

Abstract
Pancreatic cancer remains a clinical challenge, thus new therapies are urgently needed. The selective Wee1 inhibitor MK-1775 has demonstrated promising results when combined with DNA damaging agents, and more recently with CHK1 inhibitors in various malignancies. We have previously demonstrated that treatment with the pan-histone deacetylase inhibitor panobinostat (LBH589) can cause down-regulation of CHK1. Accordingly, we investigated using panobinostat to down-regulate CHK1 in combination with MK-1775 to enhance cell death in preclinical pancreatic cancer models. We demonstrate that MK-1775 treatment results in increased H2AX phosphorylation, indicating increased DNA double-strand breaks, and activation of CHK1, which are both dependent on CDK activity. Combination of MK-1775 and panobinostat resulted in synergistic antitumor activity in six pancreatic cancer cell lines. Finally, our in vivo study using a pancreatic xenograft model reveals promising cooperative antitumor activity between MK-1775 and panobinostat. Our study provides compelling evidence that the combination of MK-1775 and panobinostat has antitumor activity in preclinical models of pancreatic cancer and supports the clinical development of panobinostat in combination with MK-1775 for the treatment of this deadly disease.
Keywords: MK-1775, Panobinostat, Pancreatic cancer, CHK1, Drug combination
Introduction
Pancreatic cancer remains a very difficult disease to treat. The fourth most lethal cancer in the United States, pancreatic cancer has a grim prognosis, with a 5-year survival rate of 6% [1]. Standard chemotherapeutic care for pancreatic cancer involves treatment with gemcitabine, a nucleoside analogue, but offers only modest benefit [2]. Furthermore, combinations of other cytotoxic agents with gemcitabine have, in general, offered little improvement [3]. Thus, there is an obvious need to develop more effective therapy for this disease.
Cell cycle progression is a tightly regulated process. The cell cycle is driven by the activation of various cyclin dependent kinases (CDKs), which are regulated by a combination of cyclin expression and inhibitory phosphorylation. The Wee1 kinase is capable of phosphorylating CDK1 and CDK2 on Tyr-15 (Y15), preventing progression through G2/M and S phase, respectively [4,5]. This phosphorylation can be removed by the CDC25 phosphatases, which are in turn inactivated when phosphorylated by CHK1 [6,7]. Finally, CHK1 activity is controlled primarily by ATR kinase, which activates CHK1 by phosphorylation upon sensing replication stress or DNA damage [8,9]. Thus, both the CHK1 and the Wee1 pathways contribute to maintaining inhibitory phosphorylation of CDKs.
MK-1775, the first potent and selective inhibitor of Wee1, has been investigated primarily as an agent that can target the G2/M checkpoint to exert toxicity specifically in p53-mutant cells [10]. It has been shown that, when combined with DNA damaging agents, MK-1775 is able to abrogate the G2/M checkpoint and enhance apoptosis, although recent studies have questioned the p53-dependence of these effects [10–18]. Importantly, it has been shown that Wee1 inhibition alone can induce DNA damage, likely through the induction of replication stress secondary to overactive CDKs and inhibition of DNA repair [19].
Histone deacetylase (HDAC) inhibitors (HDACIs) have been shown in vitro to have promising anti-cancer activity, but their single-agent effectiveness in the clinic has been only modest [20–27]. However, there are many clinical trials investigating the role of HDACIs in combination therapies (NCT01242774, NCT01742793, NCT02061449, and NCT02145715, clinicaltrials.gov). Previous work from this lab has demonstrated the ability of HDACIs to synergize with standard chemotherapeutic agents, at least in part by enhancing DNA damage [28–31]. Importantly, we recently demonstrated that treatment with the pan-HDACI panobinostat (LBH589) was able to down-regulate CHK1 [28,29]. It has been reported that combined inhibition of Wee1 and CHK1 is effective at inducing cancer cell death [13,32–34], leading us to consider the combination of MK-1775 and panobinostat for the treatment of pancreatic cancer.
In this work, we use pre-clinical pancreatic cancer models to investigate the effects of the combination of MK-1775 and panobinostat, and the mechanism by which panobinostat enhances MK-1775-induced apoptosis. We demonstrate that MK-1775 alone is able to induce DNA damage and activate CHK1 in a CDK-dependent fashion. Panobinostat treatment down-regulates CHK1 and synergizes with MK-1775 to enhance apoptosis and cell growth inhibition. Importantly, we demonstrate that in some cell lines, the CHK1 pathway is able to overcome single agent Wee1 inhibition and maintain phosphorylation of CDK1. This demonstrates a potential mechanism of resistance to treatment with MK-1775 and emphasizes the importance of combinations with agents such as panobinostat.
Materials and methods
Drugs
MK-1775, panobinostat, LY2603618, and roscovitine were purchased from Selleck Chemicals (Houston, TX, USA).
Cell culture
The AsPC-1, BxPC-3, CFPAC-1, HPAC, MIAPaCa-2 and PANC-1 human pancreatic cancer cell lines were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured as previously described [35]. The cell lines were authenticated by the University of Arizona Genetics Core Facility (Tucson, AZ, USA).
In vitro cytotoxicity assays
In vitro cytotoxicities of MK-1775, panobinostat, roscovitine, and LY2603618, alone or in combination, in pancreatic cancer cell lines were measured by using MTT (3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazolium-bromide, Sigma-Aldrich, St. Louis, MO, USA) reagent, as previously described [30,36,37]. IC50 values were calculated as drug concentrations necessary to inhibit 50% growth compared to untreated control cells. The extent and direction of MK-1775 and panobinostat antitumor interactions were determined by standard isobologram analyses and by evaluating combination index (CI) values, calculated using CompuSyn software (ComboSyn, Inc., Paramus, NJ, USA), where CI < 1, CI = 1, and CI > 1 indicate synergistic, additive, and antagonistic effects, respectively.
Apoptosis and cell cycle progression
Pancreatic cancer cells were treated with the indicated drugs for up to 48 h. DNA content was determined by propidium iodide (PI) staining and flow cytometry analysis using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA), as previously described [38]. Cell cycle analysis was performed using Multicycle software (Phoenix Flow Systems, Inc., San Diego, CA, USA). Apoptotic events were expressed as the percent of cells with sub-G1 DNA content. Histograms were created using FlowJo v7.6.5 (Tree Star, Ashland, OR, USA). Apoptosis measured using annexin V/PI dual staining was performed as previously described [30,39].
Western blot analysis
Soluble proteins were extracted (in the presence of cOmplete protease and phosSTOP phosphatase inhibitors, Roche Applied Sciences, Indianapolis, IN, USA) and subjected to SDS-polyacrylamide gel electrophoresis. Separated proteins were electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes (Thermo Fisher Inc., Rockford, IL, USA) and immunoblotted with anti-Wee1 (4936), -PKMyt-1 (4282), -PARP (9542), -pCHK1(S345) (2341), -pCDC25C(S216) (9528), -pCDK1(Y15) (9111), -CDK1 (9112), -CDK2 (2546), -pH3(S10) (9701) or -γH2AX (2577, Cell Signaling Technology, Danvers, MA, USA), -CHK1 (sc8408, Santa Cruz Biotechnology, Santa Cruz, CA, USA), -pCDK2(Y15) (ab76146, Abcam, Cambridge, MA, USA), -ac-histone H4 (06–598), -histone H4 (07–108, Upstate Biotechnology, Lake Placid, NY, USA), -acetyl-α-tubulin (T7451) or -beta-actin antibody (A2228, Sigma-Aldrich), as previously described [39]. Primary antibodies were diluted 1:1000 in Odyssey Blocking Buffer (Li-Cor, Lincoln, NE, USA), except anti-beta-actin which was diluted 1:10,000. Immunoreactive proteins were visualized using the Odyssey Infrared Imaging System (Li-Cor), as described by the manufacturer.
