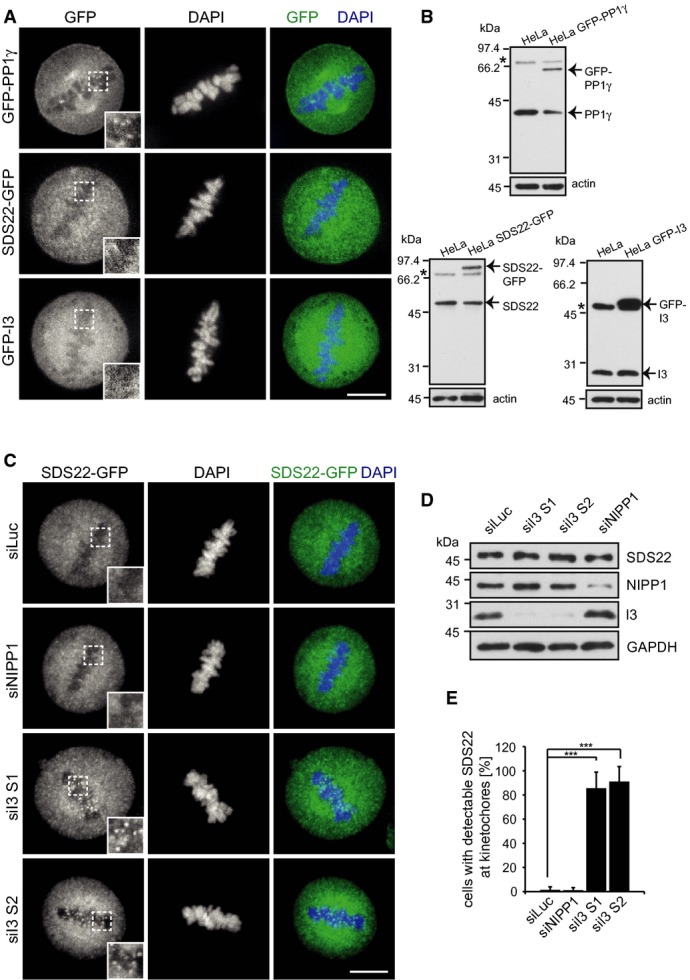
- Neither SDS22 nor I3 colocalize with PP1γ at metaphase kinetochores under control conditions. Stable HeLa cell lines expressing GFP fusions of indicated proteins were fixed and DAPI-stained. SDS22-GFP is expressed under its own promoter in BAC transgenic cells. For localization images of GFP-I3-expressing cells in all mitotic stages see also Supplementary Fig S2. Representative confocal images. Scale bar, 10 μm.
- Expression levels of GFP fusions relative to endogenous proteins in Western blots stained with specific antibodies. Actin served as a loading control. Asterisks denote non-specific bands.
- SDS22 re-localization upon I3 depletion. SDS22-GFP cells were treated with indicated siRNAs and processed as in (A). Representative images. Note the SDS22-GFP signal at kinetochores in I3, but not control-depleted cells. Scale bar, 10 μm.
- Depletion efficacy for individual siRNAs as determined by Western blot.
- Quantification of (C). Error bars indicate s.d. of three independent experiments with 25 cells per condition. ***P < 0.001.
Source data are available online for this figure.
Given that I3 forms a ternary complex with SDS22 and PP1, we next asked whether I3 may regulate SDS22 localization during mitosis. We treated SDS22-GFP cells with control or NIPP1 siRNA, or two different I3 siRNAs (Fig 4D). Again, SDS22 displayed a diffuse distribution throughout the cell after control depletion or NIPP1 depletion, and was undetectable at kinetochores (Fig 4C). Strikingly, however, depletion of I3 by either of the siRNAs led to a quantitative redistribution of SDS22 in more than 80% of cells to what appeared to be kinetochores (Fig 4C and E). This effect was suppressed by overexpression of an siRNA-resistant I3 cDNA (Supplementary Fig S3A–C). Localization of SDS22 to the kinetochore under these conditions was confirmed by staining of centromere markers with CREST serum. In I3-depleted cells, but not in control- or NIPP1-depleted cells, SDS22 was detectable at the edges of CREST-positive centromere pairs, consistent with localization to the outer kinetochore (Fig 5A, Supplementary Fig S4). These findings show that SDS22 can localize to kinetochores under certain conditions and that this localization is counteracted by I3. Moreover, they suggest that the redistribution of SDS22 to kinetochores may be the cause of the negative effect of I3 depletion on PP1 activity.
Figure 5. I3 limits association of SDS22 with KNL1-bound PP1.
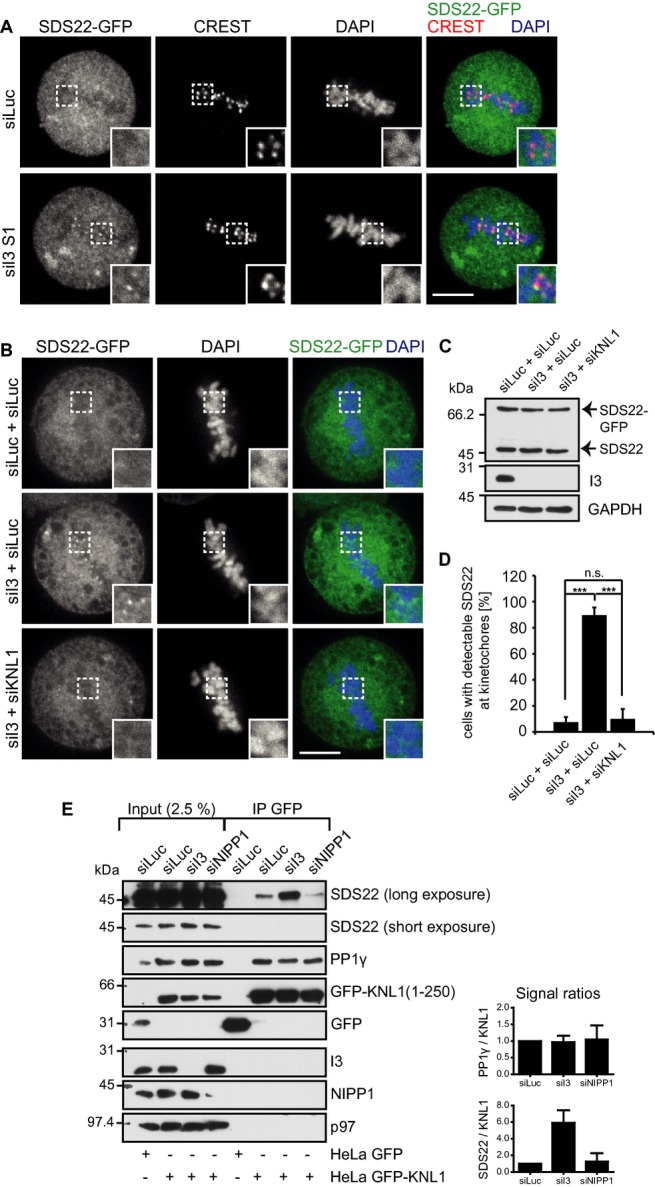
- SDS22 localizes to the outer kinetochore upon I3 depletion (see also Supplementary Fig S4). SDS22-GFP cells were treated with indicated siRNAs, fixed and stained with CREST serum. Scale bar, 10 μm. Transient expression of siRNA-resistant GFP-I3 restores the phenotype (Supplementary Fig S3).
