

Abstract
Cardiovascular disease is the leading cause of death in the western world. Heart failure is a heterogeneous and complex syndrome, arising from various etiologies, which result in cellular phenotypes that vary from patient to patient. The ability to utilize genetic manipulation and biochemical experimentation in animal models has made them indispensable in the study of this chronic condition. Similarly, proteomics has been helpful for elucidating complicated cellular and molecular phenotypes and has the potential to identify circulating biomarkers and drug targets for therapeutic intervention. In this review, the use of human samples and animal model systems (pig, dog, rat, mouse, zebrafish, and fruit fly) in cardiac research is discussed. Additionally, the protein sequence homology between these species and the extent of conservation at the level of the phospho-proteome in major kinase signaling cascades involved in heart failure are investigated.
Keywords: Animal models, Heart failure, Posttranslational modifications
1 Introduction
Cardiovascular disease (CVD) is the leading cause of death in the Western world. Each year, CVD results in approximately 700 000 deaths in the United States, and 1 900 000 deaths in the European Union (EU) 1,2. The economic burden is enormous, and the annual cost is estimated at $300 billion in the United States and €196 billion in the EU 1,2. Thus, there is a pressing need to improve diagnostic and treatment options. Proteomics can be used to explore the underlying biosignature during the development and progression of heart failure (HF). This includes characterization of signaling and post-translational modification (PTM) regulation pathways, which may lead to new drug targets for therapeutic intervention. In addition, proteomic approaches can identify and validate new circulating biomarkers to improve disease diagnosis and risk stratification, allowing for earlier and more efficacious patient treatment.
Cardiovascular proteomics began with the seminal work by Dunn's group using 2DE to separate myocardial proteins 3. At that time, the major challenge was identifying the proteins within each gel spot 3, which was revolutionized with the application of mass spectrometry (MS). Since then, the field of proteomics has matured, allowing for rapid and precise determination of absolute quantity of individual proteins as well as individually modified amino acid residues. This review will concentrate on (i) the application of proteomics to HF, (ii) the use of animal model systems (pig, dog, rat, mouse, zebrafish, and fruit fly) to study this complex condition, (iii) the determination of proteins with the highest degree of amino acid sequence homology between these different species, and (iv) the similarity of phospho-proteome and kinase signaling cascades between human and the various model organisms.
2 Human heart failure
HF is a debilitating disorder defined as a deficiency in the capability of the heart to adequately pump blood in response to systemic demands 2. The initiating and underlying causes of HF are often medical conditions such as hypertension, myocardial infarction, coronary artery disease, ischemia, myocarditis, valvular heart disease, congenital malformations, and commonly, cardiomyopathies 4,5. Hypertrophic cardiomyopathy, characterized by a thickened but nondilated left ventricle, has a prevalence of 1:500 6. Dilated cardiomyopathy, in which one or both of the ventricles are enlarged with accompanying systolic dysfunction, is less prevalent (1:2500), although it is the most frequent cause of heart transplantation 6. The least common form is restrictive cardiomyopathy, which results in impaired diastolic filling of the ventricles 6,7.
The progression toward end-stage HF involves broad cellular changes including reactivation of the fetal gene program (e.g. upregulation of atrial and brain natriuretic peptide, skeletal α-actin, β-myosin heavy chain, and fetal type cardiac ion channels), cardiomyocyte hypertrophy, mitochondrial dysfunction, and alterations in calcium handling and sarcomeric proteins that result in part from the dysregulation of intracellular kinase signaling cascades and other protein PTM pathways 4,8. The heterogeneous responses differ from patient to patient and depend on environmental and lifestyle factors, which hamper the development of new interventions. Further complications arise from the plethora of genetic mutations that can lead to inherited diseases that frequently promote HF 9–12.
The availability of both healthy and diseased human cardiac tissue samples presents a significant challenge to the study of HF. To study the progression of cardiac diseases, tissue biopsies or samples obtained during surgery are needed, but availability is limited. Furthermore, complex patient phenotypes due to overlapping pathology (e.g. diabetes or hypertension) frequently reduce experimental reproducibility and restrict tissue utility. End-stage HF samples are most commonly obtained either at the time of device implantation (such as ventricular assist devices, VADs) or during transplantation or other invasive procedures. Control tissue samples often comes from donor hearts that are not suitable for transplantation or that have other underlying issues, making these samples ill representative of a “healthy” individual. Therefore, the use of experimentally malleable animal models and their corresponding primary or derived cell lines is necessary. Lifestyle and environmental factors are controlled in most laboratory animals, and inbreeding restricts genetic variability. The underlying assumption is that the various model systems, e.g. pig/swine (Sus scrofa), dog/canine (Canis lupus familiaris), rat (Rattus norvegicus), mouse (Mus musculus), zebrafish (Danio rerio), and fruit fly (Drosophila melanogaster), recapitulate aspects (physiological, cellular, and/or molecular) of human (Homo sapiens) CVD.
This assumption is predicated on a relatively high degree of protein conservation across the animal model systems and the human cardiac proteome. Figure1A shows the number of myocardial proteins with ≥90% amino acid sequence homology between human and large (pig and dog), medium (rat and mouse), and small (zebrafish and fruit fly) models (based on proteins specifically annotated to be present in human cardiac muscle within the Uniprot database, www.uniprot.org↰). The vast majority (>98%) of protein sequences within this database are derived from the translation of coding sequences present in publicly accessible nucleic acid databases. Not all proteins are represented in all species, however, and not all entries have been observed at the transcriptional or protein level. Interestingly, the percent overlap between the human proteome and that of large or medium animal models is considerable, but this conservation drops dramatically between the human and small animal model systems in which there are few proteins with a high degree of amino acid homology. Not surprisingly, as the stringency for conservation at the amino acid level is reduced the percent overlap between human and all model systems increases dramatically.
Figure 1.
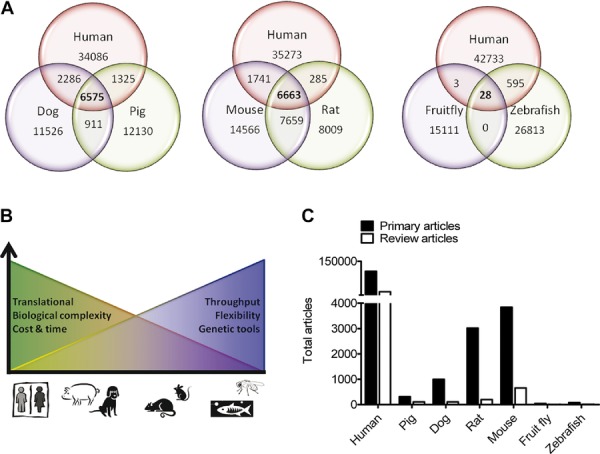
(A) Venn diagrams showing the number of proteins with ≥90% amino acid sequence homology between human, dog and pig (left); human, mouse and rat (middle); and human, zebrafish, and fruit fly (right) proteomes. CD-HIT generated protein clusters at ≥90% homology were used to identify representative proteins common across species and unique to each species, and used to create the Venn diagrams. (B) Summary of the advantages and disadvantages of the various animal models for HF research. (C) Number of peer-reviewed articles about HF on the publically available database Pubmed (http://www.ncbi.nlm.nih.gov/pubmed/), including reviews and primary articles using human, pig, dog, rat, mouse, zebrafish, or fruit fly models. The bar graph for human includes basic and clinical HF research. Pubmed was queried for all scientific papers published with key word “heart failure” from 1916 to August 2013. A total of 131 643 articles were returned.
