

Abstract
Introduction
Current methods for treating prolonged and neuropathic pain are inadequate and lead to toxicities that greatly diminish quality of life. Therefore, new approaches to the treatment of pain states are needed to address these problems.
Areas covered
The review primarily reviews approaches that have been taken in the peer-reviewed literature of multivalent ligands that interact with both μ and δ opioid receptors as agonists, and in some cases, also with pharmacophores for antagonist ligands that interact with other receptors as antagonists to block pain.
Expert opinion
Although there are a number of drugs currently on the market for the treatment of pain; none of them are 100% successful. In the authors’ opinion, it is clear that new directions and modalities are needed to better address the treatment of prolonged and neuropathic pain; one drug or class clearly is not the answer for all pain therapy. Undoubtedly, there are many different phenotypes of prolonged and neuropathic pain and this should be one avenue to further develop appropriate therapies.
Keywords: agonist, multifunctionality, multivalency, opioid, pain, receptors
1. Introduction
There is no human being in the entire world who has not faced some kind of pain at some point of time in her/his life. It is essential for our survival. According to the International Association for the Study of Pain (IASP), pain is defined as ‘an unpleasant sensory and emotional experience associated with actual or potential tissue damage or described in terms of such damage’ [1]. Pain can be classified in numerous ways and accordingly, different types of pain are discussed in the literature. Pain has significant physical, economic and social impact. Approximately 1.5 billion people around the globe suffer from chronic pain [2]. The costs associated with pain treatment are much higher than that involved for the treatment of heart disease or cancer [3]. Generally, acute pain associated with accidental injury or surgery is cured. But nearly 50% of patients who have gone through surgery face chronic pain [4]. Under-treatment of postsurgical acute pain has been found as a major reason for moderate to severe or even extreme pain in two thirds of these patients [5]. Support in favor of these statements comes from the observations made during the study on effective treatment of patients with acute pain [6,7].
In spite of having many serious side effects including respiratory depression, sedation, constipation, physical dependence and development of tolerance [8,9], opioid agonists have long been the mainstay analgesics for the treatment of various pain states because of their potency, efficacy and availability. Three classical opioid receptors, namely μ-, δ- and κ-opioid receptors (MOR, DOR and KOR, respectively), have been identified in the central nervous system (CNS) by pharmacological studies [10,11]. The common opioid drugs including morphine, codeine, oxycodone, methadone, heroin, morphine-6β-glucuronide (M6G), fentanyl, etc., which are used clinically for analgesic effects, mainly target the MOR. Most studies have confirmed that the μ-opioid receptor is primarily responsible for the antinociceptive activity. However, a number of studies have suggested that ligands with dual μ-and δ-agonist activities display better biological profiles compared to the ones acting selectively on MOR [12,13]. There is also evidence that the presence of DOR agonists can improve the analgesic efficacy of MOR agonists [14,15]. KORs, broadly found in the spinal cord, the dorsal ganglia, the periphery and the supraspinal regions, are associated with pain modulation, but are not discussed in this review due to page limitations. In addition, we do not discuss glycopeptides that target opioid receptors though they show great promise, nor other peptide conjugates that also may be useful. In addition, there is a huge amount of literature about non-peptide ligands that interact with opiate receptors that is not discussed here.
1.1 Probable paths to counteract the present problems
To overcome the difficulties in pain treatment described above, it has been proposed that new approaches to drug design are needed to deal with recent observations that in the development of prolonged and neuropathic pain states, there are critically important changes in the expressed genome in ascending and descending pain pathways, and in the CNS that result from upregulation of neurotransmitter receptors and their ligands that are stimulatory and thus can cause pain. These anti-opioid ligands and receptors need to be considered in drug design. Therefore, there is a need to develop approaches to design ligands that are multivalent and, therefore, can act at two, three or more receptors all with a single molecule. In this review, the authors will discuss some of these new approaches.
Here, it should be mentioned that the in-depth molecular-level understanding of the interactions between opioid ligands and their receptors is also very important for successful design of new drugs. Recent reports on how opioid ligands bind to their receptors by high-resolution crystal structures of three opioid receptor subtypes, that is the MOR [16], DOR [17] and KOR [18] have opened an additional opportunity to discover novel ligands targeting these G-Protein Coupled Receptors (GPCRs) that might ultimately be developed into more useful therapeutics [19,20]. However, it is important to note that the X-ray structures (conformations) of opioid receptors were occupied by antagonists and the conformation of agonist occupied receptors will clearly be different from an antagonist occupied receptor. Agonists and antagonists clearly have different structure-activity relationships (SARs) for opioid receptors.
In this review, our focus will be on agonists, which target the μ and δ opioid receptors. Some selective literature reports have been taken to discuss the drug design principles and SAR study. Emerging drug discovery approaches of multivalency/multifunctionality targeting both opioid and non-opioid targets all in a single molecule for the treatment of prolonged and neuropathic pain, also will be discussed. In addition, this review covers some selective potential drug candidates, which are in a preclinical investigational stage. Though some general aspects of non-peptide ligands for opioid receptors will be discussed, in this review the authors will focus primarily on peptide and peptidomimetic ligands for opioid receptors since the natural endogenous ligands for opioid receptors are peptides.
1.2 Literature search methodology
The literature search was carried out using SciFinder and PubMed. The phrases like ‘opioid agonist’, ‘opioid agonists for the treatment of pain’ and ‘peptide and peptidomimetic opioid ligands’ have been used during the search. Author search was conducted with well-known scientists’ names in this field. To make the review concise, we have taken our topic related publications starting from 2006 to the end of 2012, unless it was very much required for our discussion. Though there were scopes to include investigational drug candidates in Phase I and Phase II clinical trials, we left them out of this review because of the fact that we have limited access to the scientific data for those compounds.
New avenues of drug design are being developed which can provide novel ligands that are efficacious for the treatment of prolonged and neuropathic pain without the serious side effects of toxicities of current analgesics and which do not develop tolerance with prolonged use.
We strongly believe that the topic chosen and areas covered in this review are timely and very important in stimulating the scientific community to think of new approaches for management of the world’s most common health problem, that is unwanted pain in human daily life.
2. Background
Opioid agonists are the main clinically used drugs for the treatment of pain. Although they can act on all three subtypes of opioid receptors, most of the current drugs’ analgesic effects are mainly due to the activation of MOR present in the CNS. One of the key reasons of having limited a number of centrally acting drugs is due to the presence of the blood–brain barrier (BBB), which put forward some restriction for foreign molecules to enter into the brain. The BBB permits hydrophobic and selected molecules to pass through it. But hydrophobic agents are difficult to transport through blood which requires more hydrophilic nature of the drug candidates. These two opposite requirements by the blood and the BBB have made it a challenging job for scientists to discover and develop new drugs, which can be delivered into the CNS. Another very important issue associated with development of centrally acting opioid drugs is their metabolic stability. This is because of the fact that therapeutic agents should have half-lives in the acceptable range so that they can interact with their biological targets for a sufficient duration of time to produce the desired response.
3. Investigational opioid receptor agonists
To overcome the limitations of the existing opioid drugs, many approaches have been taken over the last few decades. Studies reported in the literature suggest the issues of metabolic stability and blood–brain barrier permeability should be taken into consideration at the very beginning stage of drug design. If a drug candidate is not stable enough to the enzymatic action in the physiological systems, it cannot reach the CNS. Again if it reaches the CNS, it should cross the BBB to activate the receptors in the brain. The scientific community has been trying to solve these two key issues. In the next sections, we will discuss the pharmacology and in vivo studies with some investigational opioid agonists.
3.1 μ-selective opioid agonists
Replacement of a disulfide bridge of bioactive peptides by an ethylene (–CH=CH–) or a bismethylene (–CH2–CH2–) moiety can provide a different biological profile. In an attempt to increase the metabolic stability of two dermorphin-derived cyclic tatrapeptide analogs H-Tyr-c[D-Cys-Phe-Cys]-NH2 (Ligand 4, Table 1) and H-Tyr-c[D-Cys-Phe-D-Cys]-NH2 (Ligand 7, Table 1) [21], which are agonists at MOR and DOR, Berozowska et al. [22] replaced the disulfide bridge by a bis-methylene moiety. All olefinic bond containing peptides 1, 2, and 5 (Table 1) showed considerably reduced μ and δ agonist potencies in the guinea pig ileum (GPI) and mouse vas deferens (MVD) assays, respectively. But, the –CH2–CH2–bridged peptide 3 with L-configuration in the fourth position showed comparable potency with its cysteine-containing parent compound 4 in both assays. The bis-methylene analog 6 with D-configuration in second and fourth positions became 10 – 27 less potent compared to its parent disulfide peptide 7. Berozowska et al. [23] also synthesized dicarba analogs of two cyclic opioid penta-peptides H-Tyr-c[D-Cys-Gly-Phe-D(or L)-Cys]-NH2 [24,25] evaluated for their pharmacological activities. The trans isomer 9 of H-Tyr-c[D-Allylgly-Gly-Phe-L-Allylgly]-NH2 showed a μ and δ agonist (IC50μ = 0.898 nM, IC50δ = 0.275 nM) activities with high potency (Kiμ = 0.616 nM, Kiδ = 1.25 nM) at both receptors.