- SDS22 localization to the kinetochore depends on KNL1. SDS22-GFP cells were depleted of I3 alone, or I3 and KNL1, with indicated siRNA combinations. siLuc served as a control and to adjust siRNA concentrations. The KNL1 knockdown efficiency was analyzed by visualizing PP1 localization (Supplementary Fig S5). Scale bar, 10 μm.
- Depletion efficiency of I3 was confirmed by Western blot.
- Quantification of (B). Error bars indicate s.d. of 3 independent experiments with 25 cells per condition. ***P < 0.001.
- I3 depletion stimulates SDS22 association with KNL1-bound PP1. Stable cell lines expressing GFP or a soluble GFP-KNL1 variant were treated with indicated siRNAs. GFP or GFP-KNL1 was affinity-isolated and associated proteins analyzed by Western blot with indicated antibodies. Right panel, quantification of Western blot signals normalized to siLuc. Error bars represent s.d. with n = 3. Note the increase of SDS22 association with PP1γ-KNL1 specifically upon I3 depletion, while PP1γ binding to KNL1 is unaffected.
Source data are available online for this figure.
I3 regulates association of SDS22 with KNL1-bound PP1
We next asked whether kinetochore recruitment of SDS22 was through the KNL1-bound pool of PP1 or whether it was recruited independently. In agreement with previous results (Liu et al, 2010), we found that siRNA-mediated depletion of KNL1 resulted in a nearly complete loss of the GFP-PP1γ signal from kinetochores (Supplementary Fig S5A and B), suggesting that most if not all of kinetochore-localized PP1 is anchored by KNL1. Importantly, when we combined depletion of I3 with depletion of KNL1, the redistribution of SDS22 to kinetochores caused by knockdown of I3 was abolished (Fig 5B and D). I3 knockdown was efficient in both single and double depletions as confirmed by Western blot analysis (Fig 5C).
These data suggested that I3 regulates the association of SDS22 with the KNL1-bound pool of PP1 at the kinetochore. To confirm this notion biochemically, we used a cell line stably expressing a soluble form of KNL1 fused to GFP, which covered amino acids 1–250 and included the RVXF motif as binding site for PP1 (Krenn et al, 2013). In these cells, we again depleted I3, or NIPP1 and isolated GFP-KNL1(1–250) using GFP affinity purification followed by Western blot analysis of associated proteins (Fig 5E). As expected, PP1γ was specifically co-isolated with GFP-KNL1(1–250) and absent in GFP isolates from control cells. Importantly, PP1γ association with KNL1 was not affected by either I3, NIPP1 or control depletions, because comparable amounts were isolated in all conditions (Fig 5E). In control depletions, we also detected SDS22 in the KNL1 pulldowns, suggesting that SDS22 associates with KNL1-bound PP1, although at low levels. Crucially, however, the amount of SDS22 associated with KNL1-bound PP1 was dramatically increased upon depletion of I3 (Fig 5E), which mirrored the stimulated association of SDS22 with kinetochore-bound PP1 in I3-depleted cells as seen by microscopy (Fig 4C). This increase was not observed upon depletion of NIPP1 (Fig 5E), showing that it was specific to I3 depletion. I3 was not detected in KNL1 complexes consistent with the competitive nature of KNL1 and I3 binding to PP1 through their RVXF motifs. These data confirm biochemically that I3 limits the association of SDS22 with KNL1-bound PP1, rather than association of PP1 with KNL1.
Forced localization of SDS22 to kinetochores by overexpression inhibits PP1-mediated dephosphorylation of Aurora B
To explore whether increased binding of SDS22 to kinetochore-bound PP1 could explain the negative effect of I3 depletion on PP1 activity, we transiently overexpressed mCherry fusions of SDS22 in the stable GFP-PP1γ cells. In contrast to low expressing cells, SDS22 was detectable under overexpression conditions at kinetochores without further manipulation (Fig 6A and B). Overexpressed SDS22 colocalized with PP1γ, and PP1γ localization was not affected (Fig 6A). In contrast to wild-type SDS22, two variants of SDS22 with single point mutations, E192A or W302A that abolish PP1-binding (Ceulemans et al, 2002) did not localize to kinetochores (Fig 6A), further confirming that recruitment of SDS22 to the kinetochore was through PP1. We next assessed the effect of SDS22 overexpression on the phosphorylation status of Aurora B as a readout for PP1 activity (Fig 6C and D). The SDS22-E192A and SDS22-W302A variants, which failed to localize to kinetochores, also did not affect PP1 activity with respect to the phosphorylation status of Aurora B. In contrast, overexpression of SDS22 wild-type, which we show was targeted to kinetochore-bound PP1, significantly increased Aurora B phosphorylation levels (Fig 6C and D), demonstrating that SDS22-binding to PP1 at the kinetochore correlates with inhibition of PP1 activity. Consistent with this, we observed elevated BubR1 recruitment upon SDS22 wild-type overexpression compared to control expressions (Fig 6E and F), comparable to the effect detected previously for SDS22 or I3 depletion (Fig 2F and G). We reasoned that, if I3 depletion inhibits PP1 due to excess binding of SDS22 to PP1, moderate reduction of SDS22 levels might relieve the effect of I3 depletion on PP1 activity. Indeed, short SDS22 siRNA treatment for only one day before fixation of cells, which were treated for the full period of 2 days with I3 siRNA, led to a partial but significant rescue of Aurora B phosphorylation levels (Supplementary Fig S6A and B).
Figure 6. Forced localization of SDS22 to kinetochores by overexpression inhibits PP1-mediated dephosphorylation of Aurora B.
