

Abstract
Tissue microenvironment influences the function of resident and infiltrating myeloid-derived cells. In the central nervous system (CNS), resident microglia and freshly recruited infiltrating monocyte-derived macrophages (mo-MΦ) display distinct activities under pathological conditions, yet little is known about the microenvironment-derived molecular mechanism that regulates these differences. Here, we demonstrate that long exposure to transforming growth factor-β1 (TGFβ1) impaired the ability of myeloid cells to acquire a resolving anti-inflammatory phenotype. Using genome-wide expression analysis and chromatin immunoprecipitation followed by next-generation sequencing, we show that the capacity to undergo pro- to anti-inflammatory (M1-to-M2) phenotype switch is controlled by the transcription factor interferon regulatory factor 7 (IRF7) that is down-regulated by the TGFβ1 pathway. RNAi-mediated perturbation of Irf7 inhibited the M1-to-M2 switch, while IFNβ1 (an IRF7 pathway activator) restored it. In vivo induction of Irf7 expression in microglia, following spinal cord injury, reduced their pro-inflammatory activity. These results highlight the key role of tissue-specific environmental factors in determining the fate of resident myeloid-derived cells under both physiological and pathological conditions.
Keywords: central nervous system, IRF7, myeloid cells, phenotype-switch, TGFβ
See also: N Hagemeyer & M Prinz (December 2014)
Introduction
Resident microglia are the major specialized innate immune cells of the central nervous system (CNS). Following CNS injury, both brain-resident myeloid cells (microglia) and infiltrating monocyte-derived macrophages (mo-MΦ) are present at the site of injury. These two cell populations differ in their function (Shechter et al, 2009; Mildner et al, 2011) and origin (Ginhoux et al, 2010; Yona et al, 2013); the microglia are derived from primitive yolk-sac myeloid progenitors that arise before day 8 of embryogenesis, while the mo-MΦ are derived primarily from the bone marrow. In addition, differentiation of each of these cell types requires an overlapping, though non-identical set of transcription factors (TFs) (Henkel et al, 1996; McKercher et al, 1996; Schulz et al, 2012; Gomez Perdiguero et al, 2013; Kierdorf et al, 2013).
In general, the appropriate differentiation of macrophages to a classical inflammatory activated (M1) state or alternative suppressive (M2) state is critical for tissue homeostasis and immune clearance (Gordon, 2003; Gordon & Taylor, 2005). During the process of wound healing or pathogen removal, monocytes infiltrate the damaged tissue, leading to a transient inflammatory response (M1) that is resolved either via local conversion to M2-like macrophages or through additional recruitment of anti-inflammatory cells (Auffray et al, 2007; Geissmann et al, 2008).
Following acute injury, there is an immediate and crucial phase of microglial activation in the CNS (Block et al, 2007; Hanisch & Kettenmann, 2007; Centonze et al, 2009; Maezawa & Jin, 2010); however, these cells fail to acquire an inflammation-resolving phenotype (M2-like phenotype) in a timely manner, often resulting in self-perpetuating local inflammation and tissue destruction beyond the primary insult. Under such injurious conditions, recruitment of mo-MΦ (Rapalino et al, 1998; Shechter et al, 2009; London et al, 2011) or bone-marrow-derived monocytes (Rapalino et al, 1998; Heppner et al, 2005; Simard et al, 2006; Yin et al, 2006; Shechter et al, 2009; London et al, 2011; Derecki et al, 2012) to the lesion site was found to have a pivotal role in the repair process by resolving the microglial-induced inflammation. However, why microglia, unlike mo-MΦ, fail to acquire an anti-inflammatory phenotype under such pathological conditions remains an enigma.
It is conceivable that the limited ability of resident microglia to acquire an M2-like phenotype is either an inherent aspect of the microglial differentiation program or an outcome of the unique CNS microenvironment to which they are chronically exposed, as these cells have limited capacity for self-renewal (Jung & Schwartz, 2012). In this context, it is important to note that the CNS microenvironment is characterized by enrichment of anti-inflammatory factors such as IL-13, IL-4, and members of the transforming growth factor-β (TGFβ) family, recently shown to be manifested as a signature of adult microglial markers during homeostasis (Butovsky et al, 2013). Whether and how the chronic exposure to TGFβ imprints microglial activity under pathological conditions has not been investigated. The TGFβ subfamily includes TGFβ1, TGF2, and TGF3, whose expression is abundant in the CNS (Constam et al, 1992; Flanders et al, 1998; Wyss-Coray, 2004). TGFβ1 expression by astrocytes, microglia, and neurons is up-regulated following CNS insult and is also up-regulated during aging (Finch et al, 1993). Moreover, TGFβ1 is involved in mitigating inflammation, promoting resolution (Fadok et al, 1998; Wyss-Coray et al, 2001; Huynh et al, 2002; McGeachy et al, 2007), and is highly expressed relative to the other isoforms in the spinal cord following spinal cord injury (SCI) (Shechter et al, 2013).
Here, we tested the hypothesis that the microglia, following extended exposure to TGFβ1, undergo changes in gene circuitry that render them refractory to the signals inducing the switch from pro- to anti- inflammatory phenotype under inflammatory conditions. We found that extended ex vivo exposure to TGFβ1 impaired the M1-to-M2 switch by myeloid cells. We further identified interferon regulatory factor 7 (IRF7) as a key transcription factor regulating the M1-to-M2 switch in microglia and macrophages, which is down-regulated by TGFβ1. In accordance with these results, we found that following SCI, microglia expressed reduced levels of Irf7 relative to mo-MΦ. Importantly, we demonstrate that this impairment could be reversed both in vitro and in vivo by the induction of Irf7 using IFNβ1, which abrogated the TGFβ1 imprint in vitro, and reduced the expression levels of microglia-derived pro-inflammatory cytokines in vivo following SCI.
Results
M1-to-M2 phenotype switch of newborn microglia is impaired by long exposure to TGFβ1
To test our hypothesis that although microglia differ in their origin from monocyte-derived macrophages (mo-MΦ) (Ginhoux et al, 2010; Gautier et al, 2012; Yona et al, 2013), their response under pathological conditions within the central nervous system (CNS) is dictated to a large extent by their microenvironment; we first assessed the ability of newborn-derived microglia (NB-Mg) to undergo M1-to-M2 phenotype switch. To this end, we adopted an established ex vivo model of macrophage polarization (Porta et al, 2009), in which M1 polarization, which is known to be induced by brief exposure to lipopolysaccharide (LPS, 4 h), is inhibited as a result of extended LPS pre-exposure (20 h). Under such conditions, the cells switch to an M2-like (anti-inflammatory) phenotype and remain unresponsive to further LPS challenge. Using this ex vivo assay, we compared the response of NB-Mg following 4 h LPS challenge to their response to such a challenge following a long (20 h) pre-exposure to LPS (Fig1A). Cells were harvested, and total RNA was extracted to determine expression of characteristic pro- and anti-inflammatory genes. The gene expression profile of the treated NB-Mg showed their ability to undergo M1-to-M2 phenotype switch following long LPS pre-exposure, which was highly similar to the previously documented monocyte/macrophage M2-polarization phenotype (Foster et al, 2007; Porta et al, 2009). Specifically, iNos, Tnfa, Il1b, Cxcl1, Cxcl2, and Cxcl10, M1-associated pro-inflammatory genes involved in CNS inflammation and neurodegeneration, were down-regulated in LPS-tolerant cells and barely induced following 4 h LPS re-challenge (Fig1B). Under the same experimental conditions, the prototype anti-inflammatory cytokine, Il10, was induced rather than suppressed in the LPS-tolerant NB-Mg and further elevated following re-challenge (Fig1B). These results indicate that NB-Mg, similarly to macrophages, have an inherent capacity to switch from M1 to M2 phenotype ex vivo under prolonged inflammatory conditions (e.g., long exposure to LPS).
Figure 1. M2-like phenotype acquired by NB-Mg under ex vivo conditions can be inhibited by TGFβ1 preconditioning.