Table 1.
Pharmacological data of Dicarba analogs of H-Tyr-c[D-Cys-Phe-L(or D)-Cys]-NH2 and H-Tyr-c[D-Cys-Gly-Phe-L(or D)-Cys]-NH2 at opioid receptors (OR).

| |||||||
|---|---|---|---|---|---|---|---|
| Compound number | GPI | MVD | GPI/MVD | OR binding affinity | Binding ratio | ||
|
| |||||||
| IC50 (nM) | IC50 (nM) | IC50 ratio | Kiμ (nM) | Kiδ (nM) | Kiκ (nM) | Kiμ/Kiδ/Kiκ | |
| 1 | 436 | 460 | 1/1.06 | – | – | – | – |
| 2 | 162 | 444 | 1/2.74 | – | – | – | – |
| 3 | 76.5 | 655 | 1/8.56 | – | – | – | – |
| 4 | 64.7 | 740 | 1/11.4 | 11.0 | 373 | – | 1/34/– |
| 5 | 176 | 246 | 1/1.40 | – | – | – | – |
| 6 | 208 | 786 | 1/3.78 | – | – | – | – |
| 7 | 20.0 | 28.8 | 1/1.44 | – | – | – | – |
| 8 | 1.81 | 0.496 | 3.65/1 | 2.4 | 6.55 | 200 | 1/3/83 |
| 9 | 0.898 | 0.275 | 3.27:1 | 0.616 | 1.25 | 57.6 | 1/2/94 |
| 10 | 1.02 | 3.19 | 1/3.12 | 2.34 | 5.87 | 309 | 1/3/132 |
| 11 | 1.65 | 0.603 | 2.74/1 | 1.74 | 1.61 | 40.1 | 1/1/23 |
| 12 | 11.9 | 9.91 | 1.2/1 | 2.27 | 7.04 | 498 | 1/3/219 |
| 13 | 5.63 | 4.38 | 1.3/1 | 1.05 | 1.92 | 30.4 | 1/2/29 |
| 14 | 11.2 | 3.64 | 3.1/1 | 1.17 | 3.34 | 71.5 | 1/3/61 |
| 15 | 1.30 | 0.562 | 2.31/1 | 0.550 | 0.822 | 44.9 | 1/1/82 |
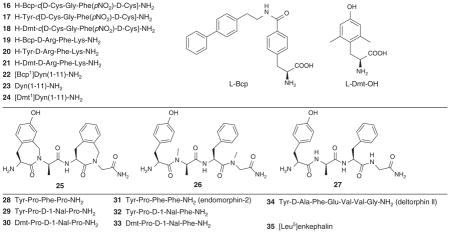
Weltrowska et al. [26] reported that analogs 16, 19 and 22 of the opioid peptides H-Tyr-c[D-Cys-Gly-Phe(pNO2)-D-Cys]-NH2 (non-selective ligand 17) [27], H-Tyr-D-Arg-Phe-Lys-NH2 (μ-selective ligand 20) [28] and dynorphin A(1-11)-NH2 (κ-selective ligand 23) containing 4′-[N-((4′-phenyl)-phenethyl)carboxamido]phenylalanine (Bcp) in place of Tyr1 maintained high μ opioid receptor binding affinity, but displayed very substantial differences in the opioid receptor profiles while compared to the corresponding Tyr1-containing parent peptides (Table 2). The cyclic peptide H-Bcp-c[D-Cys-Gly-Phe(pNO2)-D-Cys]-NH2 (16) appeared as a highly potent, μ-selective opioid agonist, whereas the Bcp1-analogue of dynorphin A(1-11)-NH2 showed partial agonism at the μ-receptor. These results suggest that the presence of bulky biphenylethyl moiety in these compounds might be involved in a hydrophobic interaction with a receptor sub site and thereby might play a role in the ligand’s ability to induce a specific receptor conformation or to bind to a separate receptor conformation in a state of conformational receptor heterogeneity.
Table 2.
Binding affinity and functional activities at ORs.
| |||||||
|---|---|---|---|---|---|---|---|
| Compound number | GPI | MVD | GPI/MVD | OR binding affinity | Binding ratio | ||
|
| |||||||
| IC50 (nM) | IC50 (nM) | IC50 ratio | Kiμ (nM) | Kiδ (nM) | Kiκ (nM) | Kiμ/Kiδ/Kiκ | |
| 16 | 0.226 | 0.319 | 1/1.4 | 0.293 | 9.99 | 6.93 | 1/34/24 |
| 17 | 0.717 | 0.211 | 3.4/1 | 0.443 | 0.389 | 1.53 | 1/0.9/3.5 |
| 18 | 0.542 | 0.182 | 3/1 | 0.247 | 0.704 | 3.77 | 1/3/15 |
| 19 | 0.984 | 4.60 | 1/4.7 | 0.948 | 102 | 1.39 | 1/108/1.5 |
| 20 | 254 | 781 | 1/3 | 1.69 | 19200 | 4230 | 1/11360/2503 |
| 21 | 1.41 | 23.1 | 1/16 | 0.143 | 2100 | 22.3 | 1/14685/156 |
| 22 | 0.591* | 1.07 | – | 3.41 | 13.9 | 1.56 | 1/4/0.5 |
| 23 | 0.376 | 15.2 | 1/40 | 0.653 | 13.5 | 0.087 | 1/21/0.13 |
| 24 | 0.502 | 0.377 | 1.3/1 | 0.322 | 0.435 | 1.18 | 1/1.4/3.7 |
| 25 | 3.64 | 30.3 | 1/8 | 20.8 | 17.8 | – | 1.2/1/– |
| 26 | 22.0 | 79.7 | 1/4 | 28.1 | 7.1 | – | 4/1/– |
| MF/Morphine | 29.3 | 155 | 1/5 | 9.77 | 112.2 | – | 1/11/– |
| 28 | 318 | 4800 | 1/15 | 19.8 | > 1000 | – | 1/> 50/– |
| 29 | 9.57 | 35.4 | 1/3.6 | 0.47 | > 1000 | 1/> 2127/– | |
| 30 | 0.45 | 0.64 | 1/1.4 | 0.00055 | 132 | – | 1/24044/– |
| 31 | 7.71 | 15.3 | 1/2 | 0.79 | > 1000 | – | 1/> 1266/– |
| 32 | 872 | 2040 | 1/2.3 | 22.3 | > 1000 | – | 1/> 45/– |
| 33 | 1.53 | 1.63 | 1/1.1 | 0.93 | > 1000 | 1/> 1075/– | |
| 34 | – | – | – | – | 0.56 | – | –/–/– |
| 35 | 246 | 11.4 | 21.6/1 | – | – | – | –/–/– |
Partial agonist.
Ballet et al. [29] compared the opioid activity of two dermorphin analogs having an almost identical structure but different structural flexibility. Conformational restriction of the aromatic amino acid residues has been a successful strategy to increase the potency, selectivity and metabolic stability of peptides [30]. Compounds 25 and 26 were derived from the parent Compound 27 to increase the lipophilicity and conformational restriction (Table 2). The main difference between 25 and 26 is in their structural rigidity. In Ligand 25, the aromatic side chains became the parts of lactam structures, which caused the conformational restriction of the ligand. In Ligand 26 they remained as such and as a result the side chains were flexible. Ligand 25 displayed comparable binding affinities for MOR and DOR (Kiμ = 20.8 nM, Kiδ = 17.8 nM), but eight times more agonist activity at μ compared to that at δ. The analog 25 showed the highest potency for in vitro GPI (IC50μ = 20.8 nM) assay.
However, both compounds (25 and 26) showed similar antinociception profile (AUC = 7820 and 8732, respectively, where AUC is the area under the curve) in the mouse tail flick test after intravenous administration of a dose of 2 mg/kg for each compound (Figure 1). This observation suggests that lipophilicity, rather than side chain flexibility, is the key decisive factor for BBB penetration.
Figure 1.

Analgesic effect as % MPE of 25 (1) and 26 (2) in the mouse tail-flick assay at different times after intravenous administration.
**P < 0.001; *P < 0.05.
Adapted from [29] with permission of the American Chemical Society.
In an effort to discover new drug candidates with better anti-nociception effect, Fichna et al. [31] synthesized [D-1-Nal3] analogs of morphiceptin and endomorphin-2, and evaluated them for their in vitro (Table 2) and in vivo activities. Ligand 30 ([Dmt1, D-1-Nal3]morphiceptin) showed very high affinity and selectivity for MOR in the receptor binding assays (Kiμ = 0.000549 nM, Kiδ = 132 nM). It displayed high agonist activities at both the μ- and δ-receptors (IC50μ = 0.45 nM, IC50δ = 0.45 nM).
A profound supraspinal analgesia (around 100-fold more potent than the endogenous MOR ligand, endomorphin-2) has been reported for [Dmt1, D-1-Nal3]morphiceptin (30) by conducting the mouse hot plate test after administration of the ligand intracerebroventricularly (i.c.v.) (Figure 2). Study of time-course revealed that the antinociceptive activity of analog 30 could persist up to 2 h (Figure 3). This study suggested [Dmt1, D-1-Nal3]morphiceptin as a promising lead compound for the design of new analgesic drug candidates.