Despite disparate predicted numbers of highly homologous myocardial proteins, each model organism has advantages and disadvantages with respect to their biological complexity, translational effectiveness, throughput, and genetic flexibility. This correlates with differences in the ease of use, cost and resources required to carry out experimental studies, and not surprisingly with the numbers of papers published with a particular model system (Fig.1B). In Fig.1C, Pubmed (http://www.ncbi.nlm.nih.gov/pubmed) queries were performed to obtain papers published involving the various species surveyed in this paper. The limitations on our numbers may be due to the shortcoming of the database used. However, the search features enabled by Pubmed provides a tool for advanced queries, which allows sorting papers as reviews, clinical, or topic-specific papers. The greatest number of published papers pertaining to myocardial and HF proteomics (clinical and basic research combined) is based on human cardiac tissue (Fig.1C).
2.1 Swine and canine models of HF
Pigs and dogs possess a high degree of overall conservation of the human cardiac proteome (Fig.1A), which is highlighted by their physiological and anatomical similarities. While the challenges of working with large animals are great, and genetic manipulation is extremely challenging, they represent the most translational models where therapeutic interventions can be readily carried out. In 1962, one of the first animal models of HF was generated by applying chronic tachycardia (rapid pacing) in a dog using an implantable pacemaker 13. Due to its size, the dog was well suited for these early studies since technology used in humans could be directly utilized. The tachypacing-induced HF model recapitulates much of the dysfunction seen in humans and is still used in research 14. This technique has since been employed to generate HF in pigs 15 and sheep 16, as well as to study various pacing-induced conditions, such as cardiac dyssynchrony 17,18. Another common approach to generate HF in large animals is via volume overload in which the chordae tendinae is severed to induce mitral regurgitation 19,20. These HF models have also proven to be very useful as preclinical support for large clinical trials. For example, validation for angiotensin II receptor blockade in HF was obtained from large animal models 21,22. State-of-the-art HF therapeutics, including gene 23,24 and stem cell-based therapies 25, use large animal models as a translational platform. Because of their size, they can be used to test implantable technologies ultimately intended for humans, such as VADs 26,27. However, these surgeries are complex, requiring sophisticated surgical suites and highly trained personnel, which leads to one of the most obvious challenges when exploiting large animals: cost. The procedures, equipment, surgical areas, personnel, housing, feeding, and other animal costs are all significantly greater than those of rodents. However, the dog heart is ∼1000 times larger than a mouse heart (∼150 mg 28 vs. ∼ 150 g 29), and many researchers harvest multiple usable samples from a single animal, which helps offset the cost burden.
Another significant drawback when using large animals for basic science research is the lack of genetic tools. Unlike mice and smaller models, there is little ability to control the genome of dogs and pigs, and there are relatively few inbred strains that can be used to reduce variability. In fact, the only genetic tools available are those available to humans, such as gene therapy.
The use of large animal models of HF predates much of the proteomics field. Compared to the smaller animal models, few cardiac proteomic studies have been conducted in pigs and dogs (Fig.1C). Nevertheless, the use of 2DE 30,31, MS 31,32, and reverse-phase protein microarrays 33 have been used to identify protein changes in HF. Additionally, cutting-edge proteomics techniques such as multiple reaction monitoring (MRM) have been used to determine changes in the phosphorylation state of single troponin I residues in the tachypaced HF dog model 34. Proteomics in large animals has also been applied to investigate other cardiovascular disorders including ischemia/reperfusion injury 35,36, myocardial stunning 37, cardiac dyssynchrony 18, and hypoxia/reoxygenation injury 38.
Despite the high degree of amino acid sequence homology, difficulties with large animal myocardial proteomics still subsist. For example, it is our experience that many antibodies that work in humans or rodents are ineffective in dogs or pigs. This most likely results from small differences in the targeted amino acid sequence, which significantly reduce antibody-binding affinity. Most commercially available antibodies are neither designed nor tested with large animal studies in mind. Moreover, protein and peptide identification using MS-based approaches is challenging because of the relatively small-annotated databases (e.g. uniprot) available for large animals compared to human and rodent. The reduced proteome coverage of these species often results in a reduction in the number of proteins resolved as well as the sequence coverage of these proteins, which can have an even larger effect on the number of PTM residues identified.
2.2 Rats and mice, the most commonly used animal models
Rodent (rat and mouse) models are extensively used in CVD research and recapitulate etiologies such as aortic stenosis and hypertrophic, restrictive, and dilated cardiomyopathies 39. There are more HF publications based on rat and mouse models than that of larger animal systems (Fig.1C). Of the various rodent models of cardiac dysfunction, it is particularly common to use surgical intervention to generate HF. In mice and rats, the aorta can be constricted using a suture or clip to elicit chronic hypertension-induced HF 40. In mouse models, the most commonly used technique is transverse aortic constriction (TAC), and in rat models of pressure overload the abdominal aorta is clipped above or below the renal arteries 41,42. Another common surgically induced model of HF is performed by either permanent occlusion of the left anterior descending coronary artery or occlusion for a specific time period followed by reperfusion to mimic myocardial infarction (MI) 43. Following surgery, the heart undergoes distinct anatomical and functional changes due to the hypertrophic growth of the ventricles and subsequent cardiac dilation. Another pathologic insult includes exposure to toxic compounds. Early studies observed that isoproterenol administered to animals produced cardiac hypertrophy, which can decompensate to HF 44. Doxorubicin is a cardiotoxin that has clinical application as a cancer therapeutic, and exposure in rat and mice results in dose-dependent dilated cardiomyopathy 45,46. An additional, distinct model of cardiomyopathy begins with inflammation of the heart (myocarditis) and is produced in rodents by exposure to pathogens including coxsackie-B3 virus, encephalomyocarditis virus, and cardiac myosin peptides 47–50. In an effort to more fully recapitulate the features of human cardiomyopathy, researchers have also developed models of HF exhibiting comorbidities such as diabetes, which may be induced by giving rodents streptozotocin and hypertension models with ischemia 51,52.