Lentivirus production and shRNA knockdown of Wee1
The pMD-VSV-G and delta 8.2 plasmids were gifts from Dr. Dong at Tulane University. Wee1 and non-target control lentiviral vectors were purchased from Sigma-Aldrich. Lentivirus production and transduction of BxPC-3 cells were carried out as previously described [28].
Establishment of a mouse pancreatic cancer xenograft model
Female BALB/c nude mice (18–22g) were purchased from Vital River Laboratories (Beijing, China). The animal study was conducted following internationally recognized guidelines and was approved by the Animal Research Committee of Norman Bethune College of Medicine, Jilin University. The BxPC-3 xenograft model was generated as previously described [35]. BxPC-3 cells were adjusted to a density of 2 × 107 cells/mL with matrigel (BD Biosciences, San Jose, CA, USA) and inoculated subcutaneously in the right side axillae (0.1 mL/mouse). Once the tumor diameter reached approximately 0.5 cm it was isolated and cut into small pieces (1 mm in diameter). The right side axillae skin of mice was punctured to form a 5 mm long sinus tract where the tumor fragment was inserted subcutaneously. When the xenografts reached a volume of 170.2 ±23.4 mm3, the mice were randomized into four groups (7 animals per group, the mean tumor volumes were 171.9 ± 10.6, 174.0 ±9.4, 163.9 ±8.7, and 171.0 ±8.5 mm3 for the vehicle control, panobinostat, MK-1775, and combination group, respectively) and treated with (i) vehicle control, (ii) 10 mg/kg panobinostat once daily on Monday and Wednesday administered by intraperitoneal injection, (iii) 20 mg/kg MK-1775 twice daily on Monday and Wednesday by oral gavage, or (iv) 10 mg/kg panobinostat once daily by intraperitoneal injection and 20 mg/kg MK-1775 by oral gavage twice daily on Monday and Wednesday, for three weeks. Tumor diameters were measured with a caliper every 2–3 days. Tumor volume was calculated according to the following formula: m12 × m2 × 0.5236 (m1: short diameter; m2: long diameter). Once a tumor in the control group reached 1500 mm3 the mice were sacrificed. Tumor growth inhibition was calculated using the equation 100%×T/C, where C = final mean tumor volume – initial tumor volume for the control and T = final mean tumor volume – initial tumor volume for the treated.
Hematoxylin & eosin (H&E), immunohistochemical, and TUNEL staining
On day 22, mice were sacrificed and tumors from 6 mice in each treatment group were excised for H&E, proliferating cell nuclear antigen (PCNA) immunohistochemical staining, as previously described [35], and TUNEL staining. The slides were analyzed using a microscope and brown staining was scored using Image-Pro Plus 6.0 (Media Cybernetics, Inc., Bethesda, MD, USA). TUNEL assay was performed using the DeadEnd™ Fluorometric TUNEL System kit (Promega, Madison, WI, USA) according to the manufacturer’s protocol. PI was used to stain the nuclei. The localized green fluorescence of the apoptotic cells and red PI-stained nuclei were visualized by fluorescence microscopy.
Statistical analysis
Differences in cell apoptosis between panobinostat, roscovitine, LY2603618 or MK-1775 treated (individually or combined) and untreated cells were compared using the pair-wise two-sample t-test. The effect of treatment on the rate of tumor growth in the animal study was assessed by the coefficients corresponding to the interactions between treatment and time in the mixed effects model [40] using the nlme package in R [41]. All other statistical analyses were performed with GraphPad Prism 5.0. In all analyses, a p<0.05 corresponded to statistical significance.
Results
MK-1775 induces DNA damage and activates CHK1
To investigate the effects of MK-1775 treatment in pancreatic cancer cells, we first determined the expression levels of Wee1, PKMyt-1, p-CDK1, CDK1, p-CDK2, and CDK2 in 6 pancreatic cell lines by Western blot. The majority of cell lines expressed variable levels for all of the proteins (Fig. 1A). Then we determined MK-1775 sensitivity by MTT assays. MK-1775 IC50s varied, ranging from 470 nM (HPAC) to 13.2 μM (ASPC-1) (Fig. 1B, Table 1).
Fig. 1.
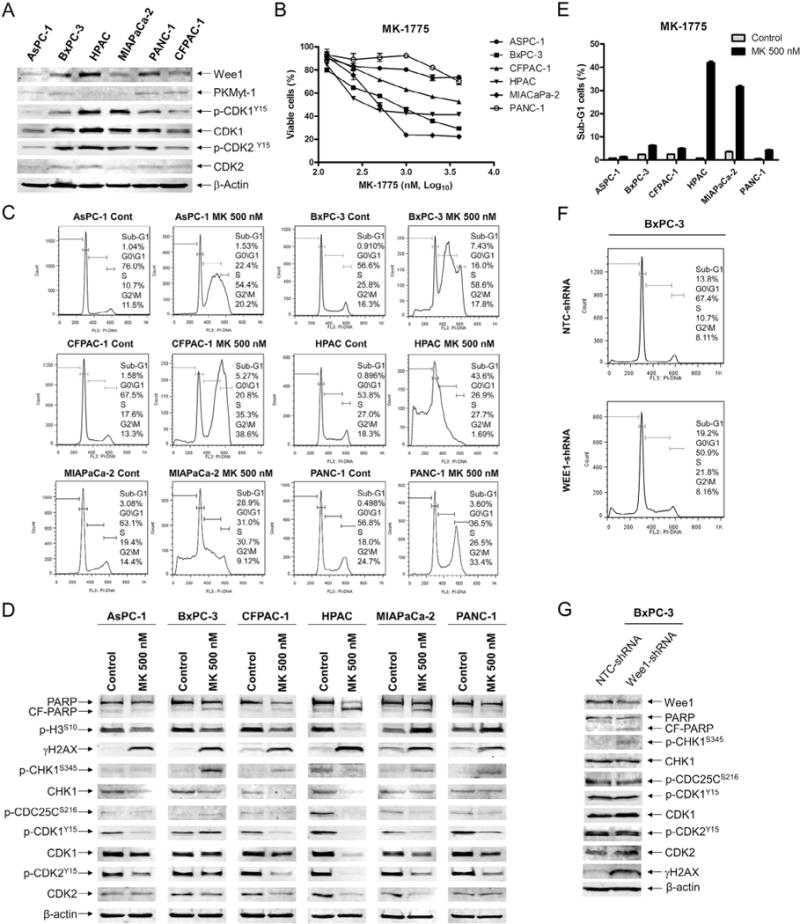
MK-1775 treatment induces DNA damage and activates the CHK1 pathway in pancreatic cancer cell lines. A, protein extracts from pancreatic cancer cell lines were subjected to Western blotting and probed with anti-Wee1, -PKMyt1, -p-CDK1, -CDK1, -p-CDK2, -CDK2, or -β-actin antibody. B, pancreatic cancer cell lines were cultured in 96-well plates at 37°C for 48 h in complete medium with variable concentrations of MK-1775 and viable cell numbers were determined using MTT reagent and a microplate reader. The data are presented as means ± standard errors from at least 3 independent experiments. C, pancreatic cancer cells were treated with vehicle control (Cont) or 500 nM MK-1775 (MK) for 48 h. The cells were fixed with ethanol, stained with PI, and subjected to flow cytometry analysis to determine cell cycle distribution. D, pancreatic cancer cells were treated with vehicle control or 500 nM MK-1775 for 48 h. Whole cell lysates were subjected to Western blotting and probed with anti-PARP, -p-H3, -γH2AX, -p-CHK1, -CHK1, -p-CDC25C, -p-CDK1, -CDK1, -p-CDK2, -CDK2, or -β-actin antibody. E, pancreatic cancer cells were treated with vehicle control or 500 nM MK-1775 for 48 h, then stained with PI and subjected to flow cytometry analysis. Apoptotic cells are expressed as the percentage of PI+ cells with sub-G1 DNA content. The data are presented as means of triplicates ± standard errors from one representative experiment. F, BxPC-3 cells were infected with Wee1 (designated Wee1-shRNA) or nontarget control (designated NTC-shRNA) shRNA lentivirus. The cells were fixed with ethanol, stained with PI, and subjected to flow cytometry analysis. G, BxPC-3 NTC- and Wee1-shRNA whole cell lysates were subjected to Western blotting and probed with anti-Wee1, -PARP, -p-CHK1, -CHK1, -p-CDC25C, -p-CDK1, -CDK1, -p-CDK2, -CDK2, -γH2AX, or -β-actin antibody. Representative Western blots and cell cycle histograms are shown.