Figure 2.

Dose–response curves determined in the hot plate test for the inhibition of paw licking (A), rearing (B), and jumping (C) induced by i.c.v. injection of morphiceptin, EM-2, [Dmt1, D-1-Nal3]morphiceptin (Analog 30 (3)), and [Dmt1, D-1-Nal3]EM-2 (Analog 33 (6)). The data represent the mean – SEM of 10 mice per group.
Adapted from [31] with permission from Elsevier.
Figure 3.

Time-course of the changes in inhibition of paw licking (A), rearing (B), and jumping (C) induced by i.c.v. injection of morphiceptin (10 μg), EM-2 (3 μg), and [Dmt1, D-1-Nal3]morphiceptin (Analog 30 (3), 0.1 μg), determined in the hot plate assay.
Adapted from [31] with permission from Elsevier.
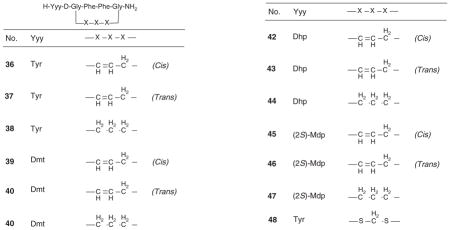
The opioid ligand H-Tyr-c[D-Cys-Phe-Phe-Cys]-NH2 (48, Table 3) cyclized through a methylene dithiother has been shown as a potent and selective MOR agonist [32]. Berezowska and coauthors prepared dicarba analogs (ligands 35 – 47, Table 3) 33 of 48 containing Tyr, 2′,6′-dimethyltyrosine (Dmt), 3-[2,6-dimethyl-4-hydroxyphenyl)propanoic acid (Dhp) or (2S)-2-methyl-3-(2,6-dimethyl-4-hydroxyphenyl) propanoic acid [(2S)-Mdp] in the position one, employing Fmoc/tBu solid-phase approach and substituting D-allylglycine and (2S)-2-amino-5-hexenoic acid in position 2 and 5, respectively, followed by ring-closing metathesis. On catalytic hydrogenation of cis and trans mixtures of isomeric olefinic peptides yielded the saturated –CH2–CH2–bridged peptides. All six Tyr1- and Dmt1-dicarba analogs (36 – 41, Table 3) showed high μ- and δ-opioid agonist potency and produced only small or no preference for MOR over DOR. The six Dhp1- and (2S)-Mdp1-dicarba analogs (42 – 47, Table 3) showed antagonist activities at MOR but, surprisingly, displayed a range of different efficacies (agonism, partial agonism or antagonism) at the DOR. The results of this study suggested that the μ versus δ receptor selectivity and the efficacy at the DOR of these cyclic ligands depend on distinct conformational features of the 15-membered peptide ring framework, which might affect the orientation of the exocyclic residue and of the Phe3 and Phe4 side chains.
Table 3.
In vitro binding affinity and functional activity data of dicarba-cyclic ligands.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound number | GPI | MVD | GPI/MVD | OR binding affinity | Binding ratio | ||||
|
| |||||||||
| IC50(nM) | Ke (nM) | IC50 (nM) | Ke (nM) | IC50 ratio | Kiμ (nM) | Kiδ (nM) | Kiκ (nM) | Kiμ/Kiδ/Kiκ | |
| 36 | 0.401 | – | 0.851 | – | 1/2 | 0.281 | 0.92 | 9.36 | 1/3/12 |
| 37 | 1.34 | – | 4.51 | – | 1/3 | 0.332 | 1.53 | 15.2 | 1/5/14 |
| 38 | 1.05 | – | 0.740 | – | 1.4/1 | 1.04 | 2.24 | 11.3 | 1/2/11 |
| 39 | 0.161 | – | 0.191 | – | 1/1.2 | 0.528 | 0.49 | 12.8 | 1/1/24 |
| 40 | 0.407 | – | 0.0444 | – | 9/1 | 0.438 | 0.29 | 4.97 | 1/1/11 |
| 41 | 0.509 | – | 0.0655 | – | 8/1 | 1.54 | 1.20 | 15.8 | 1/1/10 |
| 42 | – | 32.2 | PA (e = 0.38) | – | – | 23.5 | 86.3 | 266 | 1/4/11 |
| 43 | – | 44.3 | 239 | – | – | 16.7 | 27.0 | > 500 | 1/2/> 30 |
| 44 | – | 41.7 | 7500 | – | – | 8.95 | 39.2 | > 500 | 1/4/> 56 |
| 45 | – | 32.4 | PA | – | – | 7.44 | 7.67 | 185 | 1/1/25 |
| 46 | – | 157 | PA (e = 0.33) | – | – | 323 | 35.3 | 635 | 1/0.1/2 |
| 47 | – | 25.7 | 9.91 | 147 | – | 18.3 | 18.7 | 302 | 1/1/17 |
| 48 | – | – | – | – | – | 0.016 | 1.8 | 2.5 | 1/112/156 |
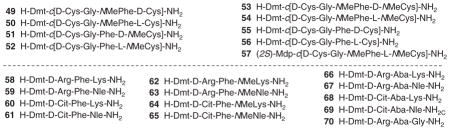
In an attempt to increase the bioavailability of the non-selective, cyclic enkephalin analogs H-Dmt-c[D-Cys-Gly-Phe-D(or L)-Cys]-NH2 (Dmt = 2′,6′-dimethyltyrosine), Weltrowska et al. [34] prepared analogs containing N-methyl group at the Phe4 and/or Cys5 residue. In comparison with the non-methylated parent peptides (55, 56), all mono- (49 – 52) and di-N-methylated (53 and 54) analogs in general showed high binding affinities at all three opioid receptors and high opioid agonist activities in functional opioid assays (Table 4). The observed results suggested that the local conformational restriction in these compounds by mono- and di-N-methylation did not greatly affect the in vitro opioid activity results. H-Dmt-c[D-Cys-Gly-NMePhe-L-NMeCys]-NH2 (54), a conformationally restricted and low-energy conformer identified from the series, showed good spatial overlap of the crucial pharmacophoric subsites with those in the proposed MOR-bound conformation of the μ-selective opioid peptide JOM-6 [H-Tyr-c(S-Et-S)[D-Cys-Phe-D-Pen]-NH2] (Pen = penicillamine) [35]. This is in agreement with the moderate μ-selectivity determined for this compound. Substitution of Dmt1 from 54 by (2S)-2-methyl-3-(2,6-dimethyl-4-hydroxyphenyl)propanoic acid [(2S)-Mdp] resulted in Ligand 57 (Table 4), which turned out to be an opioid antagonist with high opioid receptor binding affinities and was expected to show enhanced bioavailability because of its further increased lipophilicity and reduced hydrogen bonding capacity.
Table 4.
Binding and functional assay data of cyclic ligands at opioid receptors (OR).