Genetic models of cardiomyopathy have been and continue to be created by deletion or overexpression of proteins implicated in disease pathways. For example, in mice the cardiac specific overexpression of Gαq, a membrane-associated protein that can trigger signaling cascades through activation of phospholipase C, leads to hypertrophic growth and subsequent cardiac dilation and dysfunction 53. Myocarditis that occurs as a result of a pathologic signaling cascade involving TNF-α may be induced by cardiac-specific overexpression of the protein 54. Moreover, transgenic mice have been established that model hypertrophic cardiomyopathy due to introduction of an arginine403 to glutamine mutation into the α cardiac myosin heavy chain gene 55, and restrictive cardiomyopathy by expressing mutant cardiac troponin I (arginine192 to histidine mutation) 56. Genetic rat models of HF that are frequently used include spontaneously hypertensive rats, spontaneously hypertensive HF prone rats and Dahl salt-sensitive rats, which exhibit hypertension and HF with age or introduction of a high-salt diet, respectively 57–59.
Rodent models of cardiomyopathy are advantageous due to a relatively well-annotated genome and proteome and a reasonable degree of amino acid sequence homology to the human cardiac-specific proteome (Fig.1A, Supporting Information Fig. 1). Additionally, the rodent heart shares many of the complexities of the human heart including a four-chambered structure, yet there are important physiological differences in the size, heart rate (600/min in mice versus 70/min in human), and in calcium handling. Furthermore, affected human and rodent subjects differ in regards to age (rodents are relatively young), sex (male rodents are commonly used), and comorbid conditions (not commonly modeled in rodents), which may contribute to inconsistencies between human disease and rodent models. Also, the complications of strain differences may confound the exploratory work of different labs. For example, in mouse models of pressure overload, strains respond differently to TAC or MI surgeries as C57BL6 mice exhibit systolic dysfunction and remodeling whereas sv129 mice exhibit preserved systolic function at the same time-point 60,61. Thus, interpretations of protective or pathologic effects in the rodent do not always translate to clinical applications. Nonetheless, Drozdov et al. combined different murine cardiac hypertrophy genomic and proteomic datasets to study molecular patterns to distinguish pathological and physiological left ventricle HF 62. Moreover, proteomic analysis of pressure overload, myocardial infarction, myocarditis, doxorubicin toxicity interventions in addition to the genetically engineered rodents 63–66 can provide interesting new hypotheses and tremendous insight into human HF.
Recently, more awareness has been raised about the extracellular matrix (ECM) proteome. Changes in the protein concentration and modification of a family of ECM proteolytic enzymes, the matrix metalloproteinases (MMPs), have been identified in left ventricle remodeling and can be a contributing factor in the progression to HF 67. Proteomic analysis of a number of different murine models has enhanced our understanding of ECM proteins and their proteolytic enzymes 68,69. Also, new fractionation and extraction methods to improve sample preparation to study MMPs 70 and ECM proteins 71 have emerged allowing broader coverage of this insoluble subproteome of the myocyte and the heart.
2.3 The zebrafish and fruit fly: The smallest animal models used in cardiovascular research
D. melanogaster (fruit fly) has existed as a model organism for genetics research and human biology for over 100 years, beginning with the groundbreaking work of Morgan 72. D. rerio (zebrafish) has been a model for human cardiac disease for a much shorter time period than most other animal models, which helps explain the lower number of total proteomic and cardiac related publications (Fig.1C). In regard to CVD, both animals have been used as models of dilated and restrictive cardiomyopathies and, to a lesser extent, hypertrophic cardiomyopathy 73–79. Even though there is a low number of cardiac-designated proteins with over 90% amino acid homology between flies and humans (Fig.1A and Supporting Information Fig. 1A), functional conservation is often retained. This conservation can result from individual domains and structural elements of a protein, which are required for a particular function, exhibiting a high degree of amino acid sequence similarity. Other amino acid stretches of the same protein, however, may lack high homology. Since the functionally and structurally important regions may comprise relatively few amino acid residues, much of the remaining protein sequence will be under less evolutionary pressure to retain similarity. Thus, a lack of high degree of homology over the complete sequence may be misleading.
Recently, the physiological assessment of heart function in the adult fly has been greatly advanced using optical coherence tomography and high-speed digital recordings of heart contractions 80,81, making high throughput measurement of cardiac disease phenotypes more feasible. Zebrafish have been used successfully in drug discovery and chemical screening processes 82,83. However, after initial screening using these small animal models, positive hits need to be validated in larger mammalian systems, such as mice or rats and potentially pigs or dogs.
There are many aspects of the fly and zebrafish that make them preferred models for CVD research. One major benefit of Drosophila is that their maintenance is more cost effective than that of larger mammalian systems. Moreover, data acquisition is relatively rapid compared to all other animal models. The fruit fly heart is a simple linear cardiac tube consisting of a single layer of contractile myocardial cells and nonmuscular pericardial cells that align along the length of the tube. The morphological differences that exist between the fly and the human cardiovascular system (e.g. single chamber, lack of coronary circulation, etc.) limit the utility of Drosophila as an animal model for certain types of human heart disease 84–86. However, the basic cellular and molecular machinery of the cardiac fibers are highly conserved between Drosophila and vertebrates, and, moreover, the embryonic origin and tubular structure are very similar 87–90. The genome of the fly is much smaller than the human genome, although many disease-related genes found in humans have orthologous genes in the fly. Furthermore, the fly reproduces quickly and has a very short lifespan, which makes genetic crosses and establishment of transgenic lines relatively rapid. The reproductive cycle of a fly is approximately 10–12 days, and adult flies can live up to 2 or 3 months, generating hundreds of genetically identical offspring.
Unlike in Drosophila, the zebrafish heart consists of two chambers, one atrium and one ventricle, consisting of both a myocardium and an endocardium 91. In addition, the zebrafish is a vertebrate and most of its genes have human homologs. The life cycle of the zebrafish, however, is long compared to the fly; it matures within 3 months and has a lifespan up to 5 years.
Both model systems benefit from a robust array of well-developed genetic tools and currently available resources. For example, the UAS-Gal4 system is an inducible transgene expression system in which specific binding of the yeast transcription activator protein (Gal4) to an upstream activation sequence (UAS) activates downstream gene expression 92. It is commonly used in the fly to study tissue-specific gene expression (e.g. cardiomyocyte-specific expression) 92 and is also a rapidly advancing tool for zebrafish. The Vienna Drosophila Research Center (http://stockcenter.vdrc.at/control/main) has produced and maintains many publically available UAS-RNAi responder strains that target ∼90% of the entire Drosophila genome 93. Flytrap (http://flytrap.med.yale.edu/index.html) is an additional, highly valuable resource that distributes publically available transgenic fly lines that harbor a wide range of green fluorescent protein tagged proteins 94,95.
Both fish and fly model systems possess numerous unique and powerful advantages that have helped advance cardiovascular research and will continue to do so in the future. Despite their small size, which makes tissue procurement challenging, these models are being increasingly utilized for proteomics studies (e.g. MS) 96–99, including the cardiac proteome 89,100.