- Total RNA was harvested from NB-Mg following various treatments: Cells were left untreated (M), or stimulated with LPS (100 ng/ml) for 4 h [LPS(4 h), gray], or stimulated with LPS for 20 h [LPS(20 h)], or stimulated with LPS for 20 h, washed with growth medium, and re-challenged with LPS for an additional 4 h [LPS(20 h/4 h), orange], or preconditioned for 20 h with different anti-inflammatory cytokines, and then stimulated with LPS for 20 h, washed, and re-challenged with LPS for an additional 4 h [Pre(20 h)/LPS(20 h/4 h), green].
- NB-Mg were stimulated as described in (A), and RNA was analyzed by RT-qPCR for the expression of representative pro- and anti-inflammatory genes.
- NB-Mg were stimulated as described in (A) and preconditioned with TGFβ1 (100 ng/ml), IL-4 (10 ng/ml), IL-13 (10 ng/ml), or TGFβ2 (100 ng/ml). RNA was analyzed by RT-qPCR for the expression of representative pro- and anti-inflammatory genes. Results are normalized to the expression of the housekeeping gene, peptidylprolyl isomerase A (PPIA), and expressed as fold increase relative to the mRNA levels of untreated cells (M). Asterisks are relative to the [LPS(20 h/4 h)] sample, unless indicated otherwise.
- Mice were injured, and parenchymal segments of 0.5 mm from each side of the spinal cord lesion site were excised on the indicated days following SCI. Cells purified from the injured spinal cord parenchyma (n = 6) were incubated in growth medium (Materials and Methods) for 3 h and then washed and stained for the intracellular cytokine IL-10.
- Flow cytometry quantification of percentage and number of mo-MΦ and resident microglia that expressed IL-10 at day 7 following SCI. Data are normalized to 10,000 cells. Student's t-test for percent of IL-10+ cells, P = 0.034; number of IL-10+ cells, P = 0.047.
Data information: Samples were prepared in triplicates, and results are representative of two or more different experiments. *P < 0.05, **P < 0.01, ***P < 0.005. Data are represented as mean ± SEM.
Next, NB-Mg were exposed, prior to LPS treatment, to factors prevalent within the CNS microenvironment, and their subsequent ability to undergo M1-to-M2 phenotype switch was examined. We used the same LPS tolerance model, but this time, the cells were first exposed to anti-inflammatory factors, such as TGFβ1, TGFβ2, IL-4, and IL-13, and only then to LPS (Fig1A, bottom). Our premise was that long exposure to such anti-inflammatory factors would create a form of tolerance to the tested anti-inflammatory cytokines and would imprint the inability to switch from M1-to-M2 phenotype during the subsequent long LPS incubation. Of the tested factors, only incubation with TGFβ1 for 20 h, before the subsequent exposure to LPS tolerance conditions, prevented the LPS-induced polarization toward M2-like phenotype with respect to expression of key characteristic cytokines; thus, the cells exposed to TGFβ1 prior to the LPS tolerance, showed down-regulation of Il-10 expression, and increased expression of the characteristic pro-inflammatory cytokines, including Il1b, Il6, Cxcl1, and Cxcl2 (Fig1C). Importantly, the pro-inflammatory bias caused by the extended pre-exposure to TGFβ1, prior to the LPS, was not restricted to microglia; we observed a similar effect when bone-marrow-derived macrophages (BM-MΦ) were tested under the same experimental paradigm (Supplementary Fig S1A). Notably, not all pro-inflammatory genes were affected; the expression of some genes, such as iNos, Tnfa, and Cxcl10, was not affected by TGFβ1 pre-exposure in either NB-Mg or BM-MΦ (Supplementary Fig S1B). The low Il10 expression level in NB-Mg following TGFβ1 pre-exposure was reminiscent of adult microglial incompetence to secrete IL-10 in response to acute spinal cord injury (SCI), compared to high expression levels of this cytokine by mo-MΦ at the crucial phase of the repair process, day 7 (Fig1D and E). Collectively, these results support the hypothesis that a TGFβ1-enriched microenvironment, to which adult microglia are exposed prior to CNS injury, impairs their ability to acquire an inflammation-resolving phenotype and to convert into M2-like cells under severe injurious conditions; in contrast, the blood-derived monocytes are freshly recruited and thus have no experience of TGFβ1 pre-exposure.
Long exposure to TGFβ1 activates a robust gene expression program in myeloid cells, with implications to CNS pathological conditions
To understand the molecular events elicited by long exposure to TGFβ1, we measured genome-wide expression profiles, using RNA-Seq, of both BM-MΦ and NB-Mg along the time course of TGFβ1 exposure ex vivo. Globally, 2,721 and 642 genes showed expression changes (twofold change; up or down) in response to TGFβ1 in BM-MΦ and NB-Mg, respectively (Materials and Methods; Fig2A). Notably, TGFβ1 induced a response that was similar in terms of gene expression pattern in both cell types, although they are of different developmental origin, BM versus yolk sac (Ginhoux et al, 2010) (Fig2A). BM-MΦ exhibited higher responsiveness to TGFβ1 treatment compared to NB-Mg, reflected by the higher number of changed genes and the intensity of their change (P < 10−5) (Fig2B). The overall dynamic change of myeloid cell gene expression following long exposure to TGFβ1 revealed global effects of TGFβ1 on expression of genes involved in tissue repair processes (Supplementary Fig S2), as well as on up-regulation of its own receptor, Tgfbr1 (Fig2C). To test the relevance of the observed effect of preconditioning with TGFβ1, but not with TGFβ2, on the M1-to-M2 switch ex vivo (Fig1C) to the in vivo situation in adult CNS, we examined the expression levels of TGFβ receptors by adult microglia and blood monocytes. Adult microglia and blood monocytes were isolated from CX3CR1GFP/+ mice and were analyzed for their receptor levels. We found that resident adult microglia expressed higher levels of Tgfbr1 compared to blood-derived cells, whereas levels of Tgfbr2 were similar in both cell types (Fig2D). The higher expression of Tgfbr1 on adult microglia strengthened the potential relevance of TGFβ1 to the fate of these resident myeloid cells.
Figure 2. Long exposure to TGFβ1 activates a robust gene expression program in myeloid cells.

- mRNA expression profile of genes, whose expression level was elevated or reduced twofold in at least one of the time points in either TGFβ1 stimulated BM-MΦ or NB-Mg. Genes were clustered according to k-mean of 20 (red, high relative expression; white, mean expression; blue, low relative expression).
- Distributions of the number of genes expressed in BM-MΦ (blue) and NB-Mg (red) that were increased (right graph) or decreased (left graph) along the time course (Kolmogorov–Smirnov test, P-value < 10−5).
- Expression levels of Tgfbr1 in BM-MΦ (blue) and NB-Mg (red) along the time course of TGFβ1 treatment were analyzed by RNA-seq.
- Peripheral CX3CR1lowLy6C+ monocytes and resident microglia were sorted from non-injured CX3CR1GFP/+ mice, and the expression of Tgfbr1 and Tgfbr2 was analyzed using RT-qPCR. RT-qPCR results are normalized to the expression of PPIA.
- eGFP > WT chimeric mice were injured, and parenchymal segments of 0.5 mm from each side of the spinal cord lesion site were excised on different days following SCI. GFP+ mo-MΦ and GFP− resident microglia were sorted by FACS and collected directly into lysis buffer. RNA was harvested, and gene expression profile was analyzed by RNA-seq.
- Venn diagrams of genes that were up-regulated (left panel) or down-regulated (right panel) in BM-MΦ at least twofold by exposure to TGFβ1 ex vivo (red) and genes from the in vivo kinetic studies whose expression was significantly different (P-value < 0.05) between microglia and mo-MΦ, selecting those that were expressed to a higher (left panel) or lower (right panel) extent in microglia compared to mo-MΦ along the kinetics following SCI (blue), respectively. Hypergeometric test for the intersection of up-regulated genes, P-value < 10−5; hypergeometric test for the intersection of down-regulated genes, P-value = 3 × 10−3; n = 12 for all kinetic following SCI.
