

To the Editor
The analysis of big genomic data sets today engenders an all-or-nothing approach, i.e., complete, end-to-end analysis, which is time consuming and unintuitive; it also requires considerable computational expertise and costly computer infrastructure, effectively excluding many bench biologists from genome-scale analyses. We have developed and are continually expanding a web-based analysis system, iobio (http://iobio.io/), to empower all biological researchers to analyze—easily, interactively and in a visually driven manner—large biomedical data sets that are essential for their research, without onerous resource requirements. A primary example of genome-scale ‘big data’ is the BAM1 format DNA sequence alignment file. BAM files underlie diverse types of genetic analyses, acting as the universal currency of high-throughput sequence analysis. Here we report the first complete iobio web app, bam.iobio (Fig. 1; http://bam.iobio.io/), an open-source dashboard web application providing an insightful overview of the contents of these large, non–human-readable BAM files and enabling users to further analyze their alignments, all in real time.
Figure 1.
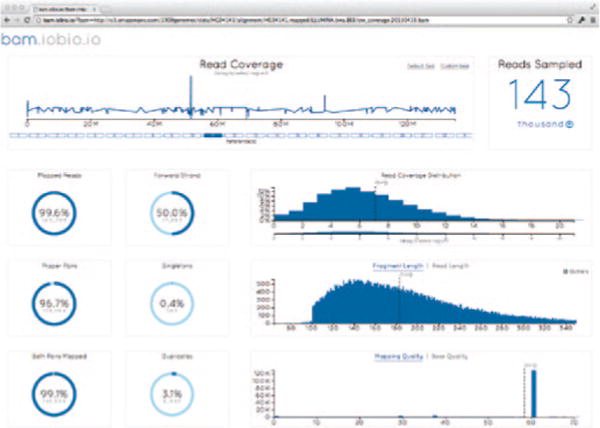
The bam.iobio.io web application. The user selects an alignment file, and the application rapidly samples the entire file to estimate genome-wide averages for a set of informative alignment metrics. Additionally, the user is able to select specific regions of interest and redo the analysis in those regions in seconds.
The user selects a BAM file either hosted remotely or from his or her own computer’s hard drive, and then our app calculates and displays, within a few seconds, crucial information about the sequence alignment: (i) the average read coverage and its distribution, (ii) the composition of the data set according to read length, (iii) the fragment-length average, distribution and outliers, (iv) the histogram of base quality values (to identify a bad sequencing run) and read duplication rate (to identify low library complexity), and (v) the histogram of mapping quality values and fraction of properly mapped read pairs (to identify poor mapping results).
Collecting such vital alignment statistics using current tools requires placing the BAM file on a Unix machine and then installing and running Unix programs such as SAMTools1 or BamTools2 on the entire BAM file. This process may take hours to complete, e.g., the 18-gigabyte BAM file in our tests took 8 hours to process (Supplementary Table 1). In contrast, our approach is to collect a random sample of the read alignments (Supplementary Fig. 1) to accurately estimate the same alignment statistics in seconds (Supplementary Fig. 2). Notably, sampling takes place where the BAM file is stored (i.e., on cloud storage or a user’s hard drive), and only the sampled data—a tiny fraction of the entire BAM file—are ever transmitted. The alignments are then streamed to data analysis web services that produce appropriate alignment statistics in seconds before transmitting these to bam.iobio for visualization (for implementation details, licensing and deployment considerations, see Supplementary Note; for system compatibility, see Supplementary Table 2). We can now analyze the same 18-gigabyte alignment file in <10 seconds. Real-time visualization allows the user to experience how the statistical distributions progressively converge and become stable as sampled alignment data are collected. The user can further explore the data interactively by selecting other chromosomes or chromosomal subregions, using the main read coverage panel for navigation.
This web app puts forward an interactive and intuitive genomic data analysis paradigm that is not achievable with existing systems, enabling users to analyze both local and remotely stored data, without tool installation or transmitting large data sets, and immediately see informative results. We are developing other real-time analysis applications: for example, to analyze multiple alignment files simultaneously using our sampling approach, and for interactive, complete analysis of genomic data in smaller genomic windows, such as in the region of a gene (demos at http://iobio.io/). We are also creating software libraries for third-party developers to build similar interactive web apps. Although large, whole-genome computation will remain essential for many tasks, we expect that web-based, visually driven, real-time tools will offer a powerful new analysis modality for bioinformatics experts and bench scientists alike.
Supplementary Material
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper (doi:10.1038/nmeth.3174).
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Li H, et al. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, Marth GT. Bioinformatics. 2011;27:1691–1692. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.