2.4 Convergence and divergence across all seven species at different levels of homology
In Supporting Information Fig. 1A we examined the conservation between the proteomes (based on 100, 90, or 80% amino acid sequence homology) of seven species: human, pig, dog, rat, mouse, zebrafish, and fruit fly. Only those proteins observed in all species, and which are also annotated as expressed in the heart based on the human Uniprot database, were included in the comparison. Interestingly, just two proteins are completely homologous (100%) between all seven species: histone 3 and its variant histone 3.3. Histones are proteins closely associated with DNA in the nucleosome core units of chromatin that are essential for life. It is therefore not surprising to have the highest conservation between almost all known species 101. Histone H3 is one of the five (H1, H2A, H2B, H3, and H4) main histone proteins and has three known sequence variants in mammalian cells, histone H3.1, histone H3.2, and histone H3.3. The variants are highly conserved and differ only by a few amino acids 102.
There are 21 proteins with ≥90% and 82 proteins with ≥80% sequence homology (Table1, see online Supporting Information for details). To determine homology, we used CD-HIT Suite 103 to cluster highly similar protein sequences across the different species with varying sequence identity cutoffs. The input used was a combined UniprotKB 104 database (release 2013–08–20) of human, rat, mouse, dog, pig, zebrafish, and fruit fly, and the generated output was a list of clusters with a representative protein for each cluster. CD-HIT was used to identify homologous and unique protein groups between species (Supporting Information Fig. 1A). Table1 lists the 82 proteins reported to be expressed in the heart (based on Uniprot annotation) and that share a ≥80% amino acid homology between all seven species.
Table 1.
Proteins common in all 7 species (human, pig, dog, rat, mouse, zebrafish, and fruit fly) with at least ≥ 80% homology
| Protein | Protein name | Protein | Protein name |
|---|---|---|---|
| accession | accession | ||
| P62191 | 26S protease regulatory subunit 4* | P62805 | Histone H4* |
| P35998 | 26S protease regulatory subunit 7* | Q16576 | Histone-binding protein |
| P62195 | 26S protease regulatory subunit 8* | Q7L9L4 | MOB kinase activator |
| O00487 | 26S proteasome regulatory subunit 14 | Q15843 | NEDD8 |
| P62266 | 40S ribosomal protein S23 | P84074 | Calcium-binding protein BDR-2 |
| P46782 | 40S ribosomal protein S5 | P0CG48 | Polyubiquitin-C* |
| P62829 | 60S ribosomal protein L23 | Q6P2Q9 | Pre-mRNA-processing-splicing factor 8* |
| P11021 | 78 kDa glucose-regulated protein | P41223 | Protein BUD31 homolog |
| P68133 | Actin* | P61326 | Protein mago nashi homolog |
| P59998 | Actin-related protein 2/3 complex subunit 4 | P61619 | Protein transport protein Sec61 subunit alpha |
| P84077 | ADP-ribosylation factor 1* | P60059 | Protein transport protein Sec61 subunit γ |
| P18085 | ADP-ribosylation factor 4 | P61236 | Protein yippee-like 3 |
| P62330 | ADP-ribosylation factor 6* | P15153 | Ras-related C3 botulinum toxin substrate |
| Q9BXS5 | AP-1 complex subunit mu-1 | P62820 | Ras-related protein Rab-1 |
| Q96CW1 | AP-2 complex subunit mu | Q15907 | Ras-related protein Rab-11 |
| P53680 | AP-2 complex subunit sigma* | P61106 | Ras-related protein Rab-14 |
| P25705 | ATP synthase subunit alpha, mitochondrial | P61019 | Ras-related protein Rab-2 |
| P62158 | Calmodulin* | Q9NRW1 | Ras-related protein Rab-6 |
| P68400 | Casein kinase II subunit alpha | P62834 | Ras-related protein Rap-1 |
| P60953 | Cell division control protein 42 homolog* | P08134 | Rho-related GTP-binding protein |
| P61201 | COP9 signalosome complex subunit 2 | P11908 | Ribose-phosphate pyrophosphokinase |
| P24468 | COUP transcription factor | P62714 | PP2A-beta* |
| P61962 | DDB1- and CUL4-associated factor 7 | P62136 | Serine/threonine-protein phosphatase PP1 |
| O00148 | DEAD box protein 39 | P62314 | Small nuclear ribonucleoprotein Sm D1 |
| Q9Y295 | DRG-1 | P83876 | Thioredoxin-like protein 4A* |
| P30876 | RNA polymerase II subunit B2 | Q15369 | Elongin-C |
| P19388 | RNA polymerase I, II, and III subunit ABC1 | P55072 | Transitional endoplasmic reticulum ATPase |
| Q96FJ2 | Dynein light chain* | Q9Y3I0 | tRNA-splicing ligase RtcB homolog |
| Q05639 | Elongation factor 1-alpha | Q9BQE3 | Tubulin alpha chain* |
| Q9H9T3 | Elongator complex protein 3 | P68371 | Tubulin beta chain* |
| P38919 | Eukaryotic initiation factor 4A-III | P62312 | U6 snRNA-associated Sm-like protein LSm6 |
| P62495 | Eukaryotic release factor 1 | P62979 | Ubiquitin-40S ribosomal protein S27a* |
| O95166 | SABA(A) receptor associated protein | P62987 | Ubiquitin-60S ribosomal protein L40* |
| P62826 | GTP-binding nuclear protein Ran | P61077 | Ubiquitin-conjugating enzyme E2* |
| P62879 | G protein subunit beta-2 | P49459 | Ubiquitin-conjugating enzyme E2 A |
| P11142 | Heat shock cognate 71kDa protein | Q9BZL1 | Ubiquitin-like protein 5 |
| Q96KK5 | Histone H2A | Q9UBQ0 | Vesicle protein sorting 29 |
| P0C0S5 | Histone H2A.Z | P27449 | V-type proton ATPase proteolipid subunit |
| P62807 | Histone H2B | P38606 | V-type proton ATPase catalytic subunit A |
| P84243 | Histone H3* | P15313 | V-type proton ATPase subunit B |
Proteins with ≥ 90% homology have been marked (*).
There is similar distribution of proteins involved in heart function and disease among these conserved proteins (Supporting Information Fig. 1B shows the functional analysis, generated using IPA (Ingenuity® Systems, www.ingenuity.com) for the proteins listed in Table1). Perhaps not surprisingly, the top functional categories (assigned by IPA, associated with the 82 proteins) include RNA, PTMs and DNA replication (Supporting Information Fig. 2C). Of these 82 proteins, 11 have been shown to be involved in a specific cardiovascular function or CVD according to their annotations (Table1).
2.5 Protein modification: The phosphorylation status in heart failure
Phosphorylation (and other PTMs) modulates cardiac function and is significantly and quantitatively changed with HF 105–107. For example, disease-dependent phosphorylation of specific cardiac sarcomeric proteins was shown as early as the 1970s to alter heart performance 108–112. MS has been successfully used to identify the exact location of specific phosphorylation sites, and more recently, the MS-based MRM (or single reaction monitoring) has been employed to quantitate both unphosphorylated and phosphorylated tryptic peptides to obtain the exact stoichiometry (extent) of protein phosphorylation.
