

Abstract
The endogenous microenvironment of the brain is an essential watchdog to guard over myeloid cell function during diseases. Limiting inflammatory reactions of activated microglia and blood-derived monocytes is a key prerequisite for the resolution of tissue insults. So far, however, it was unknown why monocytes but not microglia are able to shift to an anti-inflammatory state during inflammation. In this issue of The EMBO Journal, Cohen and colleagues identified the molecular switch underlying this fundamental functional change. The authors found that the transforming growth factor-β1 (TGFβ1) prevents activated microglia to switch to an anti-inflammatory state by regulating the expression of Irf7.
See also: M Cohen et al (December 2014)
Only after severe insults that lead to breakage of the CNS protection wall, the blood–brain barrier, the endogenous tissue macrophages, microglia, are confronted with their peripheral relatives, the monocytes. Despite being part of the same family, both cell types differ considerably not only in their ancestry but also in their functions (Prinz & Priller, 2014). As part of the prenatal myeloid lineage, microglia can be considered as the senior hostel wardens of the CNS. While microglia are yolk sac-derived and possess considerable potential for self-renewal, short-lived monocytes are the youngsters born during postnatal stages in the bone marrow (Prinz & Priller, 2014). It has been shown before, by several groups, that both myeloid cell populations have dramatically different functions during CNS diseases such as neurodegeneration (Simard et al, 2006; Mildner et al, 2011), autoimmunity (Mildner et al, 2009) or spinal cord injury (Shechter et al, 2009).
The observed functional variety of both cell types is also reflected by their differing potential for deactivation. In general, functional diversity of macrophages during diseases is reflected by several dozens of activation conditions (Xue et al, 2014) of which the M1 and M2 status are the most polarized ones. The M1 status is characterized by the release of pro-inflammatory cytokines such as interleukin-(IL) 1β, IL-6 and tumor necrosis factor (TNF) α, whereas M2-polarized macrophages act in an anti-inflammatory fashion via the release of suppressive molecules such as IL-10 (Prinz & Priller, 2014). Interestingly, even though acute injury leads to bursting microglia activation in the CNS, these cells are not able to switch to the M2-like phenotype, and monocytes are recruited to the injury site to support the repair mechanisms.
The current study by Cohen and colleagues gives an answer to this fundamental question. By applying sophisticated methods, such as next-generation RNA sequencing and Chip-seq, they identified striking differences between monocytes and microglia during disease. First, they demonstrated, in an ex vivo approach, the ability of microglia to switch from an M1- to an M2-like phenotype following extended exposure to the bacterial stimulus lipopolysaccharide (LPS). Surprisingly, pre-conditioning of both, microglia and monocytes, with TGFβ1 for 20 h precluded LPS-induced polarization. This was the first hint that the exposure of microglia to elevated levels of TGFβ1 under homeostatic conditions may impede the cells from acquiring an anti-inflammatory phenotype upon injury. TGFβ1 is a well-known brain endogenous factor produced by neurons and glial cells and is an essential survival and differentiation factor of microglia. In fact, the CNS-specific knockout of TGFβ1 leads to a disturbance in microglia development from early embryogenesis on, which results in a loss of microglia in adult mice (Butovsky et al, 2014).
Cohen and colleagues followed the TGFβ1 path and could further show that both microglia and monocytes have a similar gene expression profile after TGFβ1 exposure ex vivo even though a higher response was seen in monocytes. Interestingly, the gene expression profile of sorted microglia following spinal cord injury (SCI) showed a significant overlap with the profile of monocytes exposed to TGFβ1 ex vivo. Among the altered genes, the transcription factor Irf7, which has not been described yet for the M1-to-M2 phenotype switch, was significantly downregulated upon chronic TGFβ1 exposure. IRF7 belongs to a family of type I interferon (IFN)-induced genes such as IRF1, 2, 5, 7 and 9 that crucially modulate the course of diseases (Honda et al, 2006). In fact, the importance of the brain endogenous type I IFN system for CNS homeostasis during aging was already recently sophisticatedly shown by the same group (Baruch et al, 2014).
Cohen et al (2014) further found that the basal level of IRF7 in non-injured adult microglia was significantly lower compared to monocytes. This finding suggested Irf7 as a downstream target of TGFβ1. The importance of different IRF7 levels for myeloid cell polarization could be established by knockdown of Irf7 by small interfering RNA (siRNA) in monocytes, which consequently exhibited a pro-inflammatory phenotype after extended LPS exposure. This set of data establishes IRF7 as a new and crucial player in the regulation of pro- and anti-inflammatory phenotypes of macrophages in the CNS. Mechanistically, the authors could show that genes whose promoters bind to IRF7 showed an increased pro-inflammatory profile in microglia compared to monocytes following SCI.