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound number | GPI | MVD | GPI/MVD | OR binding affinity | Binding ratio | |||||
|
| ||||||||||
| IC50 (nM) | Keμ (nM) | Keκ(nM) | IC50 (nM) | Keδ (nM) | IC50 ratio | Kiμ(nM) | Kiδ (nM) | Kiκ (nM) | Kiμ/Kiδ/Kiκ | |
| 49 | 1.07 | – | – | 1.11 | – | 1/1 | 0.496 | 2.29 | 0.45 | 1/5/1 |
| 50 | 0.457 | – | – | 0.884 | – | 1/2 | 0.354 | 2.36 | 0.86 | 1/7/2 |
| 51 | 1.81 | – | – | 0.352 | – | 5/1 | 0.504 | 0.525 | 1.01 | 1/1/2 |
| 52 | 1.36 | – | – | 0.122 | – | 11/1 | 0.586 | 0.776 | 0.89 | 1/1/2 |
| 53 | 1.34 | – | – | 3.72 | – | 1/3 | 0.876 | 6.07 | 1.42 | 1/7/2 |
| 54 | 0.394 | – | – | 1.95 | – | 1/5 | 0.641 | 1.79 | 0.88 | 1/3/1 |
| 55 | 0.586 | – | – | 0.0530 | – | 11/1 | 0.412 | 0.202 | 0.68 | 1/0.5/1.6 |
| 56 | 0.812 | – | – | 0.115 | – | 7/1 | 0.282 | 0.306 | 0.68 | 1/1/2 |
| 57 | – | 71.0 | 151 | – | 277 | – | 14.4 | 35.9 | 29.5 | 1/2/2 |
| 58 | 1.41 | – | – | 23.1 | – | 1/16 | 0.143 | 2100 | 22.3 | 1/11400/156 |
| 59 | 2.2 | – | – | 6.2 | – | 1/2.8 | 0.23 | 15.2 | 16.2 | 1/66/70 |
| 60 | 0.31 | – | – | 1.4 | – | 1/4.5 | 0.57 | 50.0 | 20.9 | 1/88/37 |
| 61 | 0.89 | 4.7 | 1/5.3 | 0.79 | 3.98 | 98.5 | 1/5/125 | |||
| 62 | 8.3 | – | – | 7.0 | – | 1.2/1 | 0.26 | 309 | 56.1 | 1/1190/216 |
| 63 | 4.35 | – | – | 1.5 | – | 2.9/1 | 0.26 | 16.0 | 14.4 | 1/62/55 |
| 64 | 1.01 | – | – | 4.4 | – | 1/4.4 | 0.94 | 39.2 | 11.5 | 1/42/12 |
| 65 | 0.65 | – | – | 1.5 | – | 1/2.3 | 2.92 | 5.03 | > 10,000 | 1/2/> 3420 |
| 66 | 76.0 | – | – | 64 (IC25) | – | – | 0.61 | 38.60 | ND | 1/64/– |
| 67 | 1.45 | – | – | 3.1 | – | 1/2.1 | 0.43 | 7.46 | 1.12 | 1/17/3 |
| 68 | 49.0 | – | – | 1000 | – | 1/20 | 2.62 | 56.9 | ND | 1/22/– |
| 69 | 0.78 (IC35) | – | – | 1.16 | – | – | 0.93 | 3..18 | 11.1 | 1/3/12 |
| 70 | 0.32 | – | – | 0.42 | – | 1/1.3 | 0.15 | 0.60 | 118 | 1/4/787 |
Novoa et al. [36] studied the effect of variation of the net charge, lipophilicity and side chain flexibility in Dmt1-DALDA (58, Table 4) on opioid activity and biodistribution. [37]. The authors examined the impact of charges on the side chain of the second and fourth amino acid residues in the peptidic MOR lead agonist Dmt-D-Arg-Phe-Lys-NH2 ([Dmt1]-DALDA, 58). The final amide bond was N-methylated to increase the overall lipophilicity of [Dmt1]-DALDA (58 – 65, Table 4). Phenylalanine (Phe) at position three was substituted by a constrained aminobenzazepine analog to investigate the effect side chain flexibility (66 – 70). The authors conducted in vitro receptor binding and activity studies on the peptidic ligands. They also studied the ligands’ in vivo transport (brain in- and efflux, and tissue biodistribution) and antinociceptive properties after peripheral administration (ip and sc) in mice. Appreciable shifts of receptor binding, activity and transport properties were reported because of the structural modifications. Important observation was made when antinociceptive properties of these ligands were examined. While [Dmt1]-DALDA (58) and its N-methyl analog, Dmt-D-Arg-Phe-NMeLys-NH2 (62) displayed a long-lasting antinociceptive effect (> 7 h), the peptides with D-Cit2 (64) produced effective antinociception faster (maximal effect at 1 h post injection) but also lost their analgesic activity quickly when compared to [Dmt1]-DALDA (58) and [Dmt1, NMeLys4]-DALDA (62).
Guillemyn et al. has reported [38] that tetrapeptide analogs of the type H-Dmt1-Xxx2-Yyy3-Gly4-NH2 are able to cross the BBB after intravenous and subcutaneous administration and successfully activate the MOR and DOR more efficiently and over longer periods of time compared to morphine. The authors have shown a comparison in potency of antinociception (using the hot water tail flick test as the animal model) between a side chain conformationally constrained analogue containing the benzazepine ring (BVD03: H-Dmt-NMe-D-Ala-Aba-Gly-NH2, where Xxx2 = NMe-D-Ala and Yyy3 = Aba), and a ‘ring opened’ analog (BVD02: H-Dmt-NMe-D-Ala-Aba-Gly-NH2, where Xxx2 = NMe-D-Ala and Yyy3 = Phe). The results demonstrate that combination of increased lipophilicity by amide bond N-methylation and the conformational constraint introduced at the Phe3 side chain displays prolonged antinociception. Enhanced potency and maintained antinociception have been observed on substitution of N-Me-D-Ala2 by D-Arg2 in the tetrapeptide sequence as it is shown for AN81 (H-Dmt-D-Arg-Aba-Gly-NH2, where Xxx2 = D-Arg and Yyy3 = Aba) versus BVD03. However, a considerable decrease in antinociception has been reported after a regular injection of the studied opioid ligands over a time period of 5 days. In addition, the authors studied a compact opioid agonist and NK1 antagonist hybrid SBCHM01 (H-Dmt-D-Arg-Aba-Gly-NMe-3′,5′Bn(CF3)2). But it could not overcome the development of opioid induced tolerance, which is in contrast to the results manifested by a similar kind of hybrid TY005 (H-Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-O-3′,5′Bn(CF3)2) [39]. Here, we suggest that a certain distance between opioid and NK1 pharmacophores is required to display their synergistic effects.
3.2 δ/μ opioid agonists
The analgesia mediated by activation of MOR is accompanied by typical side effects of opiates. But the stimulation of DOR exhibits antinociceptive effects with reduced respiratory depression, low constipation and minimal potential for physical dependence, suggesting this opioid receptor subtype is a potential therapeutic target for the development of analgesics with better biological profiles [40–42]. As there is evidence for synergistic effects exerted through the simultaneous δ and μ receptors activation, some of the potent investigational ligands will be discussed here.
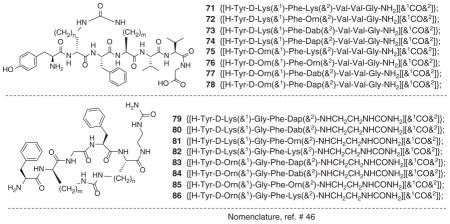
Zieleniak et al. [43] synthesized cyclic heptapeptides (71 – 78, Table 5) related to the full sequence of deltorphin. In vitro functional assay of these peptides at MOR and DOR showed high δ opioid agonist potency and high selectivity for δ receptors. Ligands 73 (IC50δ = 0.64 nM), 74 (IC50δ = 0.483 nM), 75 (IC50δ = 2.73 nM) and 76 (IC50δ = 0.814 nM) produced enhanced agonist activity in MVD assay compared to [Leu5]enkephalin (35, IC50δ = 11.4 nM, Table 2). The authors studied the conformations of these peptides using 2D-NMR in H2O/D2O and molecular dynamics, which revealed that the backbone rings had well defined conformations, while the side chains of Tyr1 and Phe3, and the exocyclic part of the C-terminal had significant conformational flexibility. These results, when compared with the previously reported data for 1 – 4 dermorphin analogs [44,45], suggested that presence of the address segment at the C-terminal played a role for δ-receptor selectivity and binding affinity of message region. The results observed during this study provided insights regarding the putative bioactive conformations and bioactivity.
Table 5.
GPI and MVD assay data for cyclic deltorphin (1 – 7) analogs and N-(ureidoethyl)amides of cyclic enkephalin analogs.
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound number | GPI | MVD | GPI/MVD | Compound number | Ring size | GPI | Relative potency | MVD | Relative potency | GPI/MVD |
|
|
|
|
|
|||||||
| IC50 (nM) | IC50 (nM) | IC50 ratio | IC50 (nM) | IC50 (nM) | IC50 ratio | |||||
| 71 | 888 | 11.8 | 75/1 | 79 | 18 | 1.97 | 125 | 1.81 | 6.30 | 1.1/1 |
| 72 | > 10,000 | 67.0 | > 149/1 | 80 | 19 | 5.55 | 44.3 | 6.0 | 1.90 | 1/1.1 |
| 73 | 65.4 | 0.640 | 102/1 | 81 | 20 | 18.1 | 13.6 | 22.7 | 0.502 | 1/1.3 |
| 74 | 25.4 | 0.483 | 53/1 | 82 | 21 | 20.2 | 12.2 | 26.6 | 0.429 | 1/1.3 |
| 75 | 1020 | 2.73 | 374/1 | 83 | 17 | 11.3 | 21.8 | 14.1 | 0.809 | 1/1.2 |
| 76 | 159 | 0.814 | 195/1 | 84 | 18 | 3.36 | 73.2 | 11.6 | 0.983 | 1/3.5 |
| 77 | 460 | 106 | 4.3/1 | 85 | 19 | 6.87 | 35.8 | 16.5 | 0.691 | 1/2.4 |
| 78 | > 10000 | 27.1 | > 369/1 | 86 | 20 | 16.1 | 15.3 | 28.2 | 0.404 | 1/1.8 |
| – | – | – | – | 35 | – | 246 | 1 | 11.4 | 1 | 21.6/1 |
Ciszewska et al. [47] prepared N-(ureidoethyl)amides of cyclic enkephalin analogs and studied for opioid receptor activities by GPI and MVD assays (79 – 86, Table 5). Depending on the size of the ring, the ligands produced diverse opioid activities. In comparison with [Leu5]enkephalin (35), all ligands (79 – 86) showed higher potency in the GPI assay (between 125 and 12 times), and 79 appeared to be almost equipotent in the GPI and MVD assay (Table 5). Ligands 79 (IC50μ = 1.97 nM, IC50δ = 1.81 nM) and 84 (IC50μ = 3.36 nM, IC50δ = 11.6 nM) containing 18-membered ring were found to the most potent. Though enkephalins are δ-selective peptides, these cyclic derivatives showed very similar potencies at MOR and DOR.