- Expression profile of genes that were down-regulated at least twofold by BM-MΦ following exposure to TGFβ1 ex vivo and were highly expressed by mo-MΦ compared to microglia (twofold) at days 3 and 7 following SCI, divided into functional groups.
- Expression profile of genes that were up-regulated by BM-MΦ at least twofold following exposure to TGFβ1 ex vivo and were highly expressed by microglia (twofold) compared to mo-MΦ at days 3 and 7 following SCI, divided into functional groups.
Data information: Data represent the average expression in two independent experiments; each experiment was performed in duplicate. Red, high relative expression; white, mean expression; blue, low relative expression.
Since previous data showed that microglia and infiltrating mo-MΦ have distinct inflammation-resolving phenotypes following SCI (Rapalino et al, 1998; Shechter et al, 2009, 2013), we isolated the activated resident microglia and the infiltrating mo-MΦ from the injured spinal cord and analyzed their global gene expression using RNA-seq. For this purpose, we used BM-chimeric mice, whose bone marrow cells were replaced with green fluorescent protein (GFP)-expressing bone marrow cells to enable accurate and pure cell separation of microglia and mo-MΦ (Supplementary Fig S3). A high degree of chimerism was achieved using two sequential irradiations, the first consisting of low-dose (300 rad) total body γ-irradiation, which induces lymphopenia and leads to lymphocyte extravasation from the lymph nodes (without inducing trafficking of immune cells to the CNS), and a second high-dose γ-irradiation (950 rad), performed using head shielding to prevent blood–brain barrier breakdown (Derecki et al, 2012). After sorting of activated resident microglia and infiltrating mo-MΦ at different time points following SCI, we determined the genome-wide expression profile of the distinct cell populations (Fig2E). Our goal was to test whether the gene expression imprint of long TGFβ1 exposure on naïve myeloid cells, such as BM-MΦ (Fig2A), is similar to the unique gene expression signature of adult microglia during recovery from SCI, as these cells are chronically exposed to the CNS microenvironment. Specifically, we tested whether genes that were highly expressed by microglia compared to mo-MΦ over the course of the response to SCI (Fig2F, left; blue; P-value < 0.05) overlapped with genes that were elevated ex vivo in BM-MΦ following long exposure to TGFβ1 (Fig2F, left; red; twofold). In parallel, we determined whether genes that were highly expressed by mo-MΦ compared to microglia over the course of the response to SCI (Fig2F, right; blue; P-value < 0.05) overlapped with genes that were reduced ex vivo by TGFβ1 in BM-MΦ that were not previously exposed to this cytokine (Fig2F, right; red; twofold). Global comparison demonstrated a significant overlap among the up-regulated genes (P < 10−5), as seen by the intersecting genes (Fig2F; Supplementary Tables S1 and S2). Next, we focused on genes that were altered by TGFβ1 and might affect the microglial phenotype during the repair process (at days 3 and 7). We identified 20 genes that were decreased by TGFβ1 treatment (twofold) ex vivo and were expressed at low levels (twofold) by microglia (Fig2G), and 41 genes that were elevated by TGFβ1 and were more highly expressed in microglia in vivo compared to mo-MΦ during days 3 and 7 following SCI (Fig2H); among them were important immune response mediators and transcription factors (TFs). The comparison between the signature of TGFβ1 on naïve myeloid cells and the CNS imprint on the resident microglia during the repair process suggests that TGFβ1 has a significant role in shaping the adult microglial response under pathology.
The transcription factor IRF7 is required for the M1-to-M2 switch and is suppressed by TGFβ1
In order to understand the mechanism underlying TGFβ1 impairment of the M1-to-M2 switch, we further analyzed our global gene expression data, seeking TFs whose expression was altered by the extended exposure to TGFβ1 (Fig3A). We first focused on clusters I and XII (Fig3A) in which the TFs were expressed by both naive BM-MΦ and NB-Mg and were similarly and significantly changed along the time course of TGFβ1 treatment. Among these TFs, we identified several candidates in cluster I: peroxisome proliferator-activated receptor γ (Pparγ), a member of the nuclear receptor family of transcription factors that mediates transcriptional activation of anti-inflammatory genes (Jiang et al, 1998; Hyong et al, 2008; Kapadia et al, 2008), and interferon regulatory factor 7 (Irf7), an essential TF for antiviral immunity (Honda et al, 2005; Amit et al, 2009), whose involvement in the regulation of the M1-to-M2 switch has not been previously reported. The expression of both TFs was down-regulated ex vivo following TGFβ1 exposure (Fig3B and C). Notably, analysis of the Irf7 expression profile in the ex vivo model following continuous exposure to LPS (Fig1A) revealed that Irf7 expression by both cell types was induced starting from the 4 h time point and remained high from 10 h onward following long exposure to LPS, during the M2-polarization period. Yet, BM-MΦ expressed higher levels of Irf7 compared to NB-Mg along the entire time course (Fig3D). Importantly, however, under the same LPS paradigm, levels of Irf7 expression in both BM-MΦ and NB-Mg were reduced in cells preconditioned with TGFβ1 (Fig3E). To substantiate the novel functional role of IRF7 in the M1-to-M2 conversion, we used small interfering RNA (siRNA) to silence Irf7 expression in BM-MΦ ex vivo (thereby reducing Irf7 levels in BM-MΦ to levels comparable to those in microglia) and tested its impact on the expression of pro- and anti-inflammatory genes (Fig3F). Importantly, Irf7 silencing decreased the levels of Il10 expression and elevated the pro-inflammatory cytokine, Il1b, and the chemokine, Cxcl1, in the LPS tolerance model (Fig3F).
Figure 3. Pro- to anti-inflammatory phenotype switch is regulated by IRF7, which is suppressed by TGFβ1.

- A Left panel: RNA-seq expression profile of transcription factor genes, whose expression level was induced or reduced by a factor of 2 on at least one of the time points in either BM-MΦ or NB-Mg stimulated with TGFβ1 (100 ng/ml) along a time course of 0, 1, 3, 6, 10, and 20 h. Genes were clustered according to their time to peak. Cluster numbers (I–XII) are noted on the right, and cluster size is indicated in parentheses; representative member genes are identified on the left; red, high relative expression; white, mean expression; blue, low relative expression. Right panel: mean relative expression profiles for each cluster were calculated at each time point of the kinetics.
- B Gene expression profile of Irf7 and Pparγ in BM-MΦ (blue) and NB-Mg (red) was analyzed from RNA-seq data. Data represent the average expression of two independent experiments; each experiment was performed in duplicate.
- C RT-qPCR analysis of Irf7 expression in BM-MΦ (blue) and NB-Mg (red) following 20 h treatment with TGFβ1 or with growth medium.
- D BM-MΦ (blue) and NB-Mg (red) were stimulated with LPS (100 ng/ml) for 20 h, RNA was harvested along the time course of 0, 1, 2, 4, 10, and 20 h of the LPS stimulation, and gene expression profile of Irf7 was analyzed by RNA-seq.
- E RT-qPCR analysis of Irf7 expression in BM-MΦ (right panel) and NB-Mg (left panel) after long stimulation with LPS followed by LPS re-challenge [LPS(20 h/4 h)], with (green) or without (orange) 20 h preconditioning with TGFβ1.
- F BM-MΦ were transfected with siRNA directed against Irf7 or scrambled, and treated for 20 h with LPS, washed, and re-challenged with LPS [LPS(20 h/4 h)]; RNA was harvested and analyzed using RT-qPCR. Results are shown as change in gene expression between siIrf7-treated cells and controls. Asterisks indicate significance of the differences between siIrf7 treatment and scrambled controls for each gene.
- G Peripheral CX3CR1lowLy6C+ monocytes and resident microglia were sorted by FACS from non-injured CX3CR1GFP/+ mice (n = 3), and the expression of Irf7 was analyzed using RT-qPCR.