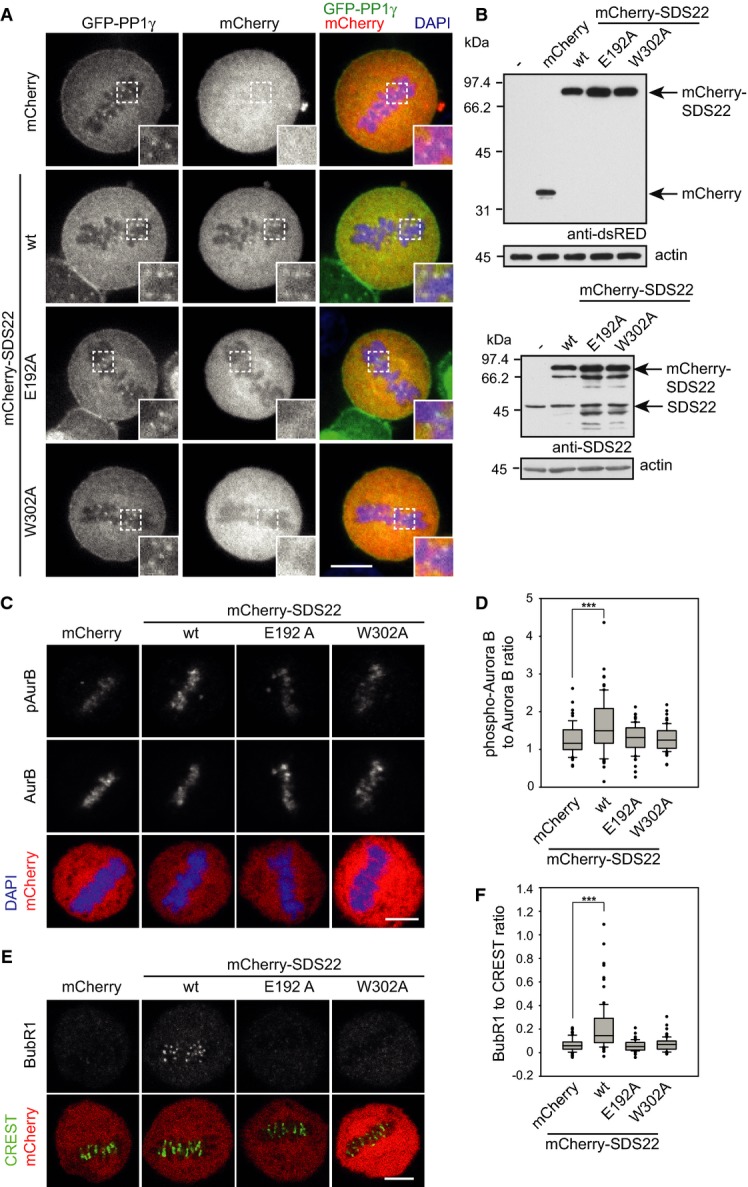
A SDS22 recruitment to kinetochores depends on PP1 binding. Stable GFP-PP1γ cells were transiently transfected with mCherry fusions of SDS22 wild-type (wt) or either of the two PP1-binding-deficient SDS22 variants E192A or W302A, fixed and DAPI-stained. Note also that PP1γ localization is not affected by SDS22 overexpression (see also Supplementary Fig S7 for I3 overexpression). Representative confocal images. Scale bar, 10 μm.
B Western blot of overexpression levels probed with dsRED (upper panel) or SDS22 (lower panel) antibodies.
C–F Overexpression of SDS22 wild-type (wt), but not PP1-binding-deficient mutants increases Aurora B phosphorylation and BubR1 levels at kinetochores. Cells were transiently transfected with indicated constructs as in (A). Fixed cells were stained with DAPI and either Aurora B-specific and phospho-T232 (phospho-Aurora B) antibodies (C) or CREST and BubR1-specific antibodies (E). Representative confocal images. Scale bars, 5 μm. Quantification of (C) and (E) is shown in (D) and (F), respectively. Box plot as in Fig 2B. Data from three independent experiments with 25 cells per condition. ***P < 0.001.
Source data are available online for this figure.
For comparison, we also overexpressed I3 by transient transfection. In contrast to SDS22, overexpressed mCherry-I3 failed to localize to the kinetochore and even induced loss of the PP1 signal from the kinetochore (Supplementary Fig S7A–C). This effect was, however, also observed upon NIPP1 overexpression, suggesting that the displacement of PP1 from kinetochores simply reflected the competitive nature of PP1 binding to the RVXF-motif proteins I3, NIPP1 and the kinetochore-anchor KNL1 rather than a regulation mechanism. Consistent with a loss of PP1 from kinetochores, overexpression of I3 wild-type or NIPP1, but not the PP1-binding-deficient I3-(V41A/W43A) variant led to an increase in Aurora B phosphorylation (Supplementary Fig S7D–F).
SDS22 inhibits dephosphorylation of Aurora B at T232 by PP1 in vitro
The data so far suggest that not only SDS22 depletion, but also increased binding of SDS22 to PP1 at the kinetochore negatively affects PP1-mediated dephosphorylation of Aurora B. To confirm that this was a direct effect of SDS22 on PP1, we reconstituted PP1-mediated dephosphorylation of Aurora B in vitro. We first asked whether PP1 directly and specifically targets Aurora B. We purified recombinant GST-tagged Aurora B in complex with the activating IN-box segment of inner centromere protein (INCENP). In this complex, Aurora B is active and autophosphorylated at T232 (Santaguida et al, 2010). As control, we phosphorylated recombinant GST-Survivin at T34 by Cdk1/CyclinB in vitro and re-purified it. We then incubated both Aurora B and Survivin with increasing concentrations of purified PP1 and monitored the degree of dephosphorylation by Western blot with pT232- or pT34-specific antibodies, respectively (Fig 7A). While Aurora B was significantly dephosphorylated by PP1 in a concentration-dependent manner, Survivin phosphorylation was largely unaffected at the same PP1 concentrations. In contrast, the non-specific lambda phosphatase dephosphorylated both targets. These data show that PP1 directly and specifically dephosphorylates pT232 of Aurora B. We therefore next explored the effect of SDS22 on this activity. We incubated Aurora B with purified PP1 either alone as before, or with increasing concentrations of SDS22. Incubation with PP1 alone again led to a specific loss of pT232, demonstrating that PP1 directly dephosphorylates Aurora B at T232 (Fig 7B). Importantly, addition of SDS22 significantly delayed Aurora B dephosphorylation in a dose-dependent manner with a half-maximal inhibition at about a 1:2 molar ratio (PP1:SDS22) at the given concentrations (Fig 7B and C). Consistent with the results in cells, these data provide direct evidence that SDS22 inhibits PP1-mediated dephosphorylation of Aurora B rather than stimulating it.
Figure 7. Binding of SDS22 to PP1 inhibits dephosphorylation of Aurora B in vitro.

A PP1 specifically dephosphorylates Aurora B pT232. Recombinant, autophosphorylated Aurora B (left panel) or in vitro phosphorylated GST-Survivin (right panel) was incubated either alone or with purified PP1 at indicated concentrations. Lambda phosphatase (λPP) was used as a positive control. Phosphorylation at T232 (Aurora B) and T34 (Survivin) was detected by Western blot with phospho-specific antibodies and equal loading confirmed by Ponceau S staining. Note PP1-mediated dephosphorylation of Aurora B, but not of Survivin.
B, C SDS22 inhibits PP1-mediated dephosphorylation of Aurora B at T232 in a dose-dependent manner. Phosphorylated GST-Aurora B was incubated with purified PP1 without or with the indicated concentration of purified SDS22 and phosphorylation monitored as in (A). Equal loading was monitored with GST antibodies. Western blot signals from three independent experiments were quantified (C) and normalized to maximum and minimum values. Error bars represent s.e.m.
Source data are available online for this figure.