MRM utilizes N15 labeled standards consisting of the equivalent synthetic peptides being targeted (note, endogenous peptides are primarily N13), which are spiked into the sample, allowing absolute quantity to be determined. Recently, this tool was used to determine the phosphorylation levels of over 10 modifiable amino acid residues of cardiac troponin I in 30 human heart samples 34. Phosphorylation of novel C-terminal sites of cardiac troponin I were increased with HF while the classical N-terminal sites (such as those phosphorylated by protein kinase A, G, or C) were decreased. In addition to serine and threonine phosphorylation, a novel tyrosine phosphorylated residue was identified in this study 34, which suggest the existence of a new kinase pathway. Furthermore, using intact protein MS analysis, Ge and colleagues have shown that phosphorylation and other disease induced modifications can occur in vivo 113,114). An intriguing, unanswered question is whether the differences in levels of phosphorylation (or other disease-induced PTMs) might serve for HF risk stratification if detected in the blood 34,115,116.
In human heart failure, phosphorylation has been the dominant PTM studied, as shown by the high number of published articles (Fig.2A), compared to those focused on O-GlcNAc or acetylation (as indexed on Pubmed). Our literature search criteria consisted of one of the three PTMs, in combination with the terms “heart failure” and “human.” Ninety-six percent of the results involve phosphorylation, 3.7% acetylation and 0.3% O-GlcNAc. Despite a minimal number of published articles, O-GlcNAc is of interest, as this enzymatically induced PTM also targets S and T residues like phosphorylation, and we are beginning to appreciate its importance in myocardial preconditioning and diabetes. It is important to note that the difference in number of publications available for each PTM does not necessarily indicate its functional importance but merely the ease and available technology to study certain modifications. This is also reflected by the difference in the extent to which various signaling pathways (TGFβ, GSK-3β, AKT, PKG, PKC, or PKA) have been investigated (Fig.2B illustrates the number of articles that have been published on HF in human or using a mouse model).
Figure 2.
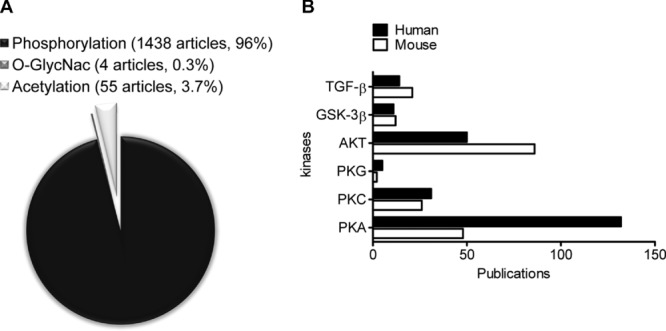
(A) Number of publications on Pubmed involving the PTMs phosphorylation, O-GlcNac, and acetylation associated with HF. In total, there were 1423, 55, and 4 articles published on phosphorylation, acetylation, and O-GlcNac modifications respectively. (B) Articles published involving phosphorylation and HF in humans (black bars) or mice (white bars) were sorted by kinase (PKA, PKC, PKG, AKT, GSK-3β, and TGF-β). Note that if there were articles containing more than one of the kinases searched for, the same article was counted more than once.
2.6 The GSK-3β/AKT and PKA pathways are highly conserved
The GSK-3β/AKT and PKA pathways have been extensively studied in HF (Fig.2B). AKT has primarily been examined in the mouse, whereas PKA has primarily been investigated in humans. We used STRING (version 9.05, 117) analysis to create a high confidence co-occurrence diagram to illustrate the presence or absence of 16 proteins within the GSK-3β/AKT pathway across species (Fig.3A). This addressed the question of how conserved key proteins are within this pathway across the seven model organisms. The intensity of the color of the red square illustrates the amount of conservation at the amino acid residue level of the homologous protein. Not surprising, GSK-3β and AKT and some of their targets (e.g. CTNNB1 (catenin β1), GYS1 (glycogen synthase 1), and SGK3 (serine/threonine protein kinase)) are highly conserved, indicating the functional importance of this pathway for cellular function throughout the animal kingdom. This implies that proteomic changes in GSK-3β/AKT signaling observed in one species will likely be relevant and translatable to humans. However, other downstream targets like SNAI1 (snail protein), APC (adenomatous polyposis coli), AXIN1 (axin protein 1), PSEN1 (presenilin 1), and FRAT1 (frat1 protein) are conserved between human, dog, rat, and mouse but not swine. This is surprising since it is believed that pig is the most translational animal model for human CVD research, but, as mentioned earlier, only functional and structural motifs need to be highly conserved within a protein in order for it to retain its biological role. We should note that our study is limited by the size and completeness of protein information for each species available in the current Uniprot database. Inadequate protein sequence coverage for any particular species will result in an underestimation of homology between all of them. The number of proteins listed in Table1 (proteins with ≥ 80% amino acid sequence homology) will most likely increase as protein annotation improves.
Figure 3.
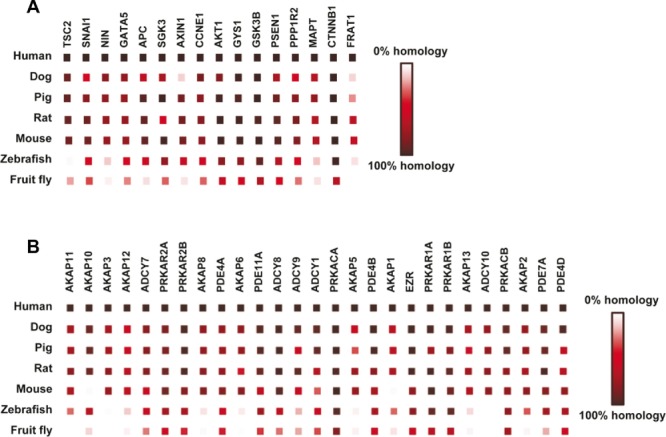
(A) High confidence score based network of the GSK-3β/AKT and PKA pathways created using STRING 117. Conservation (amino acid sequence homology) level of GSK-3β and AKT in the GSK-3β/AKT pathway across the seven species shows very high homology from human to fruit fly, suggesting their importance in cardiac function. (B) The conservation levels of the individual proteins of the PKA pathway show that all PKA subunits (PRKAR and PRACA) have a high degree of amino acid sequence homology from human to fruit fly.