- H, I GFP+ mo-MΦ and GFP− resident microglia were sorted from GFP > WT chimeric mice by FACS. RNA was harvested, and gene expression level was analyzed by RT-qPCR. (H) Irf7 expression in mo-MΦ (blue) and microglia (red) was analyzed in non-injured mice, and at 16 h, day 1, day 3, day 7, and day 14 following SCI (n = 2 at each time point). (I) Correlation between Irf7 and 1/Tgfbr1 expression level by microglia in homeostasis and following SCI (r2 = 0.84).
Data information: Data represent results of one out of two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.005. Results in (C, E–H) are normalized to the expression of PPIA. Data are represented as mean ± SEM.
To further substantiate our findings, attributing an important role to IRF7 in controlling microglial behavior at adulthood under injurious conditions, we first compared its expression levels by ‘resting’ microglia isolated from adult spinal cord parenchyma of CX3CR1GFP/+ mice relative to naïve circulating blood monocytes (CX3CR1lowLy6C+). A higher level (approximately fivefold) of Irf7 was observed in naïve monocytes as compared to healthy adult spinal cord-derived microglia (Fig3G). Quantitative PCR analyses of isolated microglia and infiltrating mo-MΦ following SCI (Fig2E) revealed an increase in Irf7 expression on day 1, both in resident microglia and in the isolated mo-MΦ, which was rapidly reduced in microglia to basal levels, unlike in mo-MΦ, which maintained high levels of Irf7 expression from day 3 onward (P-value = 0.051) (Fig3H). Interestingly, microglial behavior over the course of response to the injury revealed an inverse correlation between expression levels of Irf7 and that of the receptor to TGFβ1, Tgfbr1 (Fig3I), in line with our ex vivo observations (Figs2C, 3B and C). Overall, these results suggest that Irf7, which was down-regulated by TGFβ1 and whose expression was less pronounced in microglia during homeostasis and following SCI relative to mo-MΦ, might be a potential candidate for curtailing the M1-to-M2 circuit in myeloid cells. Therefore, IRF7 might be one of the factors that are modified in microglia by the CNS microenvironment, resulting in their inability to express the resolving phenotype under pathological conditions.
IRF7 regulates the M1-to-M2 phenotype switch via down-regulation of the pro-inflammatory genes
To identify the genes that are potentially directly regulated by Irf7, we next performed chromatin immunoprecipitation followed by massively parallel sequencing (ChIP-Seq) of LPS-treated (2 h) myeloid cells. Since over the course of the recovery process following SCI, the mo-MΦ could differentiate to M1-like phenotype, at the early stage (days 1–3 post-injury), and M2-like phenotype, at the later stage (day 7 post-injury), we searched for the intersection of genes expressed at these days by the mo-MΦ in vivo with our Irf7 ChIP-Seq data. M1-related genes (green) were characterized by high expression at the initial days following the insult, while the genes that were highly expressed at day 7 were classified as M2-related genes (orange). We found a significant intersection between the genes involved in the M1 response (that were down-regulated during the repair process) and genes whose promoters were found to bind Irf7 following LPS activation (Fig4A; P < 10−5; Supplementary Table S3). Those M1-related genes whose promoters were bound by Irf7 following LPS treatment were classified into functional groups using PANTHER gene ontology (Fig4B; Materials and Methods); among them, we focused on genes related to immune response and transcription regulation (Fig4C; Supplementary Table S3). For example, the expression level of pro-inflammatory genes whose promoters were bound by Irf7, such as Cxcl1, Cxcl2, Il1b, and Tnfa, was higher in microglia compared to mo-MΦ at days 3 and 7 following SCI, substantiating the functional link between the lower expression level of Irf7 (Fig3H) and the pro-inflammatory profile observed in microglia at day 3 onwards following SCI (Fig4D).
Figure 4. IRF7 regulates the M1-to-M2 phenotype switch by down-regulation of pro-inflammatory gene expression.

- A–C ChIP-Seq of Irf7 was performed on GM-CSF-induced bone marrow cells (stimulated 2 h with LPS or untreated controls). (A) Venn diagram of genes whose expression in vivo by mo-MΦ was either elevated (M2-related genes, orange) or decreased (M1-related genes, green) at day 7 relative to the first 3 days following SCI (P-value < 0.05) and genes whose promoters were bound by Irf7 (blue) (hypergeometric test: green–blue intersection, P-value < 10−5; orange–blue intersection P-value = 0.4). (B) Pie chart dividing the M1-related genes, whose promoters were bound by Irf7 (green–blue intersection) to functional groups using the PANTHER database of gene ontology (Materials and Methods). (C) Expression profile of transcription regulation genes (upper panel) and immune response genes (lower panel) out of M1-related genes, which were bound by Irf7 and down-regulated in mo-MΦ along the time course following SCI.
- D ChIP-Seq signal intensity of selected pro-inflammatory genes is represented by sequencing (left panel), and their in vivo expression in microglia (red) and mo-MΦ (blue) at days 3 and 7 following SCI is represented in the right panel.
To elucidate a potential direct functional link between Irf7 expression levels and the ability of the microglia to switch from M1 to M2 phenotype, we examined whether induction of Irf7 using its well-known inducer, IFNβ1, would restore the ability of microglia to acquire an M2 phenotype ex vivo (e.g., elevating the levels of Irf7 in microglia to approach those found in macrophages) (Fig5A). We first tested whether NB-Mg, which were exposed to TGFβ1, would be able to re-express Irf7 under our experimental paradigm. To this end, we exposed the cells to IFNβ1, a known inducer of Irf7. We found that the tolerant NB-Mg preconditioned with TGFβ1 were able to express Irf7 following IFNβ1 stimulation (Fig5B). Further, we observed that NB-Mg that were preconditioned with TGFβ1 and stimulated with IFNβ1 underwent M1-to-M2 phenotype switch. We found that these microglia showed high expression levels of Il10 and low expression levels of Cxcl1, Cxcl2, and Il1b, compared to NB-Mg that were only preconditioned with TGFβ1 (Fig5C). These results indicate that the effect of TGFβ1 on microglia can be abrogated by Irf7 induction, overcoming the inability to undergo an M1-to-M2 phenotype switch.
Figure 5. IFNβ1 can overcome the inability of microglial phenotype switch through elevation of IRF7.
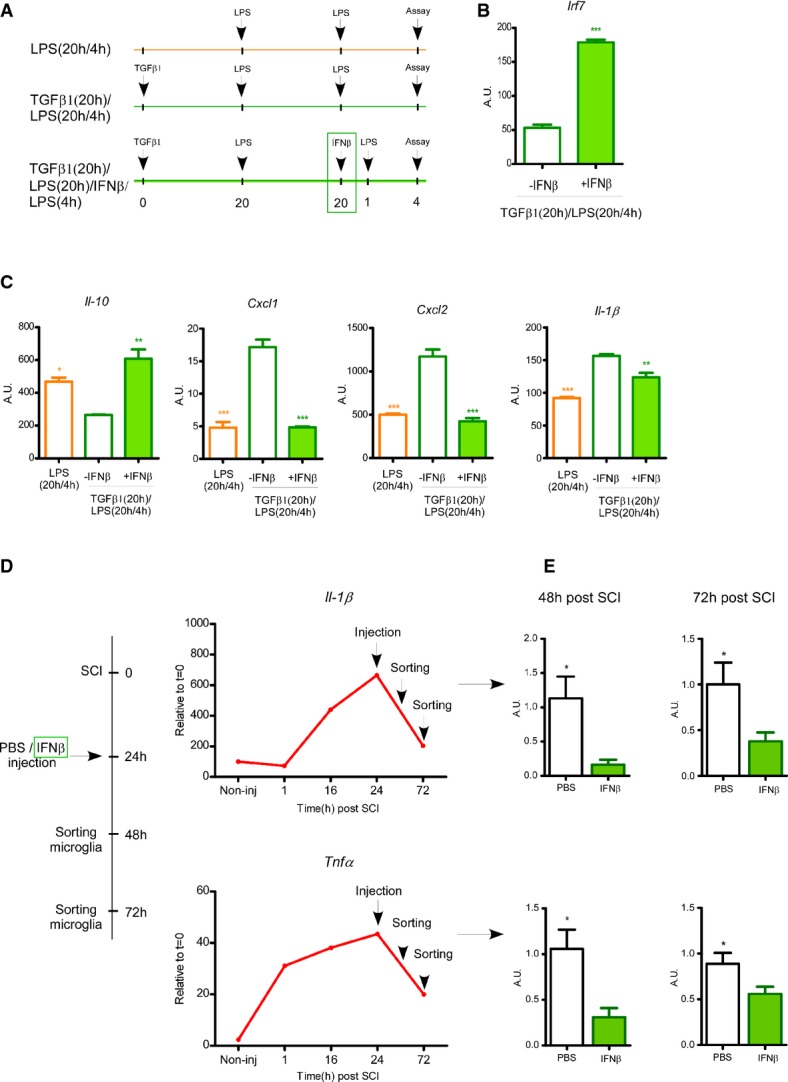
- A–C Total RNA was harvested from NB-Mg that were stimulated with LPS for 20 h, washed, and re-challenged with LPS for 4 h [LPS(20 h/4 h), empty orange bars], or preconditioned with TGFβ1 for 20 h prior to LPS (20 h/4 h) stimulation [TGFβ1(20 h)/LPS(20 h/4 h), empty green bars], or preconditioned with TGFβ1 for 20 h, and also stimulated with IFNβ1 (1,000 U/ml) starting from 1 h prior to the 4 h re-challenge with LPS [TGFβ1(20 h)/LPS(20 h)/IFNβ/LPS(4 h), filled green bars]. RNA was analyzed by RT-qPCR for the expression of (B) Irf7 and (C) representative pro- and anti-inflammatory genes. Asterisks indicate significance relative to the [TGFβ1(20 h)/LPS(20 h/4 h)] samples. Samples were prepared in triplicates, and results are representative of at least two different experiments.