I3 and SDS22 affect Aurora B inactivation during anaphase
Because SDS22 was also shown to be critical for regulation of anaphase (Wurzenberger et al, 2012), we asked whether the functional relationship described here had any relevance during late stages of mitosis. First, we again depleted I3 or NIPP1 in stable SDS22-GFP cells and analyzed the effect on SDS22 localization by fluorescence microscopy. Like in metaphase, SDS22 was again readily detectable at kinetochores in the vast majority of cells after I3 depletion with two different siRNAs even at a late stage of anaphase, while control or NIPP1 siRNAs had no effect (Fig 8A and B). We next explored the effect of SDS22 or I3 depletion on Aurora B distribution and activity (Fig 8C–E). Intriguingly, depletion of SDS22 led to persistence of Aurora B at centromeres even at late stages of anaphase, while Aurora B was completely removed from chromosomes in control or NIPP1-depleted cells as expected (Fig 8C–E). Depletion of I3, which we found to drive SDS22 localization to kinetochores, also led to persistence of active Aurora B during anaphase although to a lower degree than SDS22 depletion (Fig 8C–E). Deregulation of Aurora B during anaphase of SDS22- or I3-depleted cells correlated with a significant increase of anaphase defects such as lagging chromosomes (Fig 8F and G). These findings show that regulation of PP1 by SDS22 and I3 is also critical for proper progression through anaphase.
Figure 8. Depletion of SDS22 or I3 interferes with inactivation of Aurora B at centromeres and chromosome segregation during anaphase.

A, B I3 depletion induces SDS22 localization to kinetochores also in anaphase. Stable GFP-SDS22 cells were depleted and processed as in Fig 4C. Scale bar, 10 μm. The percentage of cells with SDS22 on anaphase kinetochores was determined (B). Error bars indicate s.d. of three independent experiments with 20 cells per condition. ***P < 0.001.
C HeLa cells were depleted of indicated proteins and processed for immunodetection of Aurora B or pT232 as in Fig 2A. Note persisting phospho-Aurora B on anaphase chromosomes in SDS22- or I3-depleted cells. Scale bar, 5 μm.
D, E Quantification of signal intensities in (C). Box plot as in Fig 2B. Data from three independent experiments with 50 cells per condition. ***P < 0.001.
F, G Representative images of anaphase cells treated with indicated siRNAs and quantification of cells with anaphase defects. Error bars indicate s.d. of 3 independent experiments with 50 cells per condition. *P < 0.05; **P < 0.01.
Discussion
The data presented here reveal a requirement of I3 for PP1 regulation at the kinetochore in mammals and establish at least 3 important principles concerning the functional links between SDS22, PP1 and I3 with regard to Aurora B regulation during chromosome segregation. First, SDS22 has a complex effect on PP1. While SDS22 is required for full PP1 activity at the kinetochore, quantitative association of SDS22 with PP1 at the kinetochore interferes with PP1 function. Second, I3 limits the amount of SDS22 associated with KNL1-bound PP1 at the kinetochore and thereby ensures PP1 activity. And third, this functional relationship is important for PP1-mediated dephosphorylation of Aurora B and for antagonizing Aurora B during establishment of bipolar spindle attachment and progression through anaphase.
A seemingly paradoxical effect of SDS22 on PP1 activity at the kinetochore
Our data are consistent with a positive role of SDS22 for PP1 at the kinetochore and its function in antagonizing Aurora B, which has been established before in yeast and mammalian cells (Peggie et al, 2002; Pedelini et al, 2007; Bharucha et al, 2008; Posch et al, 2010; Wurzenberger et al, 2012). We confirm that SDS22 depletion leads to increased Aurora B activity at the kinetochore accompanied by chromosome segregation defects, which is consistent with compromised PP1 function. In addition, we demonstrate that PP1 in fact directly dephosphorylates T232 of Aurora B.
However, our findings speak against a direct role of SDS22 that would involve quantitative localization of SDS22 at the kinetochore, for example a function as a substrate adapter or even as a recruitment factor of PP1 to the kinetochore as proposed before (Posch et al, 2010). Using the SDS22-GFP reporter at near endogenous levels, we do not detect SDS22 at the kinetochore, nor do we detect any loss of PP1 from the kinetochore upon SDS22 depletion, which is consistent with earlier data showing that KNL1 is the major anchor at the kinetochore (Liu et al, 2010).
Intriguingly, SDS22 clearly has the propensity to localize to the kinetochore. We detect SDS22 at kinetochores when SDS22 is overexpressed, which likely explains the conflicting reports in the literature about SDS22 localization (Liu et al, 2010; Posch et al, 2010). More importantly, SDS22 redistributes to the kinetochore upon I3 depletion, indicating that I3 restricts binding of SDS22 to the kinetochore. Under these conditions, SDS22 association with kinetochores is through KNL1-bound PP1, because SDS22 variants defective in PP1-binding fail to localize to the kinetochore and kinetochore recruitment of SDS22 in I3 knockdown cells is abolished upon codepletion of KNL1.
We therefore conclude that SDS22 does not facilitate PP1 localization or is even directly involved in PP1-mediated substrate dephosphorylation. On the contrary, our data suggest that SDS22 inhibits PP1 activity when quantitatively bound to PP1 at the kinetochore. This is supported by the observation that overexpression of SDS22 wild-type that localizes to kinetochores, but not of SDS22 mutants deficient in PP1-binding, inhibits PP1-mediated Aurora B dephosphorylation. Moreover, redistribution of SDS22 to kinetochore-bound PP1 upon I3 depletion correlates with increased Aurora B phosphorylation. And lastly, PP1 directly dephosphorylates pT232 of Aurora B in vitro, but this is inhibited by addition of SDS22 in a dose-dependent manner, rather than stimulated. These findings are consistent with earlier data showing that SDS22 inhibits PP1 in vitro and that overexpression of SDS22 in yeast rescues Ipl1/Aurora deficiency, suggesting that increased SDS22 levels inhibit Glc7/PP1 (Pinsky et al, 2006; Lesage et al, 2007).
The seemingly contradictory findings that SDS22 is essential for PP1 activity, yet PP1 is inhibited when SDS22 is bound to PP1 are best explained by a role of SDS22 as a chaperone for PP1 that stabilizes an inactive structural intermediate of PP1. Of note, SDS22 is one of the most ancient PP1-interacting proteins. Unlike the RVXF-motif proteins I3 or KNL1, SDS22 binds PP1 through leucine-rich motifs and this binding has a profound effect on PP1 structure (Lesage et al, 2007). This property of SDS22 is reminiscent of the regulation of the structurally related PP2A that, like PP1, requires two metal ions in the catalytic site. In the case of PP2A, the regulatory protein alpha4 binds and stabilizes a latent, inactive form of PP2A (Jiang et al, 2013). This binding may help charge PP2A with a metal ion and is essential for cellular activity of PP2A (Jiang et al, 2013). By analogy, SDS22 may stabilize discharged PP1 that dissociates from kinetochores and help reload catalytic ions, or sequester a fraction of PP1 in an inactive form as part of a regulatory mechanism. In any case, in this model and consistent with our findings, PP1 would require SDS22 for activation, but would remain inactive until SDS22 dissociates from PP1.
I3 limits SDS22 binding to kinetochore-associated PP1
The data presented here implicate I3 as a key player in the regulation of PP1 at the kinetochore in human cells and explain the functional relationship to SDS22 and PP1. Because I3, like KNL1, binds PP1 through an RVXF motif and therefore competes with KNL1 for PP1 binding (Heroes et al, 2013), an obvious expectation is that I3 would regulate the binding between PP1 and KNL1 at the kinetochore. Indeed, we find that overexpression of I3 displaces PP1 from kinetochores and thus leads to an increase of Aurora B activity, which explains the phenotype of overexpression in yeast (Pedelini et al, 2007). However, the competitive nature of I3 and KNL1 is not specific to I3 and therefore likely not physiologically relevant, because overexpression of the RVXF-motif protein NIPP1 has the same effect. Conversely, depletion of I3 does not increase PP1-KNL1 binding (Figs 3 and 5E), suggesting that I3 does not primarily regulate recruitment of PP1 to kinetochores.