The PKA pathway and 27 associated proteins were analyzed as described above. PKA is composed of two catalytic subunits (PRKACA) and two regulatory subunits (PRKAR) (Fig.3B). All PKA subunits show a high degree of amino acid sequence homology from human to fruit fly. The A-kinase anchor proteins (AKAP), which bind PKA and target it to different cellular locations, are the least conserved, while the protein phosphodiesterase (PDE) family is as conserved as the protein subunits of PKA. The PDE family controls the quantity of cAMP in a cell, which in turn regulates the activity of PKA. Perhaps these enzymes have a “more essential” role than AKAPs, or there is more redundancy within the AKAP family in which members are able to compensate for each other. Alternatively, the domains for which the individual AKAPs target may exhibit sequence variability. Practically, this information is useful when deciding which species to use when investigating a particular aspect of the PKA pathway. If the goal is to move between model systems or to associate with human cardiac disease, conservation of the pathway is necessary.
2.7 Future directions and conclusion
We believe that with improved technology the identification and characterization of proteins within the heart, as well as inclusion of the complete predicted proteomes of all species within the Uniprot database, will most likely expand the list of proteins with ≥80% amino acid sequence homology (Table1). This will certainly advance the utilization of animal models and translation to human cardiac disease. It is our hypothesis that these highly conserved proteins are critical and essential for cardiac physiology and life. Deletion of any one of these proteins should be lethal to any cell type and genetic mutations or single-nucleotide polymorphisms are likely to be disease initiating. Future investigations into the regulation of these proteins, e.g. cellular concentration, isoform expression, and PTMs, could provide insight into how cells respond to (disease) stimuli.
Each animal model has advantages and disadvantages, and the best choice for HF research will be dictated by the functional parameters of interest, the need for genetic manipulation, and the overall goal of the study. It is important to remember that the degree of protein conservation within specific pathways or disease processes can influence the extent to which the results are transferable to human pathophysiology. Regardless, quantification of proteins and their modified forms is essential to understand the underlying biology of CVD disease and HF.
Acknowledgments
This work was supported by the NIH grant P01-HL077180 (to J. E. Van Eyk); Proteomic Initiative contracts NHLBI-HV-10-05(2) (to J. E. Van Eyk) and HHSN268201000032C (to J. E. Van Eyk); and a NIH fellowship F31-HL116167 (to J. Rowell). American Federation for Aging Research Grant (to A. Cammarato), and American Heart Association grants 10SDG4180089 (to A. Cammarato) and 12POST11520006 (to V. Kooij).
The authors have declared no conflict of interest.
Glossary
- AKAP
A-kinase anchor proteins
- CVD
cardiovascular disease
- ECM
extracellular matrix
- EU
European Union
- HF
heart failure
- MI
myocardial infarction
- MMP
matrix metalloproteinase
- PDE
protein phosphodiesterase
- TAC
transverse aortic constriction
- UAS
upstream activation sequence
- VAD
ventricular assist devices
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Figure S1. Divergence and convergence across the seven species (human, pig, dog rat, mouse, zebrafish and fruit fly) at varying levels of amino acid sequence homology. A. The number of proteins with a 100%, ≥90% or ≥80% amino acid sequence homology are depicted in the bar graph. The proteins unique to a species at 100%, 90% or 80% are represented as line graphs. B. The functional analyses that were generated through the use of IPA (Ingenuity® Systems, www.ingenuity.com) for those with 80% homology showed a distribution of proteins involved in cardiovascular disease, cardiovascular development and function, or other. C. Top 5 functional categories, assigned by IPA, associated with the 82 proteins (Table 1). The number proteins assigned to the 5 categories is presented in the bar graph.
3 References
- Nichols M, Townsend N, Luengo-Fernandez R, Leal J. European cardiovascular disease statistics 2012. European Heart Network, Brussels, European Society of Cardiology, Sophia Antipolis. 2012 [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CS, Corbett JM, May AJ, Yacoub MH, Dunn MJ. A human myocardial two-dimensional electrophoresis database: protein characterisation by microsequencing and immunoblotting. Electrophoresis. 1992;13:723–726. doi: 10.1002/elps.11501301154. [DOI] [PubMed] [Google Scholar]
- Lips DJ, deWindt LJ, van Kraaij DJ, Doevendans PA. Molecular determinants of myocardial hypertrophy and failure: alternative pathways for beneficial and maladaptive hypertrophy. Eur. Heart J. 2003;24:883–896. doi: 10.1016/s0195-668x(02)00829-1. [DOI] [PubMed] [Google Scholar]
- Yancy CW, Jessup M, Bozkurt B, Masoudi FA. 2013 ACCFAHA Guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013;62:e147–e239. doi: 10.1016/j.jacc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol. Rev. 2002;82:945–980. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- Kuwahara K, Nishikimi T, Nakao K. Transcriptional regulation of the fetal cardiac gene program. J. Pharmacol. Sci. 2012;119:198–203. doi: 10.1254/jphs.12r04cp. [DOI] [PubMed] [Google Scholar]
- Herman DS, Lam L, Taylor MR, Wang L. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MR, Slavov D, Ku L, Di Lenarda A. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation. 2007;115:1244–1251. doi: 10.1161/CIRCULATIONAHA.106.646778. [DOI] [PubMed] [Google Scholar]
- Thierfelder L, Watkins H, MacRae C, Lamas R. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- Watkins H, Conner D, Thierfelder L, Jarcho JA. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat. Genet. 1995;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- Whipple GH, Sheffield LT, Woodman EG, Theophilas C, Friedman S. Reversible congestive heart failure due to chronic rapid stimulation of the normal heart. Proc. N. Engl. Cardiovasc. Soc. 1962;20:39–40. [Google Scholar]
- Chakir K, Depry C, Dimaano VL, Zhu WZ. Galphas-biased beta2-adrenergic receptor signaling from restoring synchronous contraction in the failing heart. Sci. Trans. Med. 2011;3:100ra188. doi: 10.1126/scitranslmed.3001909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paslawska U, Gajek J, Kiczak L, Noszczyk-Nowak A. Development of porcine model of chronic tachycardia-induced cardiomyopathy. Int. J. Cardiol. 2011;153:36–41. doi: 10.1016/j.ijcard.2010.08.033. [DOI] [PubMed] [Google Scholar]