- D GFP > WT chimeric mice were injured and intra-parenchymal-injected 24 h later with IFNβ1 (two injections of 800 U/25 g at the margins of the lesion site) or PBS. Parenchymal segments of the spinal cord lesion site (0.3 mm on both sides of the lesion) were excised 48 h and 72 h following SCI. Il1b and Tnfa expression in microglia was analyzed in non-injured mice, and at 1 h, 16 h, day 1, and day 3 following SCI by RNA-seq (n = 2 at each time point).
- E GFP− activated microglia were sorted by FACS at 48 h (n = 4) and 72 h (n = 10; pool of three different experiments) following SCI, RNA was extracted, and gene expression levels of Il1b (P-value (48 h) = 0.013; P-value (72 h) = 0.026) and Tnfa (P-value (48 h) = 0.019; P-value (72 h) = 0.034) were analyzed by RT-qPCR.
Data information: *P < 0.05, **P < 0.01, ***P < 0.005. (B, C, E) Results are normalized to the expression of PPIA. Data are represented as mean ± SEM.
Finally, we tested whether Irf7 induction in vivo would enable overcoming the microglia impairment to switch phenotype by down-regulating the expression levels of pro-inflammatory cytokines following SCI. To this end, spinally injured GFP > WT chimeric mice were locally injected with IFNβ1 (control mice were injected with PBS). Injections were performed directly into the parenchyma in order to elevate Irf7 expression in the inflammatory microglial cells located in close proximity to the lesion site. The time point for injection was determined based on the gene expression kinetics of inflammatory cytokines observed in microglia following SCI, in which we found that Tnfa and Il1b expression by sorted microglia peaked at day 1 and spontaneously, though not completely, resolved at day 3 following SCI (Fig5D); therefore, we injected IFNβ1 24 h after the insult in order to increase Irf7 regulatory activity during the peak of microglial inflammation. Activated microglia were sorted (Supplementary Fig S3A) 48 h and 72 h following SCI from the lesion site area of the injured spinal cords, for RNA extraction and evaluation of Tnfa and Il1b expression by RT-qPCR (Fig5D). Indeed, 12 h following IFNβ1 injection (36 h following SCI), sorted microglia from the lesion site area exhibited significantly elevated Irf7 expression levels compared to control PBS-injected mice (Supplementary Fig S3B). This induction of Irf7 expression by activated microglia was followed by significant reduction in Tnfa and Il1b expression levels observed at 48 h and 72 h following SCI, compared to microglial cells derived from the PBS-injected mice (Fig5E).
Overall, our data demonstrate that the in vivo gene expression profile of adult resident microglia following CNS insult overlaps with the expression signature of myeloid cells that were exposed to TGFβ1. Moreover, we show that IRF7 plays a critical role in M1-to-M2 conversion of myeloid cells by negatively regulating expression of inflammatory pathway genes, such as Il1b, Tnfa, Cxcl1, and Cxcl2, and up-regulating expression of anti-inflammatory genes, such as Il10. Finally, our results demonstrate that restoring Irf7 expression by IFNβ1 reactivates the circuits leading to M2 conversion by improving the resolution of pro-inflammatory cytokines expressed by microglia ex vivo and in vivo, following acute CNS insult.
Discussion
In the present study, we show that TGFβ1, known to be one of the hallmarks of the CNS microenvironment, can imprint a molecular signature on myeloid cells that impairs their ability to switch from a pro- to an anti-inflammatory state. This phenotype switch is dependent on the transcription factor Irf7, found here to be down-regulated by long exposure to TGFβ1. The relevance of these findings to microglial fate in vivo was demonstrated by comparing the signature of adult mouse microglia following injury to that of ex vivo stimulated cells.
Resident microglia are the exclusive innate immune cells of the CNS and maintain normal CNS function during homeostasis (Ziv et al, 2006; Hanisch & Kettenmann, 2007; Stevens et al, 2007; Ransohoff & Perry, 2009; Sierra et al, 2010; Aguzzi et al, 2013). However, under severe acute or chronic activation, activated microglia may become neurotoxic over time, as they fail to undergo self-resolution of their inflammatory phenotype. Under such conditions, the inflammation-resolving function in the CNS is dependent on peripheral assistance from infiltrating mo-MΦ (Simard et al, 2006; Hanisch & Kettenmann, 2007; Shechter et al, 2009; London et al, 2011; Derecki et al, 2012).
Although microglia differ from monocytes in their origin (Ginhoux et al, 2010; Gautier et al, 2012; Schulz et al, 2012), we found here that, similarly to macrophages, NB-Mg have the intrinsic capacity to acquire an anti-inflammatory polarization under extended LPS incubation, which induces M1-to-M2 conversion. Therefore, we suggest that adult microglial incompetence to switch phenotype following severe injury is a consequence of their long exposure to the CNS microenvironment.
TGFβ1 is among the molecules (Vitkovic et al, 2001; Ponomarev et al, 2007; Kierdorf & Prinz, 2013) that constitutively support adult CNS maintenance by contributing to the life-long anti-inflammatory milieu. However, in contrast to the anti-inflammatory effect of short exposure to TGFβ1 (Qian et al, 2008), the present study suggests that continuous exposure to this cytokine (Martinez-Canabal et al, 2013) has significant drawbacks under severe and potentially chronic inflammatory conditions. Thus, using genome-wide expression analysis (Amit et al, 2009, 2011), we found that long exposure to a TGFβ1-enriched milieu led to a dramatic down-regulation in the expression level of the TF, IRF7, a master regulator of IFNα/β immune response against viruses (Honda et al, 2005), which we showed here to be critical for myeloid cell conversion from a pro- to an anti-inflammatory state. While TGFβ/Smad3 signaling regulates the activity of IRF7 and induces IFNβ expression (Qing et al, 2004), in this study, we found that chronic exposure to TGFβ1 down-regulates IRF7 expression. Interestingly, Irf3 was shown elsewhere to suppress neuroinflammation (Tarassishin et al, 2013), suggesting that anti-viral circuits may negatively regulate myeloid cell inflammation, critical for preventing inflammation-induced damage. Yet, we found here that chronic exposure to TGFβ1 had a negative effect on the transcription level of Irf7, but not Irf3, consistent with the reported distinct regulation of these TFs in IFNα/β responses (Sato et al, 1998, 2000). Notably, the long ex vivo exposure of both NB-Mg and BM-MΦ to TGFβ1, found here to impair their key repair-related activities, including phagocytosis, production of ECM-degrading enzymes, and additional basic anti-inflammatory immune functions, is consistent with reports of the limited resolving activity of adult microglia under pathological conditions (Simard et al, 2006; von Bernhardi et al, 2007; Shechter et al, 2011).