In an attempt to increase the bioavailability of peptide-derived ligands at DOR and MOR in the CNS, Lee at al. [48]. introduced lipophilic aromatic amino acid Dmt in place of Tyr at position one in enkephalin-like tetrapeptide analogs with an N-phenyl-N-piperidin-4-ylpropionamide moiety at the C-terminal. SAR studies led to the discovery of non-selective ligands with high potency at μ- and δ-opioid receptors. Ligand 87 (Figure 4) containing the D-Nle2 and 4-fluoro-substituted aromatic ring in Phe4 showed high opioid activities at both the receptors (Kiδ = 0.4 nM, Kiμ = 0.02 nM; IC50δ = 0.37 nM, IC50μ = 0.26 nM). Ligand 88 (Figure 4) having the D-Ala2 and 4-chloro-substituted aromatic ring in Phe4 also produced potency at both the receptors (Kiδ = 0.14 nM, Kiμ = 0.14 nM; IC50δ = 0.70 nM, IC50μ = 2.6 nM). In addition, Ligand 88 with impressive in vitro biological profiles displayed potent in vivo antihyperalgesic and antiallodynic effects in the tail-flick assay.
Figure 4.
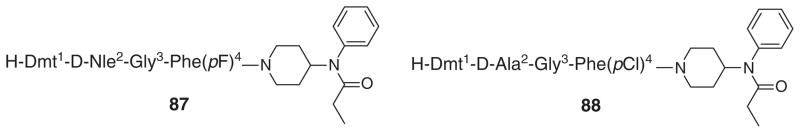
Structures of two potent opioid ligands.
4. Emerging drug design strategies: alliance of opioid agonist/NK1 or CCK antagonist activities
Most of the clinically used opioid drugs target MOR [49] to exert their antinociceptive effects. Many side effects are also known to be due to interaction of drugs with MOR. Development of addiction liability is one of the major problems in targeting MOR [50,51]. The side effects associated with the clinical use of opioid drugs as analgesics significantly decrease the patients’ quality of life. The mechanistic pathways responsible for these side effects are largely unknown. It has been observed that prolonged pain states lead to neuroplastic changes in both ascending and descending pathways in the spinal column which cause an increased release of neurotransmitters (e.g., substance P, Cholecystokinin, CCK) that enhance pain and overexpression of the corresponding receptors (NK1 and CCK) for those newly released pain-promoting ligands (see [52–54] for Substance P and NK1 receptor and [55–59] for CCK and CCK receptor). Current drugs used for the treatment of sustained pain generally can only modulate pain and cannot counteract against these induced neuroplastic modifications. Thus, it is not unexpected that current analgesics do not work effectively in these pathological conditions.
Coadministration of a δ/μ opioid agonist and a neurokinin 1 (NK1) antagonist has been reported to produce the facilitatory role of the substance P-NK1 system in opioid signal transmission [60,61]. This combination explains many important biological effects such as high potency in acute pain models and inhibition of opioid-induced tolerance in chronic tests using rats. A study reported that mice lacking NK1 receptor did not produce the rewarding properties of morphine [62]. Thus, the alliance of an agonist activity at the opioid receptors and an antagonist at NK1 receptors may have enhanced effects in the treatment of prolonged pain states that involve increased substance P activity. Poor patient compliance, complications in drug metabolism, distribution and possible drug–drug interactions limit the use of drug mixtures as therapeutics. Over the last several years, the Hruby Research Group and a few other groups have taken a new approach to combine these two activities in one ligand would have simple metabolic and pharmacokinetic properties. The anticipated activities of this kind of ligand would include potent analgesic activities in both acute pain and in neuropathic pain states without the development of tolerance [63]. The concept of drug design by this strategy is based on design of ligands with the adjacent and overlapping pharmacophores, in which the μ-/δ-opioid agonist pharmacophore is incorporated at the N-terminus, and the NK1 antagonist pharmacophore is introduced at the C-terminus of a single peptide derived ligand. The opioid pharmacophore of these hybrid molecules are designed based on the sequence of well-known opioid pharmacophore, for example. biphalin and DADLE [64,65], while the structural modifications were made on the basis of a 3′,5′-(bistrifluoromethyl)-benzyl ester of N-acylated tryptophan, a known NK1 antagonist pharmacophore at the C-terminus [66–68]. The two pharmacophores are combined either directly or through an additional address segment. The linker may also work as an address region for both pharmacophores [65,69–72]. The designed multivalent molecules may have additional advantages over a cocktail of individual drugs for easy administration such as simple ADME properties and no drug–drug interactions. A higher local concentration is also expected than that in the coadministration of two drugs because the expressions of the NK1 and opioid receptors show a significant degree of anatomical overlap in the CNS, leading to synergies in potency and efficacy [73–75]. In fact, the results published through a highly collaborative research from the groups of Hruby, Porreca, Vanderah and Lai showed that the lead multifunctional compounds, TY005 (H-Tyr1-D-Ala2-Gly3-Phe4-Met5-Pro6-Leu7-Trp8-O-[3′,5′-Bzl(CF3)2]: Kiμ = 36 nM, Kiδ = 2.8 nM, KirNK1 = 0.29 nM; IC50μ = 360 nM, IC50δ = 22 nM; KeNK1 = 25 nM; EC50μ = 32 nM, EC50δ = 2.9 nM; Emaxμ = 42%, Emaxδ = 45%) [65], and TY027 (H-Tyr1-D-Ala2-Gly3-Phe4-Met5-Pro6-Leu7-Trp8-[NH-3′,5′-Bzl(CF3)2]: Kiμ = 16.0 nM, Kiδ = 0.66 nM, KirNK1 = 7.3 nM; IC50μ = 490 nM, IC50δ = 15 nM; KeNK1 = 10 nM; EC50μ = 7.0 nM, EC50δ = 8.6 nM; Emaxμ = 55%, Emaxδ = 58%) [69] having μ-/δ-opioid agonist/NK1 antagonist activities can reverse neuropathic pain in a rodent model with BBB permeability, no sign of opioid-induce tolerance, and no development of reward liability, validating the hypothesis that a single compound possessing opioid agonist/NK1 antagonist activities is effective against neuropathic pain [39,63,76–78]. The multifunctional ligand TY027 has recently been reported [79] to show a preclinical profile of excellent antinociceptive efficacy, low abuse liability and no opioid-related emesis or constipation. In rodent models of acute and neuropathic pain, TY027 exhibited potent analgesic efficacy after central or systemic administration with a plasma half-life of over 4 h and CNS penetration. The data presented in that report showed that a multifunctional ligand can combat the pathological conditions created by chronic pain and sustained opioid use with antinociceptive efficacy in rodent models with significantly fewer side effects than morphine. In these in vivo studies it was established that peptides such as TY027 are bioavailable after peripheral administration, cross the BBB well, have minimal or none of the toxic side effects of morphine, and are potent against neuropathic pain states in animal models (e.g., see 77 – 79). In vivo studies are in progress with another potent opioid agonist/NK1 antagonist ligand TY032 (H-Dmt1-D-Ala2-Gly3-Phe4-Met5-Pro6-Leu7-Trp8-[NH-3′,5′-Bzl(CF3)2]: Kiμ = 2.0 nM, Kiδ = 0.18 nM, KirNK1 = 2.3 nM; IC50μ = 19 nM, IC50δ = 1.8 nM; KeNK1 = 7.5 nM; EC50μ = 29 nM, EC50δ = 1.0 nM; Emaxμ = 72%, Emaxδ = 94%) [80]. Thus, such rationally designed, multifunctional ligands have promising activities for treating acute and chronic pain.
Upregulation of CCK has been reported following the chronic administration of morphine to animals indicating the possibility of this endogenous peptide having pronociceptive effects [59]. Evidence is also there for neuropeptide CCK to act as an antagonist of opiate analgesia [81]. These complex interactions between opioids and endogenous CCK receptor systems have led to the suggestion that chronic and neuropathic pain can be treated by combining agonist activity at opioid receptors and antagonist activity at CCK receptor. The strategy is similar to opioid agonist/NK1 antagonist strategy. A number of reports are available in the literature focusing on the design, synthesis and SAR study of this class of ligands [82–86]. Hanlon et al. [87] studied the in vivo efficacy of novel μ-/δ-opioid agonist/CCK antagonist ligands RSA504 (H-Tyr1-D-Nle2-Gly3-Trp4-Nle5-Asp6-Phe7-NH2: Ki μ = 26 nM, Ki δ = 2.6 nM, Ki CCK-1 = 9.6 nM, KiCCK-2 = 15 nM; IC50μ = 210 nM, IC50δ = 23 nM; EC50μ = 0.46 nM, EC50δ = 4.5 nM; Emaxμ = 88%, Emaxδ = 81%), and RSA601 (H-Tyr1-D-Phe2-Gly3-D-Trp4-NMeNle5-Asp6-Phe7-NH2: Kiμ = 5.9 nM, Kiδ = 0.42 nM, KiCCK-2 = 1.1 nM; IC50μ = 71 nM, IC50δ = 24 nM; EC50μ = 530 nM, EC50δ = 29 nM; Emaxμ = 50%, Emaxδ = 14%). RSA504 and RSA601 produced antihypersensitivity to mechanical and thermal stimuli in vivo using the spinal nerve ligation model of neuropathic pain. Intrathecal administration of RSA504 and RSA601 did not display antinociceptive tolerance over 7 days of administration and did not produce motor impairment or sedation using a rotarod. These results suggested multi-targeted molecules can address the pathology of neuropathic pain. Thus ligands with δ-/μ-opioid agonist activities and CCK antagonist activity within one molecule can offer a novel approach with efficacy for neuropathic pain while lacking the side effects typically caused by conventional treatment with opioid drugs.