- Byrne MJ, Kaye DM, Mathis M, Reuter DG. Percutaneous mitral annular reduction provides continued benefit in an ovine model of dilated cardiomyopathy. Circulation. 2004;110:3088–3092. doi: 10.1161/01.CIR.0000146904.13677.E4. [DOI] [PubMed] [Google Scholar]
- Kirk JA, Holewinski RJ, Kooij V, Agnetti G. Cardiac resynchronization sensitizes the sarcomere to calcium by reactivating GSK-3beta. J. Clin. Invest. 2014;124:129–138. doi: 10.1172/JCI69253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk JA, Kass DA. Electromechanical dyssynchrony and resynchronization of the failing heart. Circ. Res. 2013;113:765–776. doi: 10.1161/CIRCRESAHA.113.300270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H, Spinale FG, Nagatsu M, Schmid PG. Effects of chronic beta-adrenergic blockade on the left ventricular and cardiocyte abnormalities of chronic canine mitral regurgitation. J. Clin. Invest. 1994;93:2639–2648. doi: 10.1172/JCI117277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallaj J, Wei CC, Hankes GH, Holland M. Beta1-adrenergic receptor blockade attenuates angiotensin II-mediated catecholamine release into the cardiac interstitium in mitral regurgitation. Circulation. 2003;108:225–230. doi: 10.1161/01.CIR.0000079226.48637.5A. [DOI] [PubMed] [Google Scholar]
- Spinale FG, Holzgrefe HH, Mukherjee R, Hird RB. Angiotensin-converting enzyme inhibition and the progression of congestive cardiomyopathy. Effects on left ventricular and myocyte structure and function. Circulation. 1995;92:562–578. doi: 10.1161/01.cir.92.3.562. [DOI] [PubMed] [Google Scholar]
- Cohn JN, Tognoni G. Valsartan Heart Failure Trial, I., A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N. Engl. J. Med. 2001;345:1667–1675. doi: 10.1056/NEJMoa010713. [DOI] [PubMed] [Google Scholar]
- Kawase Y, Ly HQ, Prunier F, Lebeche D. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 2008;51:1112–1119. doi: 10.1016/j.jacc.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Pepe M, Mamdani M, Zentilin L, Csiszar A. Intramyocardial VEGFB167 gene delivery delays the progression towards congestive failure in dogs with pacing-induced dilated cardiomyopathy. Circ. Res. 2010;106:1893–1903. doi: 10.1161/CIRCRESAHA.110.220855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ST, White AJ, Matsushita S, Malliaras K. Intramyocardial injection of autologous cardiospheres or cardiosphere-derived cells preserves function and minimizes adverse ventricular remodeling in pigs with heart failure post-myocardial infarction. J. Am. Coll. Cardiol. 2011;57:455–465. doi: 10.1016/j.jacc.2010.07.049. [DOI] [PubMed] [Google Scholar]
- Wei X, Li T, Li S, Son HS. Pre-clinical evaluation of the infant Jarvik 2000 heart in a neonate piglet model. J. Heart Lung. Transplant. 2013;32:112–119. doi: 10.1016/j.healun.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eya K, Tuzun E, Conger J, Chee HK. Effect of pump flow mode of novel left ventricular assist device upon end organ perfusion in dogs with doxorubicin induced heart failure. ASAIO J. 2005;51:41–49. doi: 10.1097/01.mat.0000150510.03339.ad. [DOI] [PubMed] [Google Scholar]
- Doevendans PA, Daemen MJ, de Muinck ED, Smits JF. Cardiovascular phenotyping in mice. Cardiovasc Res. 1998;39:34–49. doi: 10.1016/s0008-6363(98)00073-x. [DOI] [PubMed] [Google Scholar]
- Bienvenu JG, Drolet R. A quantitative study of cardiac ventricular mass in dogs. Can. J. Vet. Res. 1991;55:305–309. [PMC free article] [PubMed] [Google Scholar]
- Dohke T, Wada A, Isono T, Fujii M. Proteomic analysis reveals significant alternations of cardiac small heat shock protein expression in congestive heart failure. J. Card Fail. 2006;12:77–84. doi: 10.1016/j.cardfail.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Page B, Young R, Iyer V, Suzuki G. Persistent regional downregulation in mitochondrial enzymes and upregulation of stress proteins in swine with chronic hibernating myocardium. Circ. Res. 2008;102:103–112. doi: 10.1161/CIRCRESAHA.107.155895. [DOI] [PubMed] [Google Scholar]
- De Souza AI, Cardin S, Wait R, Chung YL. Proteomic and metabolomic analysis of atrial profibrillatory remodelling in congestive heart failure. J. Mol. Cell Cardiol. 2010;49:851–863. doi: 10.1016/j.yjmcc.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Anderson T, Wulfkuhle J, Petricoin E, 3rd, Winslow RL. High resolution mapping of the cardiac transmural proteome using reverse phase protein microarrays. Mol. Cell Proteomics. 2011;10:M111 008037. doi: 10.1074/mcp.M111.008037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Kirk JA, Ji W, Dos Remedios CG. Multiple reaction monitoring to identify site-specific troponin i phosphorylated residues in the failing human heart. Circulation. 2012;126:1828–1837. doi: 10.1161/CIRCULATIONAHA.112.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barallobre-Barreiro J, Didangelos A, Schoendube FA, Drozdov I. Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation. 2012;125:789–802. doi: 10.1161/CIRCULATIONAHA.111.056952. [DOI] [PubMed] [Google Scholar]
- Sawicki G, Jugdutt BI. Detection of regional changes in protein levels in the in vivo canine model of acute heart failure following ischemia-reperfusion injury: functional proteomics studies. Proteomics. 2004;4:2195–2202. doi: 10.1002/pmic.200300746. [DOI] [PubMed] [Google Scholar]
- Yuan C, Guo Y, Ravi R, Przyklenk K. Myosin binding protein C is differentially phosphorylated upon myocardial stunning in canine and rat hearts—evidence for novel phosphorylation sites. Proteomics. 2006;6:4176–4186. doi: 10.1002/pmic.200500894. [DOI] [PubMed] [Google Scholar]
- Fert-Bober J, Sawicki G, Lopaschuk GD, Cheung PY. Proteomic analysis of cardiac metabolic enzymes in asphyxiated newborn piglets. Mol. Cell Biochem. 2008;318:13–21. doi: 10.1007/s11010-008-9852-z. [DOI] [PubMed] [Google Scholar]
- Houser SR, Margulies KB, Murphy AM, Spinale FG. Animal models of heart failure: a scientific statement from the american heart association. Circulation Res. 2012;111:131–150. doi: 10.1161/RES.0b013e3182582523. [DOI] [PubMed] [Google Scholar]
- Litwin SE, Katz SE, Weinberg EO, Lorell BH. Serial echocardiographic-doppler assessment of left ventricular geometry and function in rats with pressure-overload hypertrophy: chronic angiotensin-converting enzyme inhibition attenuates the transition to heart failure. Circulation. 1995;91:2642–2654. doi: 10.1161/01.cir.91.10.2642. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Ross RS, Harris AN, Knowlton KU. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor EJ, Babick AP, Vasanji Z, Dhalla NS, Netticadan T. A comparative serial echocardiographic analysis of cardiac structure and function in rats subjected to pressure or volume overload. J. Mol. Cell. Cardiol. 2005;38:777–786. doi: 10.1016/j.yjmcc.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Michael LH, Entman ML, Hartley CJ, Youker KA. Myocardial ischemia and reperfusion: a murine model. Am. J. Physiol. 1995;269:H2147–H2154. doi: 10.1152/ajpheart.1995.269.6.H2147. [DOI] [PubMed] [Google Scholar]