Table 1.
Effect of MK-1775 on panobinostat sensitivities in pancreatic cancer cell lines.
| Cell line | IC50 of MK-1775 (μM) | IC50 of panobinostat (nM) in the absence or presence of MK-1775 (μM)
|
p Value# | |||
|---|---|---|---|---|---|---|
| 0 | 0.13 | 0.25 | 0.5 | |||
| ASPC-1 | 13.2 ± 1.1* | 94.6 ± 9.2 | 58.5 ± 6.3 (0.63) |
56.7 ± 4.8 (0.62) |
53.3 ± 3.7 (0.6) |
<0.05 |
| BxPC-3 | 0.8 ± 0.03 | 37.8 ± 2.2 | 16.2 ± 0.8 (0.59) |
13.2 ± 1.1 (0.66) |
11. 9 ± 1.0 (0.95) |
<0.001 |
| CFPAC-1 | 3.3 ± 0.2 | 407.6 ± 14.0 | 263.4 ± 19.9 (0.68) |
246.1 ± 7.6 (0.68) |
224.5 ± 5.0 (0.7) |
<0.005 |
| HPAC | 0.5 ± 0.01 | 81.7 ± 7.8 | 50.2 ± 1.6 (0.88) |
28.4 ± 1. 6 (0.88) |
ND | <0.05 |
| MIAPaCa-2 | 0.5 ± 0.05 | 36.0 ± 2.7 | 21.2 ± 1.5 (0.82) |
16.1 ± 1. 5 (0.91) |
ND | <0.05 |
| PANC-1 | 10.6 ± 1.1* | 569.6 ± 13.4* | 397.7 ± 23.0 (0.71) |
378.6 ± 24.3 (0.69) |
325.4 ± 21.6 (0.62) |
<0.005 |
Note: MK-1775 and panobinostat IC50s are presented as means ± standard errors from at least three independent experiments. Numbers in parentheses represent the combination index values.
ND – not determined.
indicates estimated IC50 calculated by using GraphPad Prism 5.0 software.
indicates that p values for each pair were determined using GraphPad Prism 5.0 and only the largest p value is shown for each cell line.
Next, we treated the pancreatic cancer cell lines with 500 nM MK-1775, a clinically achievable concentration [42,43], for 48 h and analyzed the cell cycle distributions. MK-1775 treatment caused S arrest or both S and G2/M arrest in all of the cell lines tested (Fig. 1C). This was accompanied by decreased p-CDK1 and p-CDK2 in all of the cell lines tested, except the BxPC-3 cells. Following MK-1775 treatment, MIAPaCa-2 and PANC-1 cells had increased levels of phosphorylated histone H3 (S10), indicating an increase of mitotic cells (Fig. 1D). Both HPAC and MIAPaCa-2 cells had a substantial amount of apoptotic cells following MK-1775 treatment, as indicated by the high percentage of cells with sub-G1 DNA content and increased PARP cleavage (Fig. 1C–E). Inhibition of Wee1 resulted in increased γH2AX levels, indicating increased DNA double-strand breaks (Fig. 1D), in all of the cell lines. In addition, most cell lines had increased p-CHK1, indicating activation of the CHK1 pathway (Fig. 1D). To determine if these were on-target effects, we used an shRNA to knockdown Wee1 in the BxPC-3 cells (designated Wee1-shRNA). This resulted in increased S phase cells and increased γH2AX and p-CHK1 levels compared to non-target control cells (designated NTC-shRNA, Fig. 1F&G), consistent with the MK-1775 treatments in the parental cell line, suggesting that the MK-1775 treatment effects were on-target.
We then treated BxPC-3 and HPAC cells with 500 nM MK-1775 to determine the effects over time. In both cell lines there was a time-dependent increase in S phase cells and apoptosis, as well as increased γH2AX, indicating increased DNA damage. Total CHK1 levels decreased over time in both cell lines (Fig. 2). Although CHK1 activation was not observed after MK-1775 48 h treatment in HPAC cells, we did detect an increased level as early as 4 h, which decreased to a level similar to baseline by 24 h (Fig. 2D). The BxPC-3 cells had a similar dynamic; however, the level at 48 h remained substantially higher than baseline (Fig. 2B). Total CDK1 and CDK2 levels remained largely unchanged over the course of the treatment in the BxPC-3 cells, whereas there was a time-dependent decrease in the HPAC cells.
Fig. 2.
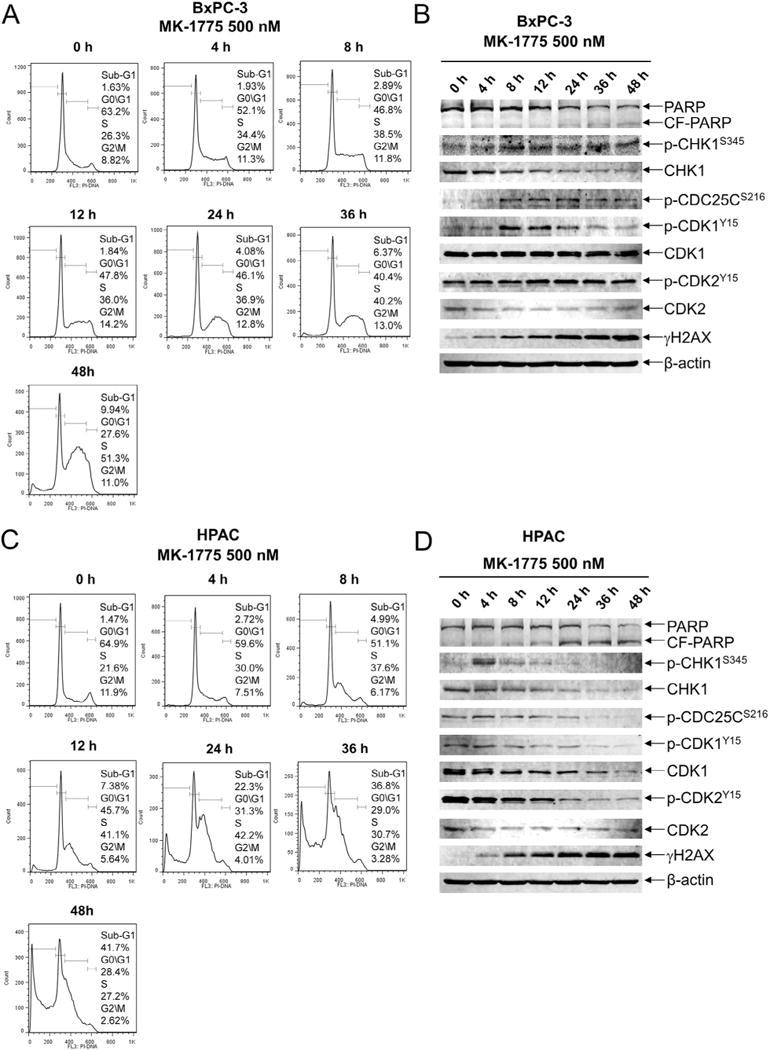
MK-1775 induces p-CHK1 at early time points. A and C, BxPC-3 and HPAC cells were treated with 500 nM MK-1775 for up to 48 h. The cells were harvested, fixed with ethanol, stained with PI, and then subjected to flow cytometry analysis. B and D, the remaining cells were lysed and the protein extracts were subjected to Western blotting, and probed with anti-PARP, -p-CHK1, -CHK1, -p-CDC25C, -p-CDK1, -CDK1, -p-CDK2, -CDK2, -γH2AX, or -β-actin antibody. Experiments were performed at least three independent times and representative Western blots and cell cycle histograms are shown.