Irf7 expression was found here to be functionally linked to the phenotype switch in both BM-MΦ and NB-Mg under inflammatory conditions. The functional link between phenotype switch from M1 to M2 polarization and expression of Irf7 was further substantiated ex vivo by both silencing Irf7 expression in M2-polarized macrophages and showing that treatment with IFNβ1, which up-regulated the IRF7 pathway, restored the phenotype switch of TGFβ1-pretreated microglia. Sorting activated microglia and mo-MΦ from the lesion site of adult spinally injured mice, as well as of naïve microglia and blood monocytes from uninjured animals, confirmed the low Irf7 expression by microglia, similar to the effect of ex vivo exposure to TGFβ1. Notably, high Irf7 expression was associated with the anti-inflammatory gene expression profile of mo-MΦ following SCI. Accordingly, in vivo intervention of IFNβ1 injection, in order to induce Irf7 expression in activated microglia, was resulted in reduced inflammatory gene expression in these cells following SCI.
Taken together, the present study identifies a novel phenomenon of TGFβ1-induced tolerance, demonstrating that long exposure to TGFβ1 induces an altered state of responsiveness to anti-inflammatory signals. Our data revealed that beyond expression of distinctive markers during homeostasis (Butovsky et al, 2013), the TGFβ1-enriched environment impaired microglial ability to switch from M1-to-M2 phenotype under inflammatory conditions, through a reduction in Irf7 expression levels. These findings suggest that the circuitry underlying the exposure of microglia to TGFβ1 within the adult CNS microenvironment might be a double-edged sword, enabling their essential functions under normal physiological conditions, but imprinting incompetence to resolve inflammation under severe pathology. Thus, the tissue microenvironment may have a major effect on the phenotype of myeloid cells residing in it, not only during homeostasis, but also in their subsequent functional response to pathology. Interventions to alter these environmental effects, such as Irf7 induction in resident microglia, might have a therapeutic benefit in reducing CNS inflammation during pathology (Fig6).
Figure 6. A model depicting the molecular mechanism explaining the perturbed M1-to-M2 switch by microglia.

- During homeostasis, the CNS microenvironment is enriched with the anti-inflammatory cytokines TGFβ1 and TGFβ2, and adult microglia express their relevant receptors. As a result of chronic exposure to a TGFβ1-enriched microenvironment, the mRNA levels of Irf7 are down-regulated in microglia, and the anti-viral program is shut off; consequently, the transcription of IRF7-induced genes is suppressed.
- A CNS insult results in the activation of resident microglia and a robust M1 response, characterized by the induction of the inflammatory program (NF-κB) and the transcription of pro-inflammatory cytokines such as Tnfa, Il1b, Cxcl1, and Cxcl2 and the down-regulation of Il10 expression. The low expression levels of Irf7, resulting from long microglial exposure to TGFβ1, prevent the switch to M2 anti-inflammatory phenotype and lead to a vicious cycle of the M1 response in adult microglia.
- Treatment of the TGFβ1-imprinted microglia, under inflammatory conditions, with IFNβ1, induces Irf7 expression and consequently the expression of IRF7-associated genes. IRF7 induction rescues the switch from M1 to M2 phenotype, possibly through inhibition of the NF-κB pathway. The up-regulation of IRF7 results in direct suppression of pro-inflammatory gene expression (e.g., Tnfa, Il1b, Cxcl1 and Cxcl2) and in indirect induction of Il10 transcripts. Black dotted lines denote pathways that are not fully characterized in this study; gray broken lines denote the suggested pathway.
Materials and Methods
Animals
Adult male C57BL/6J, Cx3cr1GFP/+(Jung et al, 2000), and eGFP mice aged 8–10 weeks or neonatal (P0–P1) C57BL/6J mice were used. Animals were supplied by the Animal Breeding Center of the Weizmann Institute of Science. All animals were handled according to the regulations formulated by the Institutional Animal Care and Use Committee (IACUC).
BM radiation chimeras
eGFP > WT BM chimeras were prepared by subjecting mice to lethal split-dose γ-irradiation (300 rad followed 48 h later by 950 rad with head protection). After 1 day following the second irradiation, the mice were injected with 5 × 106 bone marrow cells harvested from the hind limbs (tibia and femur) and forelimbs (humerus) of eGFP donor mice. BM cells were obtained by flushing the bones with Dulbecco's PBS under aseptic conditions and then collected and washed by centrifugation (10 min, 1,250 rpm, 4°C). After irradiation, mice were maintained on drinking water fortified with ciproxin for 1 week to limit infection by opportunistic pathogens. The percentage of chimerism was determined in the blood according to percentages of GFP-expressing cells out of circulating monocytes (CD115). Using this protocol, an average of 90% chimerism was achieved.
Spinal cord injury
The spinal cords of deeply anesthetized mice were exposed by laminectomy at T12, and contusive (200 kdynes) centralized injury was performed using the Infinite Horizon spinal cord impactor (Precision Systems), causing bilateral degeneration without complete penetration of the spinal cord. The animals were maintained on twice-daily bladder expression. Animals that were contused in a nonsymmetrical manner were excluded from the experimental analysis.
Intra-parenchymal injections
The spinal cords of deeply anesthetized mice were exposed 1 day following spinal cord injury, and two injections of 1 μl PBS or IFNβ1 (800 ng/ml) were performed at the margins of the lesion site, in depth of 1.2 mm and injection rate of 250 nl/min.
Flow cytometry analysis and sorting
Mice subjected to spinal cord injury were killed by an overdose of anesthetic, and their spinal cords were prepared for flow cytometric analysis by perfusion with PBS via the left ventricle. The injured sites of spinal cords were dissected from individual mice (parenchymal segments of 0.5 mm from each side of the spinal cord lesion site), and tissues were homogenized using a software-controlled sealed homogenization system (Dispomix; http://www.biocellisolation.com). Cells were analyzed on a FACS-LSRII cytometer (BD Biosciences) using FlowJo software. Isotype controls were routinely used in intracellular experiments. All samples were filtered through an 80-μm nylon mesh and blocked with Fc-block CD16/32 (BD Biosciences). Next, samples were stained using the following antibodies: FITC-conjugated CD11b, Percp Cy5.5-conjugated Ly6C, and PE-conjugated CD115 were purchased from eBioscience; PE-conjugated isotype control IgG2b(k), Pacific Blue-conjugated CD45.2, and APC-conjugated Ly6G were purchased from Biolegend; PE-conjugated IL-10 was purchased from BD Biosciences.
In sorting experiments, 500 microglia and mo-MΦ cells derived from eGFP > WT chimeras were sorted using SORP-FACS sorter (BD Biosciences) into 25 μl of lysis buffer at different time points following SCI. RNA was extracted from sorted cells, DNA libraries were produced, and sequencing was conducted, as described below.
Mixed brain glial and primary microglial cultures
Brains from neonatal (P0–P1) C57BL/6J mice were stripped of their meninges and choroid plexus in Leibovitz-15 medium (Biological Industries, Beit Ha-Emek, Israel). After trypsinization (0.5% trypsin, 10 min, 37°C, 5% CO2), the tissue was triturated. The cell suspension was washed in DMEM supplemented with 10% FCS, 1 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin. The brain glial cells were cultured at 37°C, 5% CO2 in 75-cm2 Falcon tissue-culture flasks (BD Biosciences), pre-coated with poly-D-lysine (PDL) (10 μg/ml; Sigma-Aldrich, Rehovot) for 5 h, and then washed thoroughly with sterile distilled water. The medium was replaced after 24 h in culture and every 2nd day thereafter, for a total culture period of 10–14 days. Microglia were shaken off the primary mixed brain glial cell cultures (170 rpm, 37 °C, 6 h) with maximum yields between days 10 and 14 and seeded (105 cells/ml) onto 24-well plates (1 ml/well; Corning, Corning, NY) pretreated with poly-D-lysine. Cells were grown in culture medium for microglia [RPMI-1640 medium (Sigma-Aldrich, Rehovot) supplemented with 10% FCS, 1 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin]. After seeding, NB-Mg were left untreated, stimulated with 100 ng/ml LPS (E. coli 055:B5, Sigma-Aldrich, Rehovot) for 4 h or stimulated with 100 ng/ml LPS for 20 h, washed with warm culture medium, and re-challenged with 100 ng/ml LPS for 4 h.