5. Concluding discussion
The natural ligands which modulate pain in the human are peptides (e.g., β-endorphin, enkephalins, etc.) and yet all treatments primarily use alkaloids and other heterocyclic compounds. None of these are peptides or peptide derivatives. Strangely, essentially all treatments for pain, the most ubiquitous disease, have high toxicities and for prolonged and neuropathic pain, often poor efficacies and all lead to the development of tolerance with long-term use, yet the medical profession continues to use them. Moreover, their patients often abuse current opioid drugs. As we have discussed here, there are new alternatives that in animal models of pain overcome many of the problems of current analgesics. Yet astonishingly none of these peptides have been subjected to Phase III clinical trials. Why? Are the power brokers of our pharmaceutical, medical and investment communities afraid they may work? It is mysterious and the reality is there for all to see. Hopefully, our brief and incomplete review of the opioid ligands of the past and the current ones under investigation will stimulate interest in the medical and pharmaceutical industries to further develop these ligands, and give HOPE to some of our billion fellow human beings that suffer daily from pain.
6. Expert opinion
Although there are a number of drugs for the management of pain, none of them are 100% successful in controlling it, especially when the treatment of prolonged and neuropathic pain is concerned. Several therapeutic agents including opioid analgesics, anticonvulsants, antidepressants, topical anesthetics (lidocaine patch), etc., with different biological targets and activities are currently in clinical use. These drugs can offer moderate pain relief. During the use of these drugs, there are considerable concerns of serious and unwanted side effects.
The most common feature among all available drugs and the drugs in advanced stages of clinical trials is that almost all of them target μ-opioid or other receptors or ion-channels on neurons. It is well known that most of the biological targets have subtypes, and each subtype is responsible for different kinds of biological activities. It is emerging in the literature that these biological targets have a number of conformations as well as binding sites that elicit different responses. If binding to a particular site establishes equilibrium between more than one conformations of a receptor, this can lead to several biological responses, including unwanted side effects. It is known that small molecules are generally less selective for their biological targets, often resulting in unwanted activation or deactivation of other biological systems which produce undesirable side effects. Elimination of these limitations of current drugs is an important goal. Peptide and peptidomimetic ligands have been found to be more selective for a specific target. But, some of the major problems associated with these types of ligands can include less stability in biological conditions and a reduced chance of reaching their targets. Elimination of these problems will open an extraordinary avenue in the drug discovery horizon. An approach, which might be very useful, is to design ligands that can interact with multiple biological targets with the specific desired activity at each individual target. The authors of this review suggest that such drugs with novel biological profiles will lead to better medical benefits for the treatment of pain. Molecules with high selectivity for binding sites and unique conformations can provide a new and better path to achieve these goals. The development of such conformationally and functionally biased drug candidates is a real challenge, but it can be addressed. The application of this latest approach to drug design and discovery with multivalency/ multifunctionality has been discussed here. As there is evidence for peripherally and centrally acting opioid agonists to show analgesic effects, design of drug candidates having activities at both locations can be interesting to consider.
Clearly new directions and new modalities are needed for the treatment of prolonged and neuropathic pain. Clearly also ‘one shoe’ will not fit all. Undoubtedly, there are many different phenotypes of prolonged and neuropathic pain. Hopefully, this brief review will give some stimulation to others to discover new approaches to the development of ligands to treat our most ubiquitous and poorly treated diseases, prolonged and neuropathic pain.
Article highlights.
Mixed μ/δ opioid receptor agonist ligands have no or reduced toxicities relative to μ opioid ligands such as morphine.
Highly potent μ/δ opioid ligands can have patent antinociceptive activities for neuropathic pain.
Highly potent multivalent ligands with μ/δ receptor opioid agonist activity and neurokinin-1 receptor antagonist activities have minimal toxicities of current opioid ligands in clinical use.
This box summarizes key points contained in the article.
Footnotes
Declaration of interest
The research in the V Hruby group is supported in whole or in part from grants from the US Public Health Service, NIH and NIDA. Some of the compounds have patent coverage.
Bibliography
Papers of special note have been highlighted as either of interest (●) or of considerable interest (●●) to readers.
- 1.Bonica JJ. Definitions and taxonomy of pain. In: Bonica JJ, editor. Management of pain. Lea & Febiger; Philadelphia: 1990. [Google Scholar]
- 2●.IOM (Institute of Medicine). Committee on Advancing Pain Research C, and Education; Institute of Medicine. Relieving Pain in America. A blueprint for transforming prevention, care, education, and research. The National Academies Press; Washington, DC: 2011. Recent critical outline of the treatment of pain in the United States. [PubMed] [Google Scholar]
- 3.Gaskin DJ, Richard P. The economic costs of pain in the United States. J Pain. 2012;13:715–24. doi: 10.1016/j.jpain.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367:1618–25. doi: 10.1016/S0140-6736(06)68700-X. [DOI] [PubMed] [Google Scholar]
- 5.Apfelbaum JL, Chen C, Mehta SS, Gan TJ. Postoperative pain experience: results from a national survey suggest postoperative pain continues to be undermanaged. Anesth Analg. 2003;97:534–40. doi: 10.1213/01.ANE.0000068822.10113.9E. [DOI] [PubMed] [Google Scholar]
- 6.Loftus RW, Yeager MP, Clark JA, et al. Intraoperative ketamine reduces perioperative opiate consumption in opiate-dependent patients with chronic back pain undergoing back surgery. Anesthesiology. 2010;113:639–46. doi: 10.1097/ALN.0b013e3181e90914. [DOI] [PubMed] [Google Scholar]
- 7.Buvanendran A, Kroin JS, Delia Valle CJ, et al. Perioperative oral pregabalin reduces chronic pain after total knee arthroplasty: a prospective, randomized, controlled trial. Anesth Analg. 2010;110:199–207. doi: 10.1213/ANE.0b013e3181c4273a. [DOI] [PubMed] [Google Scholar]
- 8.Benyamin R, Trescot AM, Datta S, et al. Opioid complications and side effects. Pain Physician. 2008;11:S105–20. [PubMed] [Google Scholar]
- 9.Swegle JM, Logemann C. Management of common opioid-induced adverse-effects. Am Fam Phisician. 2006;74:1347–54. [PubMed] [Google Scholar]
- 10.Dhawan BN, Cesselin F, Raghubir R, et al. International Union of Pharmacology. XIII Classification of Opioid Receptors. Pharmacol Rev. 1996;48:567–92. [PubMed] [Google Scholar]
- 11.McDonald J, Lambert DG. Opioid receptors. Contin Educ Anaesth Crit Care Pain. 2005;5:22–5. [Google Scholar]
- 12.Ananthan S. Opioid ligands with mixed mu/delta opioid receptor interactions: an emerging approach to novel analgesics. AAPS J. 2006;8:E118–25. doi: 10.1208/aapsj080114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13●.Horan PJ, Mattia A, Bilsky EJ, et al. Antinociceptive profile of biphalin, a dimeric enkephalin analog. J Pharmacol Exp Ther. 1993;265:1446–54. A thorough discussion of the in vitro and in vivo pharmacology of a μ/δ opioid agonist ligand with greatly reduced toxic side effects. [PubMed] [Google Scholar]
- 14.Horan P, Tallarida RJ, Haaseth RC, et al. Antinociceptive interactions of opioid delta receptor agonists with morphine in mice: supra- and sub-additivity. Life Sci. 1992;50:1535–41. doi: 10.1016/0024-3205(92)90144-e. [DOI] [PubMed] [Google Scholar]
- 15.Vaught JL, Takemori AE. Differential effects of leucine and methionine enlephalin on morphine-induced analgesia, acute tolerance and dependence. J Pharmacol Exp Ther. 1979;208:86–90. [PubMed] [Google Scholar]