MK-1775-induced DNA damage is dependent on CDK activity
To determine if the DNA damage induced by MK-1775 treatment is dependent on CDK activity, we treated HPAC cells with roscovitine, a CDK inhibitor, which resulted in a concentration-dependent decrease in viable cells (Fig. 3A). After MK-1775 treatment for 8 h we detected increased γH2AX and p-CHK1 and decreased p-CDK1 and p-CDK2. The addition of roscovitine attenuated the induction of both γH2AX and p-CHK1 (Fig. 3B). Treatment with MK-1775 for 48 h induced 40% apoptosis, which was reduced to about 10% by adding roscovitine (Fig. 3C). Further, MTT assays clearly demonstrated antagonistic growth inhibition by the agents (Fig. 3D&E). Similar results were obtained in the BxPC-3 cells (Fig. 3F–H). These results suggest that CDK activity is required for the antitumor activity of MK-1775 in pancreatic cancer cell lines.
Fig. 3.
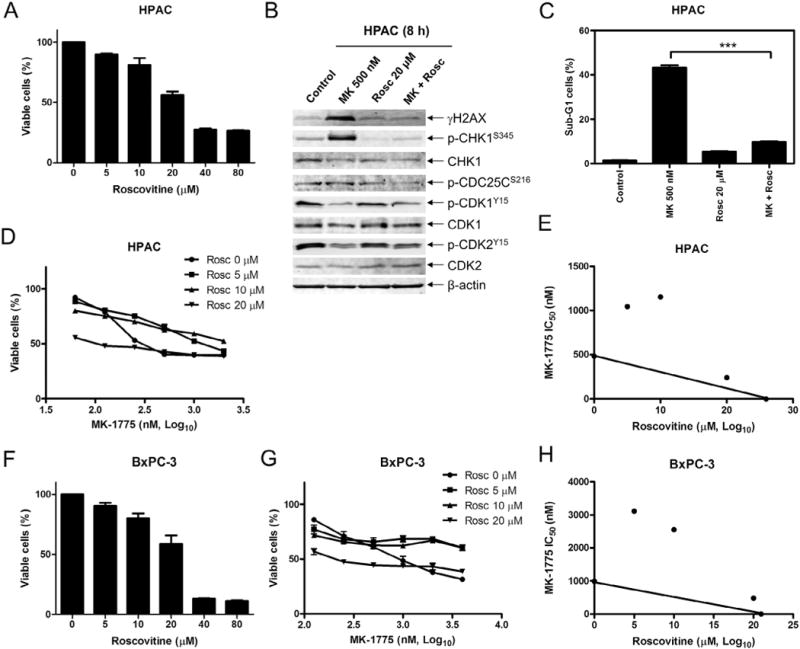
MK-1775-induced DNA damage, CHK1 phosphorylation, and cell death require active CDKs. A, HPAC cells were treated with 0–80 μM roscovitine (Rosc) for 48 h and viable cells were determined using MTT reagent and a microplate reader. The data are presented as means ± standard errors from at least 3 independent experiments. B, HPAC cells were treated with vehicle control, 500 nM MK-1775, 20μM roscovitine, or 500 nM MK-1775 plus 20 μM roscovitine for 8 h. Whole cell lysates were subjected to Western blotting and probed with anti-p-CHK1, -CHK1, -p-CDC25C, -p-CDK1, -CDK1, -p-CDK2, -CDK2, -γH2AX, or -β-actin antibody. C, HPAC cells were treated with MK-1775, roscovitine, or in combination for 48 h, stained with PI and subjected to flow cytometry analysis. Apoptotic cells were defined as the percentage of PI+ cells with sub-G1 DNA content. *** indicates p < 0.0005. D, HPAC cells were treated with variable concentrations of MK-1775 with or without 5 μM, 10μM or 20μM roscovitine. Viable cells were measured by MTT assays and the data are presented as means ± standard errors from at least 3 independent experiments. E, standard isobologram analyses of antitumor interactions between MK-1775 and roscovitine were performed in HPAC cells. The IC50 values of each drug are plotted on the axes; the solid line represents additive effect, while the points represent the concentrations of each drug resulting in 50% inhibition of growth. Points falling below the line indicate synergism, whereas those above the line indicate antagonism. F, BxPC-3 cells were treated with variable concentrations of roscovitine for 48 h and viable cells were determined by MTT assays. The data are presented as means ± standard errors from at least 3 independent experiments. G, BxPC-3 cells were treated with variable concentrations of MK-1775 with or without 5 μM, 10 μM or 20 μM roscovitine. Viable cells were measured by MTT assays and the data are presented as means ± standard errors from at least 3 independent experiments. H, standard isobologram analyses of antitumor interactions between MK-1775 and roscovitine were performed in BxPC-3 cells.
MK-1775 synergizes with LY2603618
To determine if CHK1 plays a critical role in MK-1775 sensitivity, we investigated the combination of MK-1775 with LY2603618, a CHK1 selective inhibitor, in BxPC-3 cells. Treatment with 500 nM LY2603618 (a concentration which showed minimal impact on cell growth determined by MTT assays, Fig. 4A) or MK-1775 resulted in S phase arrest, which was accompanied by a small increase in sub-G1 cells (Fig. 4B). The combined treatment resulted in substantially increased sub-G1 cells and PARP cleavage (Fig. 4B&C). In contrast to roscovitine, LY2603618 substantially increased γH2AX levels induced by MK-1775 treatment (Fig. 4C) and synergistically enhanced MK-1775-induced growth inhibition (Fig. 4D&E). These results demonstrate that simultaneous inhibition of Wee1 and CHK1 results in enhanced apoptosis and growth arrest.
Fig. 4.