Bone marrow macrophage culture
Bone marrow progenitors were harvested from C57BL/6J mice and cultured for 7 days on Petri dishes (0.5 × 106 cells/ml) in RPMI-1640 supplemented with 10% FCS, 1 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 ng/ml M-CSF (Peprotech). At day 7, cells were detached with cold PBS and replated on 24-well tissue culture plates (0.5 × 106 cells/ml; Corning, Corning, NY). On day 8, BM-MΦ were either left untreated, stimulated with 100 ng/ml LPS (E. coli 055:B5, Sigma-Aldrich, Rehovot) for 4 h or stimulated with 100 ng/ml LPS for 20 h, washed with warm culture medium, and re-challenged with 100 ng/ml LPS for 4 h.
Activation reagents
BM-MΦ and NB-Mg were preconditioned for 20 h with 100 ng/ml TGFβ1 (Peprotech), 10 ng/ml IL-4 (Peprotech), 10 ng/ml IL-13 (Peprotech), or 100 ng/ml TGFβ2 (Peprotech), washed with culture medium, stimulated for 20 h with 100 ng/ml LPS, washed again, and then re-challenged for 4 h with 100 ng/ml LPS. Cells were then washed with PBS, and total RNA was extracted. For induction of Irf7 expression, LPS-polarized NB-Mg were stimulated with 1,000 U/ml IFNβ1 (PBL Interferon Source) for 1 h prior to an additional 4 h LPS re-challenge (100 ng/ml).
RNA interference
BM-MΦ were transfected with siRNA directed against Irf7 or scrambled siRNA (Dharmacon) with Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. In brief, siRNA and Lipofectamine were diluted in Opti-MEMI Reduced Serum Medium (Invitrogen), mixed, incubated for 20 min at room temperature, and added to the BM-MΦ cultures. The cells were incubated with the transfection mixture for 5 h, and the BM-MΦ were stimulated as described above. The IRF7 siRNA consisted of four pooled 19-nucleotide duplexes. The sequences of the four duplexes were CCAACAGUCUCUACGAAGA, CCAGAUGCGUGUUCCUGUA, GAGCGAAGAGGCUGGAAGA, and GCCCUCUGCUUUCUAGUGA.
Gene expression analysis
RT-qPCR
NB-Mg and BM-MΦ were stimulated as described above and washed with PBS. Total RNA of in vitro cultured or in vivo sorted cells following SCI was extracted with the miRNeasy kit according to the manufacturer's instructions (Qiagen). For RNA extraction from the spinal cord, the excised tissues were homogenized in Tri-reagent (Sigma-Aldrich) and RNA was extracted with the RNeasy kit according to the manufacturer's instructions (Qiagen). RNA was reverse-transcribed with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), amplified using SYBR green I Master Mix (Roche), and detected by the LightCycler 480 (Roche) in duplicates. Results were normalized to the expression of the housekeeping gene, peptidylprolyl isomerase A (PPIA), and then expressed as fold up-regulation with respect to the control sample. For a list of the primers that were used in this study, refer to Supplementary Methods.
RNA sequencing
NB-Mg and BM-MΦ were harvested at different time points following TGFβ1 or LPS preconditioning. Total RNA was extracted with the miRNeasy kit according to the manufacturer's instructions (Qiagen). RNA concentrations of the samples were measured using Qubit HS RNA kit (Invitrogen), and quality was tested using TapeStation HS RNA. Total RNA (100 ng) was heat-fragmented at 94°C for 5 min into fragments with an average size of 300 nucleotides (NEBNext Magnesium RNA Fragmentation Module), and the 3′ polyadenylated fragments were enriched by selection on poly-dT beads (Dynabeads, Invitrogen). The RNA was reverse-transcribed to cDNA using smart-scribe RT kit (Clontech). Illumina-compatible adaptors were added using NEB Quick ligase, and the DNA library was amplified by PCR using P5 and P7 Illumina-compatible primers (IDT). DNA concentration was measured by Qubit DNA HS, and the quality of the library was analyzed by Tapestation (Agilent). DNA libraries were sequenced on Illumina HiSeq-1500 with average of 5.8 million aligned reads per sample.
Pre-processing of RNA-seq data
All reads were aligned to the mouse reference genome (NCBI 37, MM9) using the TopHat aligner (Trapnell et al, 2009). The raw expression levels of the genes were calculated using Scripture (Guttman et al, 2010), an ab initio software for transcriptome reconstruction. Normalization was performed using DESeq (Anders & Huber, 2010), a method based on the negative binomial distribution, with variance and mean linked by local regression. To analyze genes expressed by NB-Mg and BM-MΦ along the kinetics of TGFβ1 exposure, we identified those genes that were expressed at a threshold greater than 30 (relative to t = 0) on at least one time point along the time course, and among them, we selected only those that showed twofold or greater change in at least one time point relative to others along the kinetics. To analyze genes expressed by sorted microglia and mo-MΦ along the kinetics of following SCI, we identified those genes that were expressed at a threshold greater than 10 (relative to t = 0) on at least one time point along the time course. For further analysis, genes were categorized into functional groups using PANTHER database of gene ontology (Mi et al, 2013).
K-means clustering—Twofold changed genes were clustered by partition of n observations to k clusters in which each observation is assigned to the cluster with the nearest mean. We used the next input, k = 20 and a table log2 data of effect X(t = n) − X(t = 0) and a column of X(t−0). Clusters were manually reordered.
Chromatin immunoprecipitation (ChIP)-Seq
Whole-genome Irf7-binding profiles were obtained using high-throughput chromatin immunoprecipitation (HT-ChIP) as described before (Garber et al, 2012). Briefly, GM-CSF-treated bone-marrow-derived dendritic cells were collected following 2 h of LPS treatment or untreated control. Cells were cross-linked with formaldehyde and lysed, and chromatin was fragmented by sonication. Irf7–DNA complexes were immunoprecipitated using anti-Irf7 antibody (Bethyl laboratories). After thorough washes, reverse cross-linking, and RNase and proteinase K treatment, a sequencing library was generated, followed by Illumina sequencing HiSeq-1500 (50 base, SR). Sequenced data reads were aligned to the mouse reference genome NCBI 37 MM9 using bowtie version 4.1.2. Bowtie alignments were processed by Scripture (Guttman et al, 2010) to obtain significantly expressed transcripts for each time course. Data were filtered by peak intensity of 40.
Statistical analysis
Data were analyzed using Student's t-test to compare between two groups. One-way or two-way ANOVA tests were used to compare several groups; the Bonferroni post-test (P = 0.05) was used for follow-up pairwise comparison of groups. Kolmogorov–Smirnov test was used to compare distributions. Hypergeometric distribution test was used to compare observed and expected gene lists size. The specific tests used to analyze each set of experiments are indicated in the figure legends. The results are presented as mean ± standard error mean (SEM). *P < 0.05, **P < 0.01, ***P < 0.001.
Data deposition
RNA-seq and ChIP-seq data are deposited in the Gene Expression Omnibus (GEO) database under accession numbers GSE62698 and GSE62697, respectively. The data can be viewed from the following Web site: http://www.ncbi.nlm.nih.gov/geo.