- 16.Manglik A, Kruse AC, Kobilka TS, et al. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–6. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granier S, Manglik A, Kruse AC, et al. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–4. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu H, Wacker D, Mileni M, et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–32. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filizola M, Devi LA. Structural biology: how opioid drugs bind to receptors. Nature. 2012;485:314–17. doi: 10.1038/485314a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filizola M, Devi LA. Grand opening of structure-guided design for novel opioids. Trends Pharmacol Sci. 2013;34:6–12. doi: 10.1016/j.tips.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schiller PW, Nguyen TM-D, Maziak LA, et al. Structure-activity relationships of cyclic opioid peptide analogues containing a phenylalanine residue in the 3-position. J Med Chem. 1987;30:2094–9. doi: 10.1021/jm00394a027. [DOI] [PubMed] [Google Scholar]
- 22.Berezowska I, Chung NN, Lemieux C, et al. Cyclic dermorphin tetrapeptide analogues obtained via ring-closing metathesis. Acta Biochim Pol. 2006;53:73–6. [PubMed] [Google Scholar]
- 23.Berezowska I, Chung NN, Lemieux C, et al. Dicarba analogues of the cyclic enkephalin peptides H-Tyr-c[D-Cys-Gly-Phe-D(or L)-Cys]NH2 retain high opioid activity. J Med Chem. 2007;50:1414–17. doi: 10.1021/jm061294n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarantakis D. Analgesic polypeptide. 4148786. US. 1979
- 25.Schiller PW, Eggimann B, DiMaio J, et al. Cyclic Enkephalin analogs containing a cystine bridge. Biochem Biophys Res Commun. 1981;101:337–43. doi: 10.1016/0006-291x(81)91265-1. [DOI] [PubMed] [Google Scholar]
- 26.Weltrowska G, Nguyen TM-D, Lemieux C, et al. Potent opioid peptide agonists containing 4-[N-((4-phenyl)-phenethyl)carboxamido]phenylalanine (Bcp) in place of Tyr. Chem Biol Drug Des. 2008;72:337–40. doi: 10.1111/j.1747-0285.2008.00720.x. [DOI] [PubMed] [Google Scholar]
- 27.Schiller PW, Nguyen TM-D, DiMaio J, Lemieux C. Comparison of mu-, delta-and kappa-receptor binding sites through pharmacologic evaluation of p-nitrophenylalanine analogs of opioid peptides. Life Sci. 1983;33(Suppl 1):319–22. doi: 10.1016/0024-3205(83)90507-6. [DOI] [PubMed] [Google Scholar]
- 28.Schiller PW, Nguyen TM-D, Chung NN, Lemieux C. Dermorphin analogues carrying an increased positive net charge in their ‘message’ domain display extremely high mu opioid receptor selectivity. J Med Chem. 1889;32:698–703. doi: 10.1021/jm00123a035. [DOI] [PubMed] [Google Scholar]
- 29●.Ballet S, Misicka A, Kosson P, et al. Blood-brain barrier penetration by two dermorphin tetrapeptide analogues: role of lipophilicity vs structural flexibility. J Med Chem. 2008;51:2571–4. doi: 10.1021/jm701404s. A careful study of BBB penetration by peptide analogues. [DOI] [PubMed] [Google Scholar]
- 30.Hruby VJ. Designing peptide receptor agonists and antagonists. Nat Rev Drug Discov. 2002;1:847–58. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- 31.Fichna J, do-Rego J-C, Chung NN, et al. [Dmt1, D-1-Nal3]morphiceptin, a novel opioid peptide analog with high analgesic activity. Peptides. 2008;29:633–8. doi: 10.1016/j.peptides.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Przydial MJ, Pogozheva ID, Ho JC, et al. Design of high affinity cyclic pentapeptide ligands for kappa-opioid receptors. J Peptide Res. 2005;66:255–62. doi: 10.1111/j.1399-3011.2005.00295.x. [DOI] [PubMed] [Google Scholar]
- 33.Berezowska I, Lemieux C, Chung NN, et al. Cyclic opioid peptide agonists and antagonists obtained via ring-closing metathesis. Chem Biol Drug Des. 2009;74:329–34. doi: 10.1111/j.1747-0285.2009.00867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weltrowska G, Berezowska I, Lemieux1 C, Chung NN, Wilkes BC, Schiller PW. N-Methylated cyclic enkephalin analogues retain high opioid receptor binding affinity. Chem Biol Drug Des. 2010;75:182–8. doi: 10.1111/j.1747-0285.2009.00919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35●.Mosberg MI, Fowler CB. Development and validation of opioid ligand-receptor interaction models: the structural basis of mu vs. delta selectivity. J Peptide Res. 2002;60:329–35. doi: 10.1034/j.1399-3011.2002.21061.x. A careful molecular modeling study of μ and δ opioid receptors and their ligands. [DOI] [PubMed] [Google Scholar]
- 36.Novoa A, Dorpe SV, Wynendaele E, et al. Variation of the net charge, lipophilicity, and side chain flexibility in Dmt1-DALDA: effect on opioid activity and biodistribution. J Med Chem. 2012;55:9549–61. doi: 10.1021/jm3008079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimoyama M, Schiller PW, Shimoyama N, et al. Superior analgesic effect of H-Dmt-D-Arg-Phe-Lys-NH2 ([Dmt1]DALDA), a multifunctional opioid peptide, compared to morphine in a rat model of neuropathic pain. Chem Biol Drug Des. 2012;80:771–4. doi: 10.1111/cbdd.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guillemyn K, Kleczkowska P, Novoa A. Tourwé1, Steven Ballet. In vivo antinociception of potent mu opioid agonist tetrapeptide analogues and comparison with a compact opioid agonist - neurokinin 1 receptor antagonist chimera. Mol Brain. 2012;5:4. doi: 10.1186/1756-6606-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39●.Largent-Milnes TM, Yamamoto T, Nair P, et al. Spinal or systemic TY005, a peptidic opioid agonist/neurokinin 1 antagonist, attenuates pain with reduced tolerance. Br J Pharmacol. 2010;161:986–1001. doi: 10.1111/j.1476-5381.2010.00824.x. In vitro and in vivo studies of μ/δ agonist-NK1 antagonist ligand with greatly reduced toxicities compared to current opioid drugs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kieffer BL, Gavériaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- 41.Rapaka RS, Porreca F. Development of delta opioid peptides as nonaddicting analgesics. Pharm Res. 1991;8:1–8. doi: 10.1023/a:1015809702296. [DOI] [PubMed] [Google Scholar]
- 42.Coop A, Rice KC. Role of delta-opioid receptors in biological processes. Drug News Perspect. 2000;13:481–7. [PubMed] [Google Scholar]
- 43.Zieleniak A, Rodziewicz-Motowidlo S, Rusak L, et al. Deltorphin analogs restricted via a urea bridge: structure and opioid activity. J Pept Sci. 2008;14:830–7. doi: 10.1007/978-0-387-73657-0_212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Filip K, Oleszczuk M, Pawlak D, et al. Potent side-chain to side-chain cyclized dermorphin analogues containing a carbonyl bridge. J Pept Sci. 2003;9:649–57. doi: 10.1002/psc.510. [DOI] [PubMed] [Google Scholar]
- 45.Filip K, Oleszczuk M, Wójcik J, et al. Cyclic enkephalin and dermorphin analogues containing a carbonyl bridge. J Pept Sci. 2005;11:347–52. doi: 10.1002/psc.613. [DOI] [PubMed] [Google Scholar]
- 46.Spengler J, Jiménez J-C, Burger K, et al. Abbreviated nomenclature for cyclic and branched homo- and heterodetic peptides. J Pept Res. 2005;65:550–5. doi: 10.1111/j.1399-3011.2005.00254.x. [DOI] [PubMed] [Google Scholar]
- 47.Ciszewska M, Kwasiborska M, Nowakowski M, et al. Deltorphin analogs restricted via a urea bridge: structure and opioid activity. J Pept Sci. 2009;15:312–18. [Google Scholar]
- 48.Lee YS, Kulkarani V, Cowell SM, et al. Development of potent mu and delta opioid agonists with high lipophilicity. J Med Chem. 2011;54:382–6. doi: 10.1021/jm100982d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCurdy CR, Prisinzano TE. Opioid receptor ligands. In: Abraham DJ, Rotella DR, editors. Burger’s medicinal chemistry, drug discovery, and development. 7. John Wiley & Sons, Inc; New York: 2010. pp. 569–735. [Google Scholar]
- 50.DeLander GE, Portoghese PS, Takemori AE. Role of spinal mu opioid receptors in the development of morphine tolerance and dependence. J Pharmacol Exp Ther. 1984;231:91–6. [PubMed] [Google Scholar]
- 51.Fields HL. Understanding how opioids contribute to reward and analgesia. Reg Anesth Pain Med. 2007;32:242–6. doi: 10.1016/j.rapm.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 52.Mantyh PW, Allen CJ, Ghilardi JR, et al. Rapid endocytosis of a G protein-coupled receptor: substance P evoked internalization of its receptor in the rat striatum in vivo. Proc Natl Acad Sci USA. 1995;92:2622–6. doi: 10.1073/pnas.92.7.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalso E. Improving opioid effectiveness: from ideas to evidence. Eur J Pain (Amsterdam, Neth) 2005;9:131–5. doi: 10.1016/j.ejpain.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 54.King T, Ossipov MH, Vanderah TW, et al. Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals. 2005;14:194–205. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- 55.Stanfa L, Dickenson A, Xu XJ, Wiesenfeld-Hallin Z. Cholecystokinin and morphine analgesia: variations on a theme. Trends Pharm Sci. 1994;15:65–6. doi: 10.1016/0165-6147(94)90279-8. [DOI] [PubMed] [Google Scholar]
- 56.Wiesenfeld-Hallin Z, Xu XJ. The role of cholecystokinin in nociception, neuropathic pain and opiate tolerance. Regul Pept. 1996;65:23–8. doi: 10.1016/0167-0115(96)00068-7. [DOI] [PubMed] [Google Scholar]