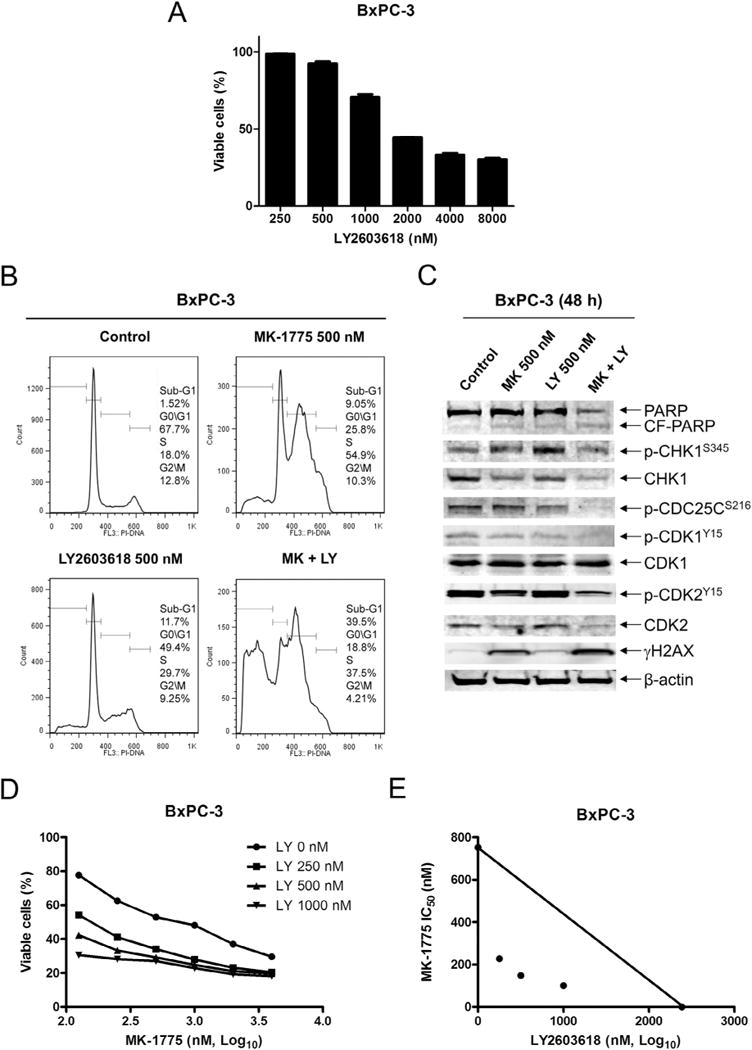
MK-1775 synergizes with LY2603618 in BxPC-3 cells. A, BxPC-3 cells were treated with variable concentrations of LY2603618 for 48 h and viable cells were determined by MTT assays. The data are presented as means ± standard errors from at least 3 independent experiments. B, BxPC-3 cells were treated with vehicle control, MK-1775 (MK), LY2603618 (LY) or MK-1775 plus LY2603618 for 48 h, stained with PI and subjected to flow cytometry analysis. C, protein extracts were subjected to Western blotting and probed with anti-PARP, -p-CHK1, -CHK1, -p-CDC25C, -p-CDK1, -CDK1, -p-CDK2, -CDK2, -γH2AX, or -β-actin antibody. D, BxPC-3 cells were treated with variable concentrations of MK-1775 with or without 0.25 μM, 0.5 μM or 1 μM LY2603618. Viable cells were measured by MTT assays and the data are presented as means ± standard errors from at least 3 independent experiments. E, standard isobologram analyses of antitumor interactions between MK-1775 and LY2603618 in BxPC-3 cells were performed.
Panobinostat synergizes with MK-1775
We have previously demonstrated that panobinostat treatment suppresses CHK1 expression in acute myeloid leukemia and neuroblastoma [28,29]. Based on these studies and our current study demonstrating synergistic antitumor activity of combined CHK1 and Wee1 inhibition, we hypothesized that panobinostat would synergize with MK-1775 in pancreatic cancer through suppression of CHK1. Panobinostat treatments caused growth inhibition in the pancreatic cancer cell lines, with IC50s ranging from 36.0 nM (MIAPaCa-2) to 569.6 nM (PANC-1, Fig. 5A, Table 1). Treatment of the cell lines with 40 nM panobinostat for 48 h caused a decrease in S phase cells and an increase in sub-G1 cells (Fig. 5B&C). Panobinostat treatment resulted in increased γH2AX levels (Fig. 5D). Furthermore, p-CHK1, CHK1, or both were decreased after treatment and this was accompanied by decreased p-CDC25C, CDK1, CDK2, p-CDK1, and p-CDK2 in all but the HPAC cells. In contrast, panobinostat treatment resulted in increased p-CDC25C and p-CDK2 in the HPAC cells (Fig. 5D).
Fig. 5.
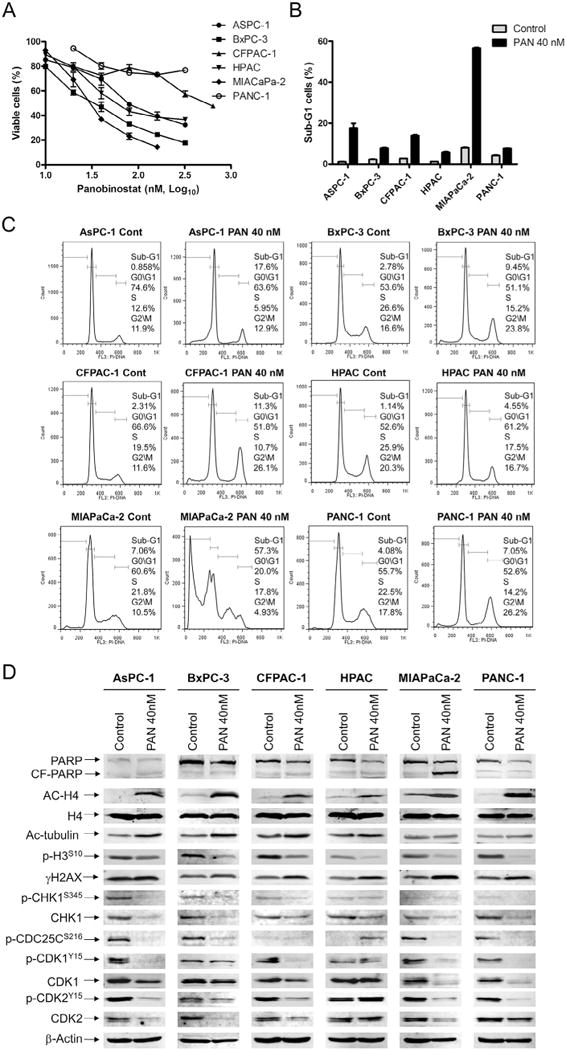
Panobinostat down-regulates CHK1 in pancreatic cancer cells. A, pancreatic cancer cell lines were cultured in 96-well plates at 37°C for 48 h in complete medium with variable concentrations of panobinostat and viable cell numbers were determined using MTT reagent and a microplate reader. The data are presented as means ± standard errors from at least 3 independent experiments. B–D, pancreatic cancer cells were treated with vehicle control or 40 nM panobinostat for 48 h, fixed with ethanol, stained with PI, and subjected to flow cytometry analysis. The sub-G1 data are presented as means of triplicates ± standard errors from one representative experiment (B). Representative cell cycle histograms are shown (C). Protein lysates were extracted from the remaining cells, subjected to Western blotting, and then probed with anti-PARP, -p-H3, -γH2AX, -ac-H4, -H4, -ac-tubulin, -p-CHK1, -CHK1, -p-CDC25C, -p-CDK1, -CDK1, -p-CDK2, -CDK2, or -β-actin antibody (D).
Consistent with our hypothesis, panobinostat synergized with MK-1775 in all of the cell lines, as indicated by CI values less than 1.0 (Table 1, CI values ranged from 0.60 to 0.95). For BxPC-3 cells, panobinostat enhanced MK-1775-induced apoptosis, as measured by PI staining and cell cycle analyses (Fig. 6A–C), annexin V/PI double staining and flow cytometry analyses (Fig. 6E&F), and PARP cleavage (Fig. 6D). CI values indicate that the enhancement on apoptosis was synergistic (Fig. 6B&F). Panobinostat treatment was able to abrogate the S phase cell cycle checkpoint, which was activated by MK-1775 treatment (Fig. 6C), and abolish p-CHK1 induced by MK-1775 (Fig. 6D). In addition, CHK1, p-CDC25C, p-CDK1, and p-CDK2 protein levels were lower after the combined drug treatment than after MK-1775 treatment alone.
Fig. 6.