Acknowledgments
We thank S. Schwarzbaum for proofreading and M. Azoulay for handling the animals. This work was supported by the European Research Council (E.R.C.) Award given to M.S. (Grant no. 232835) and by the EU Seventh Framework Program HEALTH-2011 given to M.S. (Grant no. 279017). M. Schwartz holds the Maurice and Ilse Katz Professorial Chair in Neuroimmunology. Research in I. Amit laboratory is supported by a grant from the European Research Council (309788), the Israeli Science Foundation (1782/11) and the Human Frontiers Science Program, Career Development Award.
Author contributions
MC, OM, IA, and MS conceived and designed this study. MC and OM performed all the experiments and conducted data analysis. AS was involved in the early discussion and the setup of the in vitro LPS model. ED and ZBI contributed for the transcriptome data analysis. HKS and DAJ contributed for the libraries preparation of the RNA-seq. RBG performed the ChIP-seq assay. MC, OM, IA, and MS wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
Supplementary Figures
Supplementary Information
Review Process File
References
- Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339:156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, Schubert LA, Birditt B, Shay T, Goren A, Zhang X, Smith Z, Deering R, McDonald RC, Cabili M, Bernstein BE, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Regev A, Hacohen N. Strategies to discover regulatory circuits of the mammalian immune system. Nat Rev Immunol. 2011;11:873–880. doi: 10.1038/nri3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- von Bernhardi R, Ramirez G, Toro R, Eugenin J. Pro-inflammatory conditions promote neuronal damage mediated by Amyloid Precursor Protein and decrease its phagocytosis and degradation by microglial cells in culture. Neurobiol Dis. 2007;26:153–164. doi: 10.1016/j.nbd.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2013;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A, Musella A, D'Amelio M, Cavallucci V, Martorana A, Bergamaschi A, Cencioni MT, Diamantini A, Butti E, Comi G, Bernardi G, Cecconi F, Battistini L, Furlan R, Martino G. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci. 2009;29:3442–3452. doi: 10.1523/JNEUROSCI.5804-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constam DB, Philipp J, Malipiero UV, ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J Immunol. 1992;148:1404–1410. [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Investig. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch CE, Laping NJ, Morgan TE, Nichols NR, Pasinetti GM. TGF-beta 1 is an organizer of responses to neurodegeneration. J Cell Biochem. 1993;53:314–322. doi: 10.1002/jcb.240530408. [DOI] [PubMed] [Google Scholar]
- Flanders KC, Ren RF, Lippa CF. Transforming growth factor-betas in neurodegenerative disease. Prog Neurobiol. 1998;54:71–85. doi: 10.1016/s0301-0082(97)00066-x. [DOI] [PubMed] [Google Scholar]
- Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, Robinson J, Minie B, Chevrier N, Itzhaki Z, Blecher-Gonen R, Bornstein C, Amann-Zalcenstein D, Weiner A, Friedrich D, Meldrim J, Ram O, Cheng C, Gnirke A, Fisher S, et al. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol Cell. 2012;47:810–822. doi: 10.1016/j.molcel.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma'ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ Immunological Genome C. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Auffray C, Palframan R, Wirrig C, Ciocca A, Campisi L, Narni-Mancinelli E, Lauvau G. Blood monocytes: distinct subsets, how they relate to dendritic cells, and their possible roles in the regulation of T-cell responses. Immunol Cell Biol. 2008;86:398–408. doi: 10.1038/icb.2008.19. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Perdiguero E, Schulz C, Geissmann F. Development and homeostasis of “resident” myeloid cells: the case of the microglia. Glia. 2013;61:112–120. doi: 10.1002/glia.22393. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, Rinn JL, Lander ES, Regev A. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Henkel GW, McKercher SR, Yamamoto H, Anderson KL, Oshima RG, Maki RA. PU.1 but not ets-2 is essential for macrophage development from embryonic stem cells. Blood. 1996;88:2917–2926. [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, Becher B, Aguzzi A. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Investig. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyong A, Jadhav V, Lee S, Tong W, Rowe J, Zhang JH, Tang J. Rosiglitazone, a PPAR gamma agonist, attenuates inflammation after surgical brain injury in rodents. Brain Res. 2008;1215:218–224. doi: 10.1016/j.brainres.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Schwartz M. Non-identical twins - microglia and monocyte-derived macrophages in acute injury and autoimmune inflammation. Front Immunol. 2012;3:89. doi: 10.3389/fimmu.2012.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci. 2008;13:1813–1826. doi: 10.2741/2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C, Muller DN, Luckow B, Brocker T, Debowski K, Fritz G, Opdenakker G, Diefenbach A, Biber K, Heikenwalder M, Geissmann F, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16:273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Prinz M. Factors regulating microglia activation. Front Cell Neurosci. 2013;7:44. doi: 10.3389/fncel.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London A, Itskovich E, Benhar I, Kalchenko V, Mack M, Jung S, Schwartz M. Neuroprotection and progenitor cell renewal in the injured adult murine retina requires healing monocyte-derived macrophages. J Exp Med. 2011;208:23–39. doi: 10.1084/jem.20101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Canabal A, Wheeler AL, Sarkis D, Lerch JP, Lu WY, Buckwalter MS, Wyss-Coray T, Josselyn SA, Frankland PW. Chronic over-expression of TGFbeta1 alters hippocampal structure and causes learning deficits. Hippocampus. 2013;23:1198–1211. doi: 10.1002/hipo.22159. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, Maki RA. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15:5647–5658. [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schlevogt B, Kierdorf K, Bottcher C, Erny D, Kummer MP, Quinn M, Bruck W, Bechmann I, Heneka MT, Priller J, Prinz M. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. J Neurosci. 2011;31:11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27:10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, Dieli F, Ghisletti S, Natoli G, De Baetselier P, Mantovani A, Sica A. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci USA. 2009;106:14978–14983. doi: 10.1073/pnas.0809784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Wei SJ, Zhang D, Hu X, Xu Z, Wilson B, El-Benna J, Hong JS, Flood PM. Potent anti-inflammatory and neuroprotective effects of TGF-beta1 are mediated through the inhibition of ERK and p47phox-Ser345 phosphorylation and translocation in microglia. J Immunol. 2008;181:660–668. doi: 10.4049/jimmunol.181.1.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing J, Liu C, Choy L, Wu RY, Pagano JS, Derynck R. Transforming growth factor beta/Smad3 signaling regulates IRF-7 function and transcriptional activation of the beta interferon promoter. Mol Cell Biol. 2004;24:1411–1425. doi: 10.1128/MCB.24.3.1411-1425.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Rapalino O, Lazarov-Spiegler O, Agranov E, Velan GJ, Yoles E, Fraidakis M, Solomon A, Gepstein R, Katz A, Belkin M, Hadani M, Schwartz M. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med. 1998;4:814–821. doi: 10.1038/nm0798-814. [DOI] [PubMed] [Google Scholar]
- Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998;441:106–110. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, Kim KW, Klein E, Kalchenko V, Bendel P, Lira SA, Jung S, Schwartz M. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. 2013;38:555–569. doi: 10.1016/j.immuni.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Raposo C, London A, Sagi I, Schwartz M. The glial scar-monocyte interplay: a pivotal resolution phase in spinal cord repair. PLoS One. 2011;6:e27969. doi: 10.1371/journal.pone.0027969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Tarassishin L, Bauman A, Suh HS, Lee SC. Anti-viral and anti-inflammatory mechanisms of the innate immune transcription factor interferon regulatory factor 3: relevance to human CNS diseases. J Neuroimmune Pharmacol. 2013;8:132–144. doi: 10.1007/s11481-012-9360-5. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitkovic L, Maeda S, Sternberg E. Anti-inflammatory cytokines: expression and action in the brain. NeuroImmunoModulation. 2001;9:295–312. doi: 10.1159/000059387. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T. Transforming growth factor-beta signaling pathway as a therapeutic target in neurodegeneration. J Mol Neurosci. 2004;24:149–153. doi: 10.1385/JMN:24:1:149. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Yin Y, Henzl MT, Lorber B, Nakazawa T, Thomas TT, Jiang F, Langer R, Benowitz LI. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat Neurosci. 2006;9:843–852. doi: 10.1038/nn1701. [DOI] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9:268–275. doi: 10.1038/nn1629. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Information
Review Process File