In contrast and rather unexpectedly, depletion of I3 leads to quantitative association of SDS22 with PP1 on KNL1 at kinetochores and thus to a concomitant loss of PP1 activity at kinetochores. This redistribution can be recapitulated biochemically, as I3 depletion leads to increased association of SDS22 with PP1 when the latter is bound to a soluble form of KNL1. This is specific to I3 and not a trivial effect of any RVXF-motif protein, because depletion of NIPP1 does not cause this change in SDS22 distribution. The best explanation is the fact that I3 forms a ternary complex with SDS22-PP1 in solution, and it does so preferentially over binding to PP1 alone (Lesage et al, 2007; Pedelini et al, 2007). I3 therefore specifically sequesters SDS22-PP1 and prevents association of the SDS22-PP1 complex to kinetochores. While keeping SDS22-PP1 in solution, active PP1 from the SDS22-free pool can be recruited to kinetochores. We therefore propose a model (Fig 9) in which SDS22 binds PP1 in solution and stabilizes it in an inactive form. This could help recharge PP1 with catalytic metal ions or be part of a regulatory mechanism. While SDS22 is bound to PP1, I3 binds SDS22-PP1 and blocks its association with KNL1 until SDS22 is dissociated and PP1 is fully activated. The SDS22-free and active PP1 can then bind KNL1 and efficiently antagonize Aurora B at the kinetochore.
Figure 9. Model figure for the role of SDS22 and I3 in the regulation of kinetochore-bound PP1.
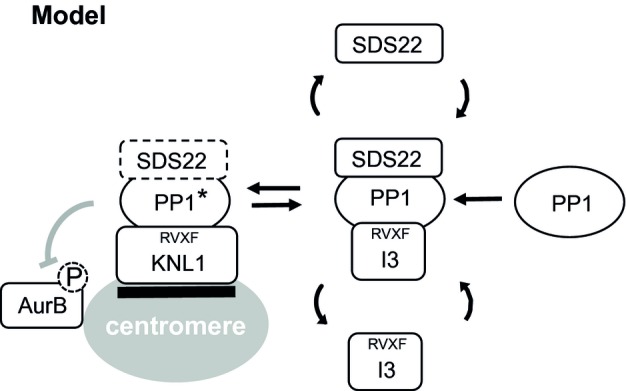
PP1 dynamically exchanges between a KNL1-bound state at the kinetochore and a SDS22-I3-bound state in solution. SDS22 activates PP1, for example, by contributing to the incorporation of metals at the active site, but keeps it inactive while bound. I3 prevents SDS22-PP1 from binding to KNL1 and may facilitate SDS22 dissociation from PP1. Lack of I3 causes (inactive) SDS22-PP1 to bind KNL1, which then fails to antagonize Aurora B. PP1* denotes active PP1.
A role for SDS22 and I3 in anaphase?
The relationship of SDS22 and I3 revealed here represents an additional layer of complexity in the regulation of PP1 at the kinetochore. We show that it is critical for balancing Aurora B activity as evidenced by increased pT232 autophosphorylation after SDS22 or I3 depletion. Therefore, controlling PP1 in this manner is important for successful establishment of bipolar chromosome alignment. In addition, we also demonstrate that it is critical in anaphase, as depletion of SDS22 or I3 leads to lagging chromosomes. With regard to SDS22, this finding is in line with a previous report showing increased Aurora B substrate phosphorylation and anaphase defects in SDS22-depleted cells (Wurzenberger et al, 2012). However, in addition, we detect active Aurora B at centromeres in anaphase upon SDS22 or I3 depletion and therefore correlate the anaphase defects with persistent Aurora B activity at centromeres. Binding of Aurora B to centromeres is mediated by phosphorylation at threonine-3 of histone H3 (H3T3) through its partner Survivin (Kelly et al, 2010) and controlled by a positive feedback loop that is governed by Aurora B and prevents H3T3 dephosphorylation by a different PP1 complex with the RVXF-motif protein Repo-Man (Qian et al, 2013). At anaphase onset, PP1-Repo-Man dephosphorylates H3T3 and helps dissociate Aurora B from centromeres (Qian et al, 2011). We therefore speculate that attenuation of Aurora B activity by the SDS22-PP1-I3 circuit until anaphase onset is important for PP1-Repo-Man to functionally take over and timely dissociate Aurora B from centromeres. Persistent Aurora B activity and anaphase defects upon SDS22 or I3 depletion would therefore reflect a role of SDS22 and I3 at the transition into anaphase that ensures timely removal of Aurora B from centromeres rather than a role specifically during anaphase.
Material and Methods
Plasmids and constructs
NIPP1-GFP and GFP-SDS22 plasmids were described before (Jagiello et al, 2000; Lesage et al, 2007). For GFP-I3 expression, the human I3 cDNA was cloned from the image clone 40029469 into a pEGFP-C1 vector. mCherry-SDS22 and mCherry-I3 were cloned by swapping the sequence coding for the fluorophore using the restriction sites BsrGI/NheI and AgeI/EcoRI, respectively. pcDNA5 FRT/TO GFP-I3 was generated by transferring GFP-I3 from pEGFP-C1 GFP-I3 via the restriction sites HindIII/BamHI into an empty pcDNA5 FRT/TO vector. The mutant forms of mCherry-SDS22 (E192A), mCherry-SDS22 (W302A), GFP-I3 (V41A/W43A) and the siRNA-resistant mCherry-I3 (resist) as well as GFP-I3 (resist) were generated by Quikchange mutagenesis (primers: SDS22 (E192A): 5′-CAACTACAGATGCTAGCGCTGGGATCTAACCGC-3′, SDS22 (W302A): 5′-GAGCTGCAAGAGTTCGCGATGAACGACAATCTCC-3′, I3 (V41A/W43A): 5′-GCCAGAGAAAAAGGCAGAAGCGACAAGTGACACTG-3′, I3 (resist): 5′-GCTCATCCAAATGCTGTTGTATCTACGAGAAACCTCGGG-3′). pET41 GST-Survivin was kindly provided by Shirley Knauer (University of Duisburg-Essen).