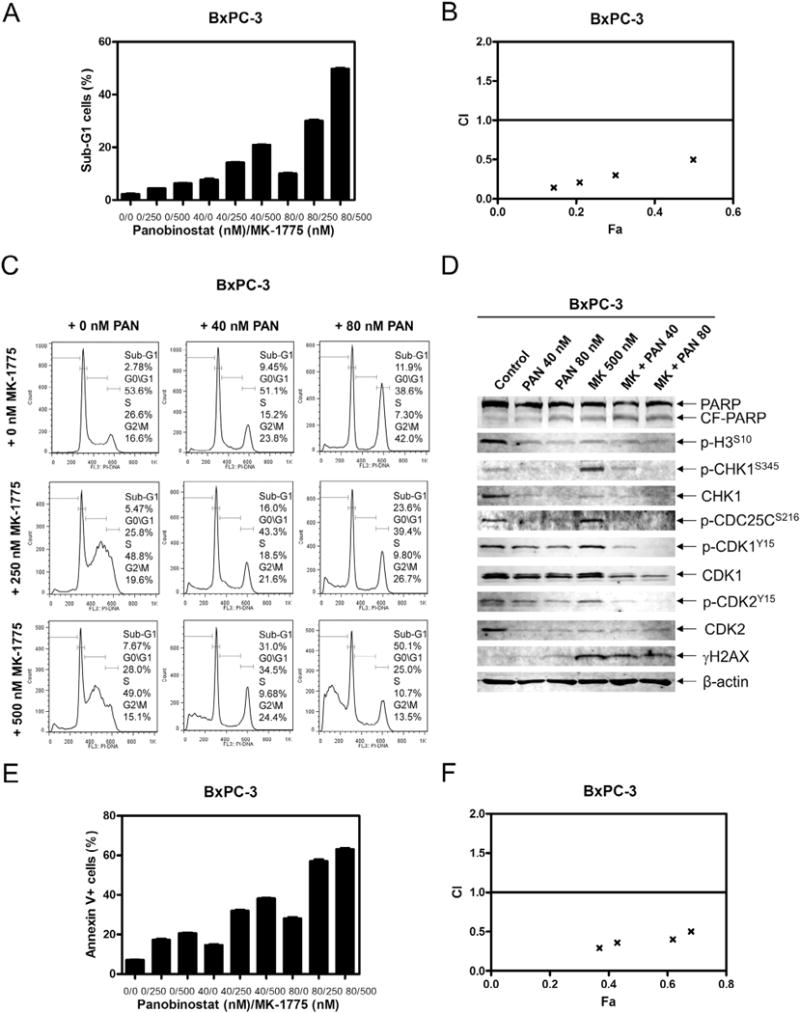
Panobinostat synergizes with MK-1775 in BxPC-3 cells. A, BxPC-3 cells were treated with panobinostat and MK-1775, alone or combined, for 48 h and subjected to PI staining and flow cytometry analysis. B, CI vs. Fa plot (combination index vs. fraction affected) for the apoptosis data presented in Panel A. The CI values were calculated using CompuSyn software. C, BxPC-3 cells were treated with panobinostat and/or MK-1775 for 48 h and subjected to PI staining and flow cytometry analysis. D, protein extracts were subjected to Western blotting and probed with anti-PARP, -p-H3, -p-CHK1, -CHK1, -p-CDC25C, -p-CDK1, -CDK1, -p-CDK2, -CDK2, -γH2AX, or -β-actin antibody. E, BxPC-3 cells were treated with panobinostat and MK-1775 for 48 h, then stained with annexin V/PI and subjected to flow cytometry analysis. The data are presented as means ± standard errors from one representative experiment. F, CI vs. Fa plot (combination index vs. fraction affected) for the apoptosis data presented in Panel E. The CI values were calculated using CompuSyn software.
Finally, we examined the in vivo effects of MK-1775 and panobinostat. Mice bearing BxPC-3 xenograft tumors received twice a week for three weeks: vehicle, MK-1775 (20 mg/kg) twice daily, panobinostat (10 mg/kg) once daily, or MK-1775 and panobinostat. As shown in Fig. 7A, body weights remained relatively the same throughout the study, regardless of treatment group, indicating that the drugs were well tolerated. Treatment with either MK-1775 or panobinostat alone had modest delay of externally measurable tumor growth (30.9% and 37.8% on day 20, respectively). In contrast, the combined drug treatment resulted in significant delay of tumor growth during the treatment period compared to single drug treatment (Fig. 7B), with 58.7% tumor growth inhibition on day 20. To determine if the combined treatment affected tumor growth rate, a mixed effects linear model was applied to the tumor volume for each experimental group. Statistically significant difference in the rate of change in tumor volume was detected. Compared to the control group, the panobinostat, MK-1775, and combination had p values of 0.0076, 0.0368, and <0.0001, respectively.
Fig. 7.
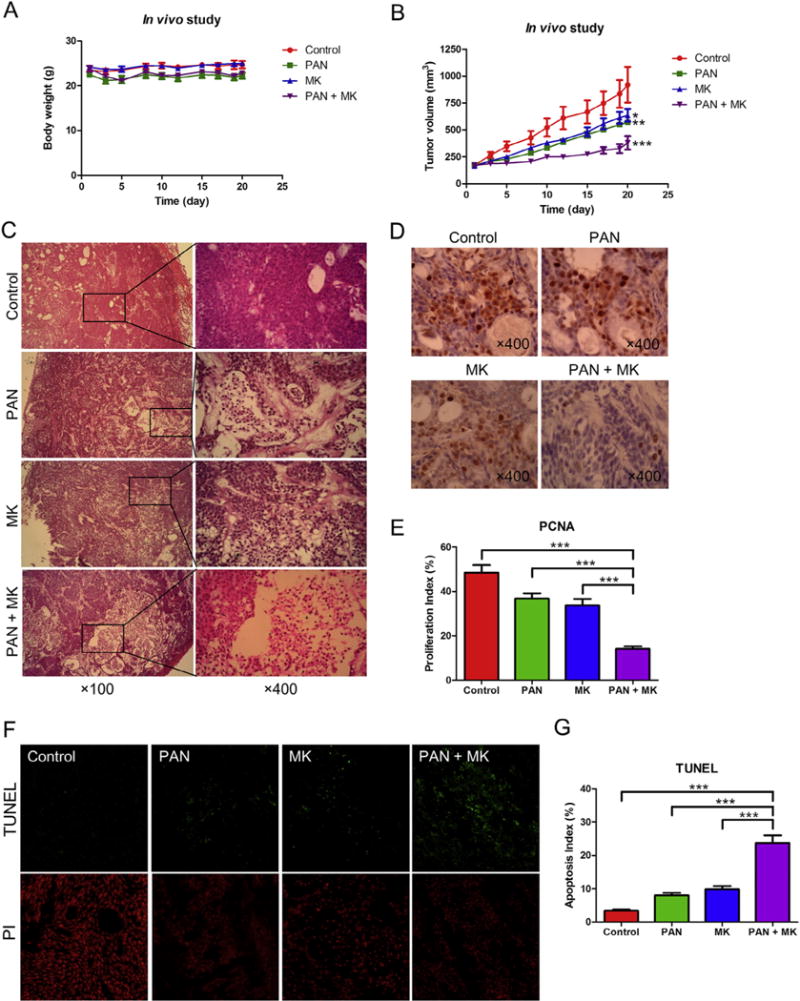
Panobinostat enhances the antitumor activity of MK-1775 in a BxPC-3 xenograft model. A, body weights were measured every 3–4 days. B, tumor volumes were calculated according to the following formula: m12 × m2× 0.5236 (m1: short diameter; m2: long diameter). C–E, tumor specimens were fixed in 10% formalin, embedded in paraffin, and cut into 4 μM-thick slides for H&E (C) and PCNA staining (D). The proliferation index was calculated as proliferation index = PCNA positive cells/observed cells × 100% and graphed as means ± standard errors (E). F, apoptosis was measured using the TUNEL assay in the tumor specimens. G, apoptosis index was calculated as TUNEL-positive cells/observed cells × 100% and graphed as means ± standard errors. *** indicates p < 0.0005.