As a proof of principle, the authors further demonstrated that the exposure of microglia to IFNβ, a known inducer of Irf7, could induce the switch from M1- to M2-phenotype in the LPS tolerance model. Additionally, the injection of IFNβ to SCI-induced GFP → WT chimeric mice showed that activated microglia isolated from the lesion site had elevated IRF7 levels as well as significantly reduced TNFα and Il-1β expression levels (Fig1). These data strongly suggest that the CNS endogenous microenvironment crucially modulated the activation status of microglia upon injury.
Figure 1. IRF7 regulates M1-to-M2 shift in microglia and monocytes differentially.
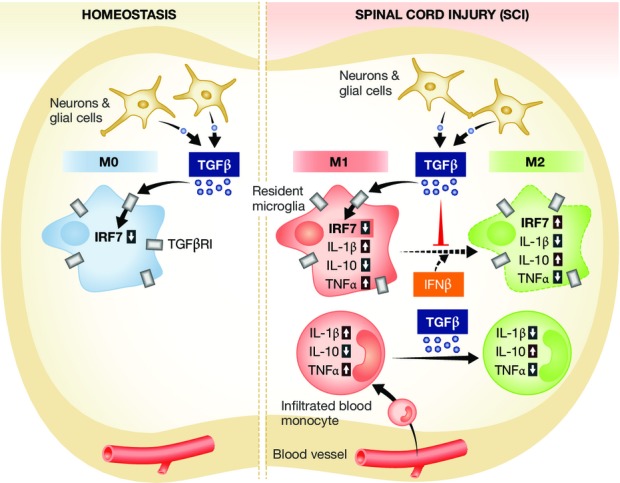
Under homeostatic conditions, microglia are exposed to constitutively high TGFβ1 levels, which lead to a decreased level of IRF7 (see left panel) keeping microglia in a M0 status. Upon spinal cord injury (SCI, right panel), the blood–brain barrier is disrupted and monocytes can infiltrate into the damaged spinal cord. Consequently, both microglia and monocytes shift to a pro-inflammatory (M1) phenotype. Sustained inflammation results in a shift of monocytes to an anti-inflammatory (M2) phenotype, whereas microglia are kept in M1-polarization caused by the increased TGFβ1 exposure and further decrease of IRF7. However, IFNβ-induced upregulation of Irf7 enables microglia to switch to the M2 phenotype.
Even though a recent study already identified high amounts of TGFβ1 constitutively expressed in the CNS and firmly proved the importance of TGFβ1 for microglia development (Butovsky et al, 2014), the current study by Cohen and colleagues went further and focused on clinically relevant pathophysiological conditions. Thereby, they identified the constant high levels of TGFβ1 in the microglial milieu as the main reason for a reduced phenotype switch under pathological conditions. Microglia are kept in a pro-inflammatory state, which is caused by constitutive low levels of the factor IRF7 as a consequence of high TGF1β levels (Fig1). The value of this study is the identification of IRF7 as a key molecule for the phenotype switch that could potentially be used as new target for future clinical interventions. However, further disease models will be mandatory to understand the complex role of the TGFβ1–IRF7 axis in the inflamed or otherwise damaged CNS.
References
- Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, Amit I, Schwartz M. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014;346:89–93. doi: 10.1126/science.1252945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-[beta]-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Matcovitch O, David E, Barnett-Itzhaki Z, Keren-Shaul H, Blecher-Gonen R, Jaitin DA, Sica A, Amit I, Schwartz M. Chronic exposure to TGF1ß regulates myeloid-cell inflammatory response in an IRF7 dependent manner. EMBO J. 2014;33:2906–2921. doi: 10.15252/embj.201489293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T. Type I inteferon gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Mildner A, Mack M, Schmidt H, Brück W, Djukic M, Zabel MD, Hille A, Priller J, Prinz M. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. 2009;132:2487–2500. doi: 10.1093/brain/awp144. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schlevogt B, Kierdorf K, Böttcher C, Erny D, Kummer MP, Quinn M, Brück W, Bechmann I, Heneka MT, Priller J, Prinz M. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. J Neurosci. 2011;31:11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien J-P, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]