Antibodies
For immunofluorescence (IF) staining, anti-AurB pT232 antibodies (Rockland) were used 1:1,000, anti-AurB antibodies (BD) were used 1:500, anti-BubR1 antibodies (Millipore) were used 1:1,000 and anti-centromere serum CREST (Antibody Inc.) was used 1:400. Anti-Dsn1 pS109 antibodies were a kind gift from Iain Cheeseman and were used as described elsewhere (Welburn et al, 2010). For Western blotting, anti-NIPP1 (Sigma-Aldrich), anti-SDS22 (Santa Cruz), anti-PP1γ (Santa Cruz) and anti-Survivin pT34 (Novus Biologicals) antibodies were diluted 1:500. Anti-actin (Santa Cruz), anti-dsRED (Clontech) and anti-GFP (Roche) antibodies were diluted 1:1,000. Anti-AurB pT232 antibodies (Rockland) were used 1:2,500. Anti-GAPDH (Sigma-Aldrich), anti-HSC70 (Santa Cruz) and anti-GST antibodies (Santa Cruz, sc-459) were used 1:10,000. Anti-I3 serum was raised in rabbits against a C-terminal peptide (aa 115–126, DPSQPPPGPMQH) and affinity-purified (Eurogentec) and used 1:500. Anti-p97 (HME 8) (Meyer et al, 2000) serum was diluted 1:2,000. Alexa Fluor-conjugated secondary antibodies (Invitrogen) for IF were used 1:600 and HRP-coupled secondary antibodies (Bio-Rad) for Western blotting were used 1:10,000.
Cell culture, transfection and drug treatment
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma) with 10% fetal calf serum in the presence of penicillin/streptomycin. HeLa cells stably expressing H2B-mRFP and IBB-GFP, or H2B-mCherry were kind gifts from Daniel Gerlich (IMBA, Austria). A stable cell line expressing I3 was generated by transfecting HeLa H2B-mCherry cells with pEGFP-C1-I3. Both cell lines were maintained in medium supplemented with 500 μg/ml G418 and 0.5 μg/ml puromycin. HeLa Flp-In TR cells were a kind gift from Patrick Meraldi. A stable cell line expressing GFP-I3 (resist) was generated by transfecting HeLa Flp-In TR cells with pcDNA5 FRT/TO GFP-I3 (resist) and maintained in medium containing tetracycline-free fetal calf serum, 4 μg/ml blasticidin and 300 μg/ml hygromycin. Protein expression was induced by 1 μg/ml doxycycline for 40 h. The HeLa SDS22-GFP BAC cell line (kindly provided by Antony Hyman) (Poser et al, 2008) was maintained in medium supplemented with 400 μg/ml G418. HeLa GFP-PP1γ cells were described elsewhere (Trinkle-Mulcahy et al, 2003). HeLa cells expressing either GFP or GFP-KNL1(1-250) were maintained as described (Krenn et al, 2013). Protein expression was induced by 1 μg/ml doxycycline for 24 h. For transfections, JetPEI (Polyplus) was used according to the manufacturer's protocol. The medium was replaced 6 h after transfection, and cells were analyzed 48 h post-transfection.
RNAi experiments
The siRNA oligomers I3 S1 (GUAGAAUGGACAAGUGACA), I3 S2 (CUGCUGTAUUUAUGAGAAA), NIPP1 (GGAACCTCACAAGCCTCAGCAAATT, Invitrogen), SDS22 (AGAGTTCTGGATGAACGACAA, Qiagen) and Luc (CGUACGCGGAAUACUUCGATT, Microsynth), as a negative control, were used at a final concentration of 10 nM. Cells were transfected with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. After 6 h, the medium was replaced and cells were analyzed 48 h post-transfection. For experiments using HK Flp-In GFP-I3 (resist) cells, the medium was replaced after 4 h with medium containing 1 μg/ml doxycycline to induce the expression of GFP-I3. The siRNA targeting KNL1 (mixture of CACCCAGUGUCAUACAGCCAAUAUU and UCUACUGUGGUGGAGUUCUUGAUAA) (Krenn et al, 2013) was used at a final concentration of 60 nM, and cells were analyzed after 24 h. For the double-depletion experiment involving KNL1 siRNA, cells were first transfected with Luc or Inh3 S2 siRNA, after 24 h transfected with KNL1 or Luc siRNA, and after additional 24 h analyzed. For the double-depletion rescue experiments, cells were first treated with Luciferase or I3 S2 siRNA 48 h, and then with SDS22 or Luciferase siRNA 24 h before fixation and analysis. For experiments involving RNAi silencing and DNA transfection, first an siRNA transfection was done and after 24 h the DNA was transfected. The cells were grown for additional 48 h before analysis. For experiments involving RNAi silencing and GFP-KNL1 expression, GFP-KNL1 was induced 24 h after siRNA treatment, and cells grown for additional 24 h before harvesting.
Immunofluorescence and live-cell imaging
For immunofluorescence, HeLa cells were grown on glass coverslips. Forty-eight hours post-transfection, cells were fixed in 4% paraformaldehyde in PBS for 15 min and permeabilized with 0.1% Triton X-100 for 10 min. After blocking in 3% BSA in PBS for 1 h, the coverslips were incubated in primary antibody (diluted in 3% BSA in PBS) for 1 h, followed by incubation in secondary antibody (diluted in 3% BSA in PBS) for 45 min. Coverslips were mounted in Mowiol (Calbiochem) containing 0.5 μg/ml DAPI. HeLa SDS22-GFP cells were seeded in 8-well μ-slides (ibidi) and fixed with 4% paraformaldehyde, followed by incubation in PBS supplemented with 0.2% DAPI for at least 30 min. For immunofluorescence, HeLa SDS22-GFP cells were grown in 8-well μ-slides and treated like HeLa cells, but not mounted in Mowiol. HeLa GFP-PP1γ cells were treated like HeLa SDS22-GFP cells but permeabilized while fixing by addition of 0.1% Triton X-100. For time-lapse imaging, HeLa H2B-mRFP and IBB-GFP cells were grown in 8-well μ-slides and medium was changed to imaging medium (DMEM without phenol red supplemented with 10% FCS) 30 min prior imaging. During imaging, cells were supplied with 5% CO2 at 37°C. All imaging except for SDS22 overexpression imaging to monitor effects on phospho-AurB and BubR1 (Fig 6C–F) was performed on an inverted spinning disk confocal microscope (Nikon Eclipse Ti with a Yokogawa CSU X-1 spinning disk unit) using a 100×/1.49 NA oil immersion or a 20×/0.75 NA air objective. Images were acquired with an Andor iXon X3 EMCCD camera. SDS22 overexpression imaging (Fig 6C–F) was performed on a TCS SP5 AOBS system (Leica Microsystems) supported by LASAF software (Leica Microsystems) and equipped with an HCX PL APO ×63/1.4NA oil-immersion objective, as well as PMT and HyD detectors.
Immunoprecipitation and Western blotting
For cell extracts, cells were harvested in extraction buffer (150 mM KCl, 50 mM Tris pH 7.4, 5 mM MgCl2, 5% glycerol, 1% Triton X-100, 2 mM β-mercaptoethanol supplemented with Roche complete EDTA-free protease inhibitor and Roche PhosSTOP phosphatase inhibitor) and incubated on ice for 20 min. After centrifugation for 15 min, the protein concentration in the supernatant was determined in a BCA assay (Interchim). GFP-tagged proteins were affinity-isolated using anti-GFP nanobodies coupled to Sepharose beads (Qian et al, 2013). Affinity-purified nanobodies were coupled to NHS-activated Sepharose 4 Fast Flow (GE) (2 mg per ml bead slurry). For immunoprecipitations, cell extracts containing 500–1,000 μg total cellular protein in 400 μl extraction buffer supplemented with 1 μg/ml BSA were incubated with 30 μl anti-GFP nanobody Sepharose bead slurry for 2 h at 4°C with rotation and then washed three times with extraction buffer. Finally, proteins were eluted from the beads by boiling the samples in SDS sample buffer. Samples were resolved by SDS-PAGE and analyzed by Western blotting.