To further investigate the in vivo effects of MK-1775 and panobinostat treatment, tumors were analyzed by H&E, immunohistochemical, and TUNEL staining. Individual drug treatment resulted in increased tumor necrosis, which was further increased following combination treatment, as indicated by H&E staining (Fig. 7C). Proliferation was substantially lower in the combination group compared to the individual drug treatment groups, as indicated by lower PCNA staining and significantly lower proliferation index values (Fig. 7D&E). Single drug treatment resulted in increased apoptosis, as measured by TUNEL assay and calculation of apoptotic indices, which was significantly increased in the combined drug treatment (Fig. 7F&G). These data highlight the potential for using combined MK-1775 and panobinostat for the treatment of pancreatic cancer.
Discussion
Pancreatic cancer’s poor prognosis highlights the need for new therapies. Recent studies have demonstrated that MK-1775 treatment results in increased phosphorylation of both H2AX and CHK1 [13,33,34], which we confirmed in six pancreatic cancer cell lines. We found that the increased phosphorylation of CHK1 and H2AX following MK-1775 treatment was dependent on CDK activity. Furthermore, we found that a CHK1 selective inhibitor (LY2603618) enhanced MK-1775 sensitivity in a synergistic manner in pancreatic cancer cell lines. Although others have demonstrated similar synergistic interaction between MK-1775 and a CHK1 inhibitor, we used a different CHK1 inhibitor than that which has been reported [13,32–34,44]. Additionally, we demonstrated that panobinostat treatment can be used as an alternate means to inactivate the CHK1 pathway and has synergistic antitumor activity in combination with MK-1775 in pancreatic cancer cell lines. Finally, in vivo cooperative antitumor activity of the two agents was demonstrated in a BxPC-3 xenograft mouse model.
As discussed in Guertin et al. [34], inhibition of CHK1 or Wee1 increases CDK1/2 activity, therefore the combination of a CHK1 and Wee1 inhibitor could result in additive increase of CDK1/2. Although they were unable to detect further decrease in p-CDK1/2 beyond single agent MK-1775 treatment, we did see further decrease in p-CDK2 by the addition of LY2603618. This difference may be due to the use of a different CHK1 inhibitor (MK-8776). Another study which investigated the combination of a CHK1 inhibitor (AR458323) and MK-1775 also found that both drugs reduced inhibitory phosphorylation of CDK1/CDK2 in an erythroleukemia cell line [33]. However, in addition to further decrease of p-CDK2, they also found further decrease of p-CDK1. These differences may be due to the differences in the CHK1 inhibitors used and/or the different malignancies that cell lines were derived from. In addition, we found that panobinostat treatment also decreased p-CDK2 levels below what was detected with MK-1775 treatment alone. Although MK-1775 treatment alone did not appear to decrease p-CDK1 levels in BxPC-3 cells, p-CDK1 levels were decreased by panobinostat treatment and were even further decreased in the combined treatment. Our in vivo data further confirm that panobinostat enhances MK-1775 antitumor activity, which is in line with the results presented by Davies et al. [33]. They demonstrated cooperative antitumor activity between MK-8776 and MK-1775 in vivo using colorectal cancer and ovarian carcinoma mouse xenograft models. While we demonstrate that panobinostat inactivates the CHK1 pathway, we cannot rule out the involvement of other pathways as panobinostat affects a multitude of other targets [45]. Panobinostat has also been demonstrated to induce cell death and inhibit tumor growth in various malignancies through regulation of gene expression and protein acetylation [46–48]. We previously demonstrated that panobinostat treatment results in suppression of CHK1, Rad51, and BRCA1 in acute myeloid leukemia cells [28]. In this study, panobinostat in combination with MK-1775 was more potent than the combination of LY2603618 with MK-1775, as evident by the different amount of apoptosis induced by the combination despite similar apoptosis induction by the single drug treatments (Figs. 4B & 6C). Thus, it is possible that panobinostat may affect expression of additional proteins involved in DNA damage repair, apoptosis, and/or cell cycle progression. Therefore, inhibition of the CHK1 pathway may not be the only pathway involved in the mechanism of action of the combination of panobinostat and MK-1775.
Based on published reports and our own results, we propose a mechanism of action for the combined MK-1775 and panobinostat treatment in Fig. 8. MK-1775 inhibits Wee1, resulting in an increased pool of active CDK1/CDK2, which leads to DNA damage, activation of ATM/ATR, and activation of CHK1. Active CHK1 inactivates CDC25s, leading to decreased pools of active CDK1/2, cell cycle arrest and eventually DNA repair and cell survival. In addition, active CHK1 may directly increase Wee1 activity [32,49]. Combining panobinostat with MK-1775 would prevent CHK1 activation, thus leading to more DNA damage and increased cell death.
Fig. 8.
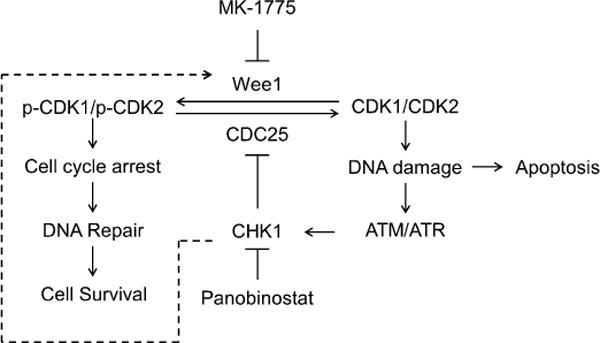
Proposed mechanism for the antitumor interactions between MK-1775 and panobinostat in pancreatic cancer. Inhibition of Wee1 reduces phosphorylation of CDK1/CDK2, allowing CDK1/CDK2 to remain active, which leads to DNA damage. DNA damage triggers activation of ATM/ATR, which then activates CHK1. Active CHK1 inhibits CDC25s, leading to decreased removal of the inhibitory phosphorylation on CDK1/CDK2, resulting in increased p-CDK1/p-CDK2. This limits the amount of active CDK1/CDK2 and the resulting DNA damage following MK-1775 treatment. Panobinostat inhibits activation of CHK1, thus maintaining the active CDK1/CDK2 pools, enhancing DNA damage, and eventually leading to apoptosis. Others have suggested that CHK1 can activate Wee1, as indicated by the dashed line.
In conclusion, we demonstrate that panobinostat synergistically/cooperatively enhances MK-1775 efficacy in pancreatic cancer cell lines and a BxPC-3 xenograft mouse model. Our results suggest that panobinostat inactivates the CHK1 pathway and activates CDK1 and CDK2, thus allowing MK-1775-induced DNA damage to accumulate and subsequently induce apoptosis. Our results support the clinical development of MK-1775 and panobinostat combination for the treatment of pancreatic cancer.
Acknowledgments
This study was supported by a Start-up Fund from Jilin University, Changchun, China, and a grant from the National Natural Science Foundation of China, NSFC 31271477. Mr. JTC is a predoctoral trainee supported by T32 CA009531 from the National Cancer Institute. The funders had no role in study design, data collection, analysis and interpretation of data, decision to publish, or preparation of the manuscript.