- 57.Wiesenfeld-Hallin Z, de Arauja Lucas G, Alster P, et al. Cholecystokinin/opioid interactions. Brain Res. 1999;848:78–89. doi: 10.1016/s0006-8993(99)01978-2. [DOI] [PubMed] [Google Scholar]
- 58.Heinricher MM, Neubert MJ. Neural basis for the hyperalgesic action of cholecystokinin in the rostral ventromedial medulla. J Neurophysiol. 2004;92:1982–9. doi: 10.1152/jn.00411.2004. [DOI] [PubMed] [Google Scholar]
- 59.Zhou Y, Sun YH, Zhang ZW, Han JS. Increased release of immunoreactive cholecystokinin octapeptide by morphine and potentiation of mu-opioid analgesia by CCKB receptor antagonist L-365,260 in rat spinal cord. Eur J Pharmacol. 1993;234:147–54. doi: 10.1016/0014-2999(93)90948-h. [DOI] [PubMed] [Google Scholar]
- 60.Misterek K, Maszczynska I, Dorociak A, et al. Spinal co-administration of peptide substance P antagonist increases antinociceptive effect of the opioid peptide biphalin. Life Sci. 1994;54:939–44. doi: 10.1016/0024-3205(94)00494-3. [DOI] [PubMed] [Google Scholar]
- 61.Powell KJ, Quirion R, Jhamandas K. Inhibition of neurokinin-1-substance P receptor and prostanoid activity prevents and reverses the development of morphine tolerance in vivo and the morphine-induced increase in CGRP expression in cultured dorsal root ganglion neurons. Eur J Neurosci. 2003;18:1572–83. doi: 10.1046/j.1460-9568.2003.02887.x. [DOI] [PubMed] [Google Scholar]
- 62.Ripley TL, Gadd CA, De Felipe C, et al. Lack of self-administration and behavioural sensitisation to morphine, but not cocaine, in mice lacking NK1 receptors. Neuropharmacology. 2002;43:1258–68. doi: 10.1016/s0028-3908(02)00295-2. [DOI] [PubMed] [Google Scholar]
- 63.Hruby VJ. Organic chemistry and biology: chemical biology through the eyes of collaboration. J Org Chem. 2009;74:9245–64. doi: 10.1021/jo901767e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horan PJ, Mattia A, Bilsky EJ, et al. Antinociceptive profile of biphalin, a dimeric enkephalin analog. J Pharmacol Exp Ther. 1993;265:1446–54. [PubMed] [Google Scholar]
- 65.Yamamoto T, Nair P, Davis P, et al. Design, synthesis and biological evaluation of novel bifunctional C-terminal modified peptides for delta/mu opioid receptor agonists and neurokinin–1 receptor antagonists. J Med Chem. 2007;50:2779–86. doi: 10.1021/jm061369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cascieri MA, Macleod AM, Underwood D, et al. Characterization of the interaction of N-acyl-L-tryptophan benzyl ester neurokinin antagonists with the human neurokinin-1 receptor. J Biol Chem. 1994;269:6587–91. [PubMed] [Google Scholar]
- 67.MacLeod AM, Merchant KJ, Cascieri MA, et al. N-Acyl-L-tryptophan benzyl esters: potent substance P receptor antagonists. J Med Chem. 1993;36:2044–5. doi: 10.1021/jm00066a015. [DOI] [PubMed] [Google Scholar]
- 68.Millet R, Goossens L, Goossens JF, et al. Conformation of the tripeptide Cbz-Pro-Leu-Trp-OBzl(CF3)2 deduced from two-dimensional 1H-NMR and conformational energy calculations is related to its affinity for NK1-receptor. J Pept Sci. 2001;7:323–30. doi: 10.1002/psc.326. [DOI] [PubMed] [Google Scholar]
- 69●●.Yamamoto T, Nair P, Jacobsen NE, et al. The importance of micelle-bound states for the bioactivities of bifunctional peptide derivatives for delta/mu opioid receptor agonists and neurokinin 1 receptor antagonists. J Med Chem. 2008;51:6334–47. doi: 10.1021/jm800389v. Extensive study of the conformational properties of multivalent ligands with μ/δ receptor agonist-NK1 receptors antagonists within the presence of membranes using NMR and computational methods and their biological activities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamamoto T, Nair P, Jacobsen NE, et al. Improving metabolic stability by glycosylation: bifunctional peptide derivatives that are opioid receptor agonists and neurokinin 1 receptor antagonists. J Med Chem. 2009;52:5164–75. doi: 10.1021/jm900473p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamamoto T, Nair P, Vagner J, et al. A structure-activity relationship study and combinatorial synthetic approach of C-terminal modified bifunctional peptides that are delta/mu opioid receptor agonists and neurokinin 1 receptor antagonists. J Med Chem. 2008;51:1369–76. doi: 10.1021/jm070332f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamamoto T, Nair P, Davis P, et al. The biological activity and metabolic stability of peptidic bifunctional compounds that are opioid receptor agonists and neurokinin 1 receptor antagonists with a cystine moiety. Bioorg Med Chem. 2009;17:7337–43. doi: 10.1016/j.bmc.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hokfelt T, Kellerth J-O, Nilsson G, Pernow B. Experimental immunohistochemical studies on the localization and distribution of substance P in cat primary sensory neurons. Brain Res. 1975;100:235–52. doi: 10.1016/0006-8993(75)90481-3. [DOI] [PubMed] [Google Scholar]
- 74.Marchand JE, Kream RM, Substance P. Somatostatin levels in rhumatioid arthesis, molecular physiology. In: Leeman SE, Krause JE, Lembeck F, editors. Substance P and related peptides: cellular and molecular physiology. New York Academy of Science; New York: 1990. pp. 437–8. [Google Scholar]
- 75.Kondo I, Marvizon JC, Song B, et al. Inhibition by spinal mu- and delta-opioid agonists of afferent-evoked substance P release. J Neurosci. 2005;25:3651–60. doi: 10.1523/JNEUROSCI.0252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Largent-Milnes TM, Yamamoto T, Davis P, et al. Dual acting opioid agonist/NK1 antagonist reverses neuropathic pain and does not produce tolerance. Society for Neuroscience; San Diego, CA: 2007. Poster 725. Visceral Pain: Transmitters and Receptors. [Google Scholar]
- 77.Largent-Milnes TM, Yamamoto T, Nair P, et al. Dual acting opioid agonist/ NK1 antagonist peptide reverses neuropathic pain in an animal model without demonstrating common opioid unwanted side effects. International Association for the Study of Pain/12th World Congress on Pain; Glasgow, Scotland. 2008. [Google Scholar]
- 78.Largent-Milnes TM, Yamamoto T, Campos CR, et al. Dual acting opioid agonist/NK1 antagonist does not produce antinociceptive tolerance or reward in an animal model of neuropathic pain. Society for Neuroscience; Chicago, IL: 2009. Program/Poster: 255.10/U7. [Google Scholar]
- 79●●.Largent-Milnes TM, Brookshire SW, Skinner DP, Jr, et al. Building a better analgesic: multifunctional compounds that address injury-induced pathology to enhance analgesic efficacy while eliminating unwanted side effects. J Pharmacol Exp Ther. 2013;347:7–19. doi: 10.1124/jpet.113.205245. Comprehensive in vivo analysis of a multivalent ligand with analgesic activities and minimal toxicities and other side effects. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yamamoto T, Nair P, Largent-Milnes TM, et al. Discovery of a potent and efficacious peptide derivative for delta/mu opioid agonist/neurokinin 1 antagonist activity with a 2′,6′-dimethyl-LTyrosine: In Vitro, In Vivo, And NMR-based structural studies. J Med Chem. 2011;54:2029–38. doi: 10.1021/jm101023r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Faris PL, Komisaruk BR, Watkins LR, Mayer DJ. Evidence for the neuropeptide cholecystokinin as an antagonist of opiate analgesia. Science. 1983;219:310–12. doi: 10.1126/science.6294831. [DOI] [PubMed] [Google Scholar]
- 82.Agnes RS, Ying J, Kovér KE, et al. Structure-activity relationships of bifunctional cyclic disulfide peptides based on overlapping pharmacophores at opioid and cholecystokinin receptors. Peptides. 2008;29:1413–23. doi: 10.1016/j.peptides.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee YS, Agnes RS, Davis P, et al. Partial retro-inverso, retro, and inverso modifications of hydrazide linked bifunctional peptides for opioid and Cholecystokinin (CCK) receptors. J Med Chem. 2007;50:165–8. doi: 10.1021/jm061268p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Agnes RS, Lee YS, Davis P, et al. Structure-activity relationships of bifunctional peptides based on overlapping pharmacophores at opioid and cholecystokinin receptors. J Med Chem. 2006;49:2868–75. doi: 10.1021/jm050921q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee YS, Agnes RS, Badghisi H, et al. Design and synthesis of novel hydrazide-linked bifunctional peptides as δ/μ opioid receptor agonists and CCK-1/ CCK-2 receptor antagonists. J Med Chem. 2006;49:1773–80. doi: 10.1021/jm05085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hruby VJ, Agnes RS, Davis P, et al. Design of novel peptide ligands which have opioid agonist activity and CCK antagonist activity for the treatment of pain. Life Sci. 2003;73:699–704. doi: 10.1016/s0024-3205(03)00390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hanlon KE, Herman DS, Agnes RS, et al. Novel peptide ligands with dual acting pharmacophores designed for the pathophysiology of neuropathic pain. Brain Res. 2011;1395:1–11. doi: 10.1016/j.brainres.2011.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]