In vitro dephosphorylation assays
Autophosphorylated GST-Aurora B-INCENP was purified as described (Santaguida et al, 2010). GST-Survivin was phosphorylated with purified cyclin B/CDK1 (kindly provided by Yanzhuang Wang, University of Michigan) in 50 mM Tris/HCl pH 7.5, 10 mM MgCl2, 0.1 mM EDTA supplemented with 1 mM DTT and 2 mM ATP at 30°C for 30 min and re-purified by GSTrap FF column (GE Healthcare). For specificity assays, 12.5 pmol of Aurora B-INCENP or GST-Survivin was incubated with 0.33 or 1.67 pmol recombinant rabbit skeletal muscle PP1 (NEB) or with 40 pmol Lambda phosphatase (NEB) in 1× NEBuffer for PMP, supplemented with 1 mM MnCl2 at 30°C for 15 min. For inhibition experiments, PP1 was purified from rabbit skeletal muscle as described previously (DeGuzman & Lee, 1988). EGFP-SDS22 was expressed in HEK293 cells, affinity-purified using anti-GFP nanobodies and eluted from the matrix by TEV cleavage to generate tag-free full-length SDS22 protein. 5 pmol of autophosphorylated GST-Aurora B-INCENP was incubated with 0.5 pmol PP1 without or with the indicated concentration of SDS22 in 20 mM Tris/HCl pH 7.5 supplemented with 0.1 mg/ml BSA and 2 mM DTT at 30°C for 15 min. Reactions were stopped in SDS sample buffer and analyzed by Western blot with phospho-specific antibodies and visualized with ECL reagent (Perkin Elmer Life Sciences) on a LAS400 imaging system (GE Healthcare). Signal intensities were quantified using ImageJ and normalized by subtracting the pT232 signal of GST-Aurora B-INCENP dephosphorylated by PP1 in the absence of SDS22 (normalized Aurora B pT232 signal = 0) followed by setting the pT232 signal of maximally phosphorylated GST-Aurora B-INCENP (500 nM SDS22) to 1. The normalized PP1 activity was determined by subtracting the normalized Aurora B pT232 signal from 1. A variable slope dose–response curve was calculated using GraphPad Prism 5.
Quantification and statistics
Fluorescence intensities were quantified using Cell Profiler. For phospho-AurB and AurB stainings, the DAPI image was used to build a mask for measuring only the fluorescence signal on chromatin. For CREST, Dsn1 pS109 and BubR1 stainings, a second mask covering kinetochores was built using the CREST image. The intensity of the signal at kinetochores was measured, and the signal on remaining areas of the chromatin mask subtracted as background. GFP-PP1γ localization to kinetochores was quantified by measuring the difference of the fluorescence signal on single kinetochores to the fluorescence signal in the cytoplasm of the same cell. The obtained values were normalized to the expression level of GFP-PP1γ in each cell. Significance was calculated with SigmaPlot software (Systat) using Mann–Whitney U-tests. This non-parametric test procedure was used since the assumptions of normal distribution of the sample values and unchanged variances between treated and untreated samples were not satisfied. Box plots show medians, lower and upper quartiles (line and box), 10th and 90th percentiles (whiskers), and outliers (•). Western blot signal intensities from scanned X-ray films were quantified using ImageJ.
Acknowledgments
We thank L. Trinkle-Mulcahy for providing the GFP-PP1γ cell line and I. Cheeseman for Dsn1 antibodies. This work was supported by DFG grants Me 1626/3 and GRK1431 to HM, the SFB1093 to HM and AM, and a Flemish Concerted Research Action (GOA 10/18) to MB.
Author contributions
AE, JS and MW performed cell-based and biochemical assays. BL and MBe performed and helped with phosphatase assays. VK generated KNL1 cell lines. MBo and AM helped with the design of experiments and manuscript. HM devised the project and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Andrews PD, Ovechkina Y, Morrice N, Wagenbach M, Duncan K, Wordeman L, Swedlow JR. Aurora B regulates MCAK at the mitotic centromere. Dev Cell. 2004;6:253–268. doi: 10.1016/s1534-5807(04)00025-5. [DOI] [PubMed] [Google Scholar]
- Bharucha JP, Larson JR, Gao L, Daves LK, Tatchell K. Ypi1, a positive regulator of nuclear protein phosphatase type 1 activity in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19:1032–1045. doi: 10.1091/mbc.E07-05-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceulemans H, Vulsteke V, De Maeyer M, Tatchell K, Stalmans W, Bollen M. Binding of the concave surface of the Sds22 superhelix to the alpha 4/alpha 5/alpha 6-triangle of protein phosphatase-1. J Biol Chem. 2002;277:47331–47337. doi: 10.1074/jbc.M206838200. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Anderson S, Jwa M, Green EM, Kang J, Yates JR, 3rd, Chan CS, Drubin DG, Barnes G. Phospho-regulation of kinetochore-microtubule attachments by the Aurora kinase Ipl1p. Cell. 2002;111:163–172. doi: 10.1016/s0092-8674(02)00973-x. [DOI] [PubMed] [Google Scholar]
- Ciferri C, Pasqualato S, Screpanti E, Varetti G, Santaguida S, Dos Reis G, Maiolica A, Polka J, De Luca JG, De Wulf P, Salek M, Rappsilber J, Moores CA, Salmon ED, Musacchio A. Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell. 2008;133:427–439. doi: 10.1016/j.cell.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGuzman A, Lee EY. Preparation of low-molecular-weight forms of rabbit muscle protein phosphatase. Methods Enzymol. 1988;159:356–368. doi: 10.1016/0076-6879(88)59036-5. [DOI] [PubMed] [Google Scholar]
- Emanuele MJ, Lan W, Jwa M, Miller SA, Chan CS, Stukenberg PT. Aurora B kinase and protein phosphatase 1 have opposing roles in modulating kinetochore assembly. J Cell Biol. 2008;181:241–254. doi: 10.1083/jcb.200710019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gimeno MA, Munoz I, Arino J, Sanz P. Molecular characterization of Ypi1, a novel Saccharomyces cerevisiae type 1 protein phosphatase inhibitor. J Biol Chem. 2003;278:47744–47752. doi: 10.1074/jbc.M306157200. [DOI] [PubMed] [Google Scholar]
- Heroes E, Lesage B, Gornemann J, Beullens M, Van Meervelt L, Bollen M. The PP1 binding code: a molecular-lego strategy that governs specificity. FEBS J. 2013;280:584–595. doi: 10.1111/j.1742-4658.2012.08547.x. [DOI] [PubMed] [Google Scholar]
- Huang HS, Pozarowski P, Gao Y, Darzynkiewicz Z, Lee EY. Protein phosphatase-1 inhibitor-3 is co-localized to the nucleoli and centrosomes with PP1gamma1 and PP1alpha, respectively. Arch Biochem Biophys. 2005;443:33–44. doi: 10.1016/j.abb.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Jagiello I, Van Eynde A, Vulsteke V, Beullens M, Boudrez A, Keppens S, Stalmans W, Bollen M. Nuclear and subnuclear targeting sequences of the protein phosphatase-1 regulator NIPP1. J Cell Sci. 2000;113(Pt 21):3761–3768. doi: 10.1242/jcs.113.21.3761. [DOI] [PubMed] [Google Scholar]