Abbreviations
- PAN
panobinostat
- CI
combination index
- MK
MK-1775
- Cont
control
- Rosc
roscovitine
Footnotes
Conflict of interest
The authors declare no competing financial interests.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955–1957. doi: 10.1126/science.1384126. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14:1878–1891. doi: 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 7.Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 8.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 11.Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524–539. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 12.Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, et al. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. 2011;17:5638–5648. doi: 10.1158/1078-0432.CCR-11-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaudhuri L, Vincelette ND, Koh BD, Naylor RM, Flatten KS, Peterson KL, et al. CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica. 2014;99:688–696. doi: 10.3324/haematol.2013.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther. 2013;12:1442–1452. doi: 10.1158/1535-7163.MCT-13-0025. [DOI] [PubMed] [Google Scholar]
- 15.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9:514–522. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 16.Kreahling JM, Foroutan P, Reed D, Martinez G, Razabdouski T, Bui MM, et al. Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PLoS ONE. 2013;8:e57523. doi: 10.1371/journal.pone.0057523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799–2806. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Linden AA, Baturin D, Ford JB, Fosmire SP, Gardner L, Korch C, et al. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol Cancer Ther. 2013;12:2675–2684. doi: 10.1158/1535-7163.MCT-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krajewska M, Heijink AM, Bisselink YJ, Seinstra RI, Sillje HH, de Vries EG, et al. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene. 2013;32:3001–3008. doi: 10.1038/onc.2012.296. [DOI] [PubMed] [Google Scholar]
- 20.Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105:959–967. doi: 10.1182/blood-2004-05-1693. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;112:981–989. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111:1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 23.Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res. 2006;12:4628–4635. doi: 10.1158/1078-0432.CCR-06-0511. [DOI] [PubMed] [Google Scholar]
- 24.Gimsing P, Hansen M, Knudsen LM, Knoblauch P, Christensen IJ, Ooi CE, et al. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur J Haematol. 2008;81:170–176. doi: 10.1111/j.1600-0609.2008.01102.x. [DOI] [PubMed] [Google Scholar]
- 25.Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007;109:2781–2790. doi: 10.1182/blood-2006-05-021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klimek VM, Fircanis S, Maslak P, Guernah I, Baum M, Wu N, et al. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res. 2008;14:826–832. doi: 10.1158/1078-0432.CCR-07-0318. [DOI] [PubMed] [Google Scholar]
- 27.Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, et al. The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer. 2006;106:112–119. doi: 10.1002/cncr.21552. [DOI] [PubMed] [Google Scholar]
- 28.Xie C, Drenberg C, Edwards H, Caldwell JT, Chen W, Inaba H, et al. Panobinostat enhances cytarabine and daunorubicin sensitivities in AML cells through suppressing the expression of BRCA1, CHK1, and Rad51. PLoS ONE. 2013;8:e79106. doi: 10.1371/journal.pone.0079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang G, Edwards H, Caldwell JT, Buck SA, Qing WY, Taub JW, et al. Panobinostat synergistically enhances the cytotoxic effects of cisplatin, doxorubicin or etoposide on high-risk neuroblastoma cells. PLoS ONE. 2013;8:e76662. doi: 10.1371/journal.pone.0076662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie C, Edwards H, Xu X, Zhou H, Buck SA, Stout ML, et al. Mechanisms of synergistic antileukemic interactions between valproic acid and cytarabine in pediatric acute myeloid leukemia. Clin Cancer Res. 2010;16:5499–5510. doi: 10.1158/1078-0432.CCR-10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie C, Edwards H, Lograsso SB, Buck SA, Matherly LH, Taub JW, et al. Valproic acid synergistically enhances the cytotoxicity of clofarabine in pediatric acute myeloid leukemia cells. Pediatr Blood Cancer. 2012;59:1245–1251. doi: 10.1002/pbc.24152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carrassa L, Chila R, Lupi M, Ricci F, Celenza C, Mazzoletti M, et al. Combined inhibition of Chk1 and Wee1: in vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle. 2012;11:2507–2517. doi: 10.4161/cc.20899. [DOI] [PubMed] [Google Scholar]
- 33.Davies KD, Cable PL, Garrus JE, Sullivan FX, von Carlowitz I, Huerou YL, et al. Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol Ther. 2011;12:788–796. doi: 10.4161/cbt.12.9.17673. [DOI] [PubMed] [Google Scholar]
- 34.Guertin AD, Martin MM, Roberts B, Hurd M, Qu X, Miselis NR, et al. Unique functions of CHK1 and WEE1 underlie synergistic anti-tumor activity upon pharmacologic inhibition. Cancer Cell Int. 2012;12:45. doi: 10.1186/1475-2867-12-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen S, Wang G, Niu X, Zhao J, Tan W, Wang H, et al. Combination of AZD2281 (Olaparib) and GX15-070 (Obatoclax) results in synergistic antitumor activities in preclinical models of pancreatic cancer. Cancer Lett. 2014;348:20–28. doi: 10.1016/j.canlet.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 36.Xu X, Xie C, Edwards H, Zhou H, Buck SA, Ge Y. Inhibition of histone deacetylases 1 and 6 enhances cytarabine-induced apoptosis in pediatric acute myeloid leukemia cells. PLoS ONE. 2011;6:e17138. doi: 10.1371/journal.pone.0017138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ge Y, Dombkowski AA, LaFiura KM, Tatman D, Yedidi RS, Stout ML, et al. Differential gene expression, GATA1 target genes, and the chemotherapy sensitivity of Down syndrome megakaryocytic leukemia. Blood. 2006;107:1570–1581. doi: 10.1182/blood-2005-06-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang G, He J, Zhao J, Yun W, Xie C, Taub JW, et al. and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PLoS ONE. 2012;7:e52095. doi: 10.1371/journal.pone.0052095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edwards H, Xie C, LaFiura KM, Dombkowski AA, Buck SA, Boerner JL, et al. RUNX1 regulates phosphoinositide 3-kinase/AKT pathway: role in chemotherapy sensitivity in acute megakaryocytic leukemia. Blood. 2009;114:2744–2752. doi: 10.1182/blood-2008-09-179812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38:963–974. [PubMed] [Google Scholar]
- 41.Pinheiro J, Bates B, DebRoy S, Sarkar D. R Package Version 3. 2005 [Google Scholar]
- 42.Tibes R, Bogenberger JM, Chaudhuri L, Hagelstrom RT, Chow D, Buechel ME, et al. RNAi screening of the kinome with cytarabine in leukemias. Blood. 2012;119:2863–2872. doi: 10.1182/blood-2011-07-367557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leijen S, Schellens JH, Shapiro G, Pavlick AC, Tibes R, Demuth T, et al. A phase I pharmacological and pharmacodynamic study of MK-1775, a Weel tyrosine kinase inhibitor, in monotherapy and combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2010;28:3067. [Google Scholar]
- 44.Russell MR, Levin K, Rader J, Belcastro L, Li Y, Martinez D, et al. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013;73:776–784. doi: 10.1158/0008-5472.CAN-12-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 46.Brazelle W, Kreahling JM, Gemmer J, Ma Y, Cress WD, Haura E, et al. Histone deacetylase inhibitors downregulate checkpoint kinase 1 expression to induce cell death in non-small cell lung cancer cells. PLoS ONE. 2010;5:e14335. doi: 10.1371/journal.pone.0014335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scuto A, Kirschbaum M, Kowolik C, Kretzner L, Juhasz A, Atadja P, et al. The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph- acute lymphoblastic leukemia cells. Blood. 2008;111:5093–5100. doi: 10.1182/blood-2007-10-117762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther. 2011;10:2034–2042. doi: 10.1158/1535-7163.MCT-11-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rothblum-Oviatt CJ, Ryan CE, Piwnica-Worms H. 14-3-3 binding regulates catalytic activity of human Wee1 kinase. Cell Growth Differ. 2001;12:581–589. [PubMed] [Google Scholar]