- Jiang L, Stanevich V, Satyshur KA, Kong M, Watkins GR, Wadzinski BE, Sengupta R, Xing Y. Structural basis of protein phosphatase 2A stable latency. Nat Commun. 2013;4:1699. doi: 10.1038/ncomms2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AE, Ghenoiu C, Xue JZ, Zierhut C, Kimura H, Funabiki H. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science. 2010;330:235–239. doi: 10.1126/science.1189505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Holland AJ, Lan W, Cleveland DW. Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP-E. Cell. 2010;142:444–455. doi: 10.1016/j.cell.2010.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn V, Overlack K, Primorac I, van Gerwen S, Musacchio A. KI motifs of human Knl1 enhance assembly of comprehensive spindle checkpoint complexes around MELT repeats. Curr Biol. 2013;24:29–39. doi: 10.1016/j.cub.2013.11.046. [DOI] [PubMed] [Google Scholar]
- Lampson MA, Renduchitala K, Khodjakov A, Kapoor TM. Correcting improper chromosome-spindle attachments during cell division. Nat Cell Biol. 2004;6:232–237. doi: 10.1038/ncb1102. [DOI] [PubMed] [Google Scholar]
- Lan W, Zhang X, Kline-Smith SL, Rosasco SE, Barrett-Wilt GA, Shabanowitz J, Hunt DF, Walczak CE, Stukenberg PT. Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr Biol. 2004;14:273–286. doi: 10.1016/j.cub.2004.01.055. [DOI] [PubMed] [Google Scholar]
- Lesage B, Beullens M, Pedelini L, Garcia-Gimeno MA, Waelkens E, Sanz P, Bollen M. A complex of catalytically inactive protein phosphatase-1 sandwiched between Sds22 and inhibitor-3. Biochemistry. 2007;46:8909–8919. doi: 10.1021/bi7003119. [DOI] [PubMed] [Google Scholar]
- Liu D, Vleugel M, Backer CB, Hori T, Fukagawa T, Cheeseman IM, Lampson MA. Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J Cell Biol. 2010;188:809–820. doi: 10.1083/jcb.201001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows JC, Shepperd LA, Vanoosthuyse V, Lancaster TC, Sochaj AM, Buttrick GJ, Hardwick KG, Millar JB. Spindle checkpoint silencing requires association of PP1 to both Spc7 and kinesin-8 motors. Dev Cell. 2011;20:739–750. doi: 10.1016/j.devcel.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G. A complex of mammalian Ufd1 and Npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000;19:2181–2192. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuytten M, Beke L, Van Eynde A, Ceulemans H, Beullens M, Van Hummelen P, Fuks F, Bollen M. The transcriptional repressor NIPP1 is an essential player in EZH2-mediated gene silencing. Oncogene. 2008;27:1449–1460. doi: 10.1038/sj.onc.1210774. [DOI] [PubMed] [Google Scholar]
- Pedelini L, Marquina M, Arino J, Casamayor A, Sanz L, Bollen M, Sanz P, Garcia-Gimeno MA. YPI1 and SDS22 proteins regulate the nuclear localization and function of yeast type 1 phosphatase Glc7. J Biol Chem. 2007;282:3282–3292. doi: 10.1074/jbc.M607171200. [DOI] [PubMed] [Google Scholar]
- Peggie MW, MacKelvie SH, Bloecher A, Knatko EV, Tatchell K, Stark MJ. Essential functions of Sds22p in chromosome stability and nuclear localization of PP1. J Cell Sci. 2002;115:195–206. doi: 10.1242/jcs.115.1.195. [DOI] [PubMed] [Google Scholar]
- Pinsky BA, Kotwaliwale CV, Tatsutani SY, Breed CA, Biggins S. Glc7/protein phosphatase 1 regulatory subunits can oppose the Ipl1/aurora protein kinase by redistributing Glc7. Mol Cell Biol. 2006;26:2648–2660. doi: 10.1128/MCB.26.7.2648-2660.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posch M, Khoudoli GA, Swift S, King EM, Deluca JG, Swedlow JR. Sds22 regulates aurora B activity and microtubule-kinetochore interactions at mitosis. J Cell Biol. 2010;191:61–74. doi: 10.1083/jcb.200912046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poser I, Sarov M, Hutchins JR, Heriche JK, Toyoda Y, Pozniakovsky A, Weigl D, Nitzsche A, Hegemann B, Bird AW, Pelletier L, Kittler R, Hua S, Naumann R, Augsburg M, Sykora MM, Hofemeister H, Zhang Y, Nasmyth K, White KP, et al. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat Methods. 2008;5:409–415. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Beullens M, Lesage B, Bollen M. Aurora B defines its own chromosomal targeting by opposing the recruitment of the phosphatase scaffold Repo-Man. Curr Biol. 2013;23:1136–1143. doi: 10.1016/j.cub.2013.05.017. [DOI] [PubMed] [Google Scholar]
- Qian J, Lesage B, Beullens M, Van Eynde A, Bollen M. PP1/Repo-man dephosphorylates mitotic histone H3 at T3 and regulates chromosomal aurora B targeting. Curr Biol. 2011;21:766–773. doi: 10.1016/j.cub.2011.03.047. [DOI] [PubMed] [Google Scholar]
- Rosenberg JS, Cross FR, Funabiki H. KNL1/Spc105 recruits PP1 to silence the spindle assembly checkpoint. Curr Biol. 2011;21:942–947. doi: 10.1016/j.cub.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. doi: 10.1038/nrm2257. [DOI] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol. 2010;190:73–87. doi: 10.1083/jcb.201001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L, Andrews PD, Wickramasinghe S, Sleeman J, Prescott A, Lam YW, Lyon C, Swedlow JR, Lamond AI. Time-lapse imaging reveals dynamic relocalization of PP1gamma throughout the mammalian cell cycle. Mol Biol Cell. 2003;14:107–117. doi: 10.1091/mbc.E02-07-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L, Lamond AI. Mitotic phosphatases: no longer silent partners. Curr Opin Cell Biol. 2006;18:623–631. doi: 10.1016/j.ceb.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Welburn JP, Vleugel M, Liu D, Yates JR, 3rd, Lampson MA, Fukagawa T, Cheeseman IM. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol Cell. 2010;38:383–392. doi: 10.1016/j.molcel.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurzenberger C, Held M, Lampson MA, Poser I, Hyman AA, Gerlich DW. Sds22 and Repo-Man stabilize chromosome segregation by counteracting Aurora B on anaphase kinetochores. J Cell Biol. 2012;198:173–183. doi: 10.1083/jcb.201112112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi Y, Honda T, Tanno Y, Watanabe Y. Two histone marks establish the inner centromere and chromosome bi-orientation. Science. 2010;330:239–243. doi: 10.1126/science.1194498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.