- Balazs T, Herman EH. Toxic cardiomyopathies. Ann. Clin. Lab. Sci. 1976;6:467–476. [PubMed] [Google Scholar]
- Szenczi O, Kemecsei P, Holthuijsen MF, van Riel NA. Poly(ADP-ribose) polymerase regulates myocardial calcium handling in doxorubicin-induced heart failure. Biochem. Pharmacol. 2005;69:725–732. doi: 10.1016/j.bcp.2004.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado RM, 3rd, Nawar MA, Zewail AM, Kar B. Cyclooxygenase-2 inhibitor treatment improves left ventricular function and mortality in a murine model of doxorubicin-induced heart failure. Circulation. 2004;109:1428–1433. doi: 10.1161/01.CIR.0000121354.34067.48. [DOI] [PubMed] [Google Scholar]
- Heymans S, Pauschinger M, De Palma A, Kallwellis-Opara A. Inhibition of urokinase-type plasminogen activator or matrix metalloproteinases prevents cardiac injury and dysfunction during viral myocarditis. Circulation. 2006;114:565–573. doi: 10.1161/CIRCULATIONAHA.105.591032. [DOI] [PubMed] [Google Scholar]
- Fairweather D, Rose NR. Coxsackievirus-induced myocarditis in mice: a model of autoimmune disease for studying immunotoxicity. Methods. 2007;41:118–122. doi: 10.1016/j.ymeth.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakisaka Y, Niwano S, Niwano H, Saito J. Structural and electrical ventricular remodeling in rat acute myocarditis and subsequent heart failure. Cardiovasc. Res. 2004;63:689–699. doi: 10.1016/j.cardiores.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Nishio R, Sasayama S, Matsumori A. Left ventricular pressure-volume relationship in a murine model of congestive heart failure due to acute viral myocarditis. J. Am. Coll. Cardiol. 2002;40:1506–1514. doi: 10.1016/s0735-1097(02)02166-6. [DOI] [PubMed] [Google Scholar]
- Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- Jiqiu C, Elie RC, Li Fan L, Thomas JL. A new model of congestive heart failure in rats. Am. J. Physiol. 2011;301:H994–H1003. doi: 10.1152/ajpheart.00245.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP. Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc. Natl. Acad. Sci. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, McTiernan CF, Frye CS, Demetris AJ, Feldman AM. Cardiac-specific overexpression of tumor necrosis factor-alpha causes lethal myocarditis in transgenic mice. J. Card Fail. 1997;3:117–124. doi: 10.1016/s1071-9164(97)90045-2. [DOI] [PubMed] [Google Scholar]
- Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS. A mouse model of familial hypertrophic cardiomyopathy. Science. 1996;272:731–734. doi: 10.1126/science.272.5262.731. [DOI] [PubMed] [Google Scholar]
- Du J, Zhang C, Liu J, Sidky C, Huang XP. A point mutation (R192H) in the C-terminus of human cardiac troponin I causes diastolic dysfunction in transgenic mice. Arch. Biochem. Biophys. 2006;456:143–150. doi: 10.1016/j.abb.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Dahl LK, Heine M, Tassinari L. Role of genetic factors in susceptibility to experimental hypertension due to chronic excess salt ingestion. Nature. 1962;194:480–482. doi: 10.1038/194480b0. [DOI] [PubMed] [Google Scholar]
- Pfeffer JM, Pfeffer MA, Fishbein MC, Frohlich ED. Cardiac function and morphology with aging in the spontaneously hypertensive rat. Am. J. Physiol. 1979;237:H461–H468. doi: 10.1152/ajpheart.1979.237.4.H461. [DOI] [PubMed] [Google Scholar]
- Michael O'Donnell J, Narayan P, Bailey MQ, Abduljalil AM. 31P-NMR analysis of congestive heart failure in the SHHFMcc-facp rat heart. J. Mol. Cell. Cardiol. 1998;30:235–241. doi: 10.1006/jmcc.1997.0587. [DOI] [PubMed] [Google Scholar]
- Barrick CJ, Rojas M, Schoonhoven R, Smyth SS, Threadgill DW. Cardiac response to pressure overload in 129S1/SvImJ and C57BL/6J mice: temporal- and background-dependent development of concentric left ventricular hypertrophy. Am. J. Physiol. 2007;292:H2119–H2130. doi: 10.1152/ajpheart.00816.2006. [DOI] [PubMed] [Google Scholar]
- Gao X-M, Ming Z, Su Y, Fang L. Infarct size and post-infarct inflammation determine the risk of cardiac rupture in mice. Int. J. Cardiol. 2010;143:20–28. doi: 10.1016/j.ijcard.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Drozdov I, Didangelos A, Yin X, Zampetaki A. Gene network and proteomic analyses of cardiac responses to pathological and physiological stress. Circ. Cardiovasc. Genet. 2013;6:588–597. doi: 10.1161/CIRCGENETICS.113.000063. [DOI] [PubMed] [Google Scholar]
- Gratia S, Kay L, Michelland S, Sève M. Cardiac phosphoproteome reveals cell signaling events involved in doxorubicin cardiotoxicity. J. Proteomics. 2012;75:4705–4716. doi: 10.1016/j.jprot.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Hammer E, Phong TQ, Steil L, Klingel K. Viral myocarditis induced by Coxsackievirus B3 in A.BYSnJ mice: analysis of changes in the myocardial proteome. Proteomics. 2010;10:1802–1818. doi: 10.1002/pmic.200900734. [DOI] [PubMed] [Google Scholar]
- Cieniewski-Bernard C, Mulder P, Henry JP, Drobecq H. Proteomic analysis of left ventricular remodeling in an experimental model of heart failure. J. Proteome Res. 2008;7:5004–5016. doi: 10.1021/pr800409u. [DOI] [PubMed] [Google Scholar]
- Gramolini AO, Kislinger T, Alikhani-Koopaei R, Fong V. Comparative proteomics profiling of a phospholamban mutant mouse model of dilated cardiomyopathy reveals progressive intracellular stress responses. Mol. Cell Proteomics. 2008;7:519–533. doi: 10.1074/mcp.M700245-MCP200. [DOI] [PubMed] [Google Scholar]
- Spinale FG, Janicki JS, Zile MR. Membrane-associated matrix proteolysis and heart failure. Circ. Res. 2013;112:195–208. doi: 10.1161/CIRCRESAHA.112.266882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamilpa R, Lopez EF, Chiao YA, Dai Q. Proteomic analysis identifies in vivo candidate matrix metalloproteinase-9 substrates in the left ventricle post-myocardial infarction. Proteomics. 2010;10:2214–2223. doi: 10.1002/pmic.200900587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abonnenc M, Nabeebaccus AA, Mayr U, Barallobre-Barreiro J. Extracellular matrix secretion by cardiac fibroblasts: role of microRNA-29b and microRNA-30c. Circ. Res. 2013;113:1138–1147. doi: 10.1161/CIRCRESAHA.113.302400. [DOI] [PubMed] [Google Scholar]
- de Castro Bras LE, Deleon KY, Ma Y, Dai Q. Plasma fractionation enriches post-myocardial infarction samples prior to proteomics analysis. Int. J. Proteomics. 2012;2012:397103. doi: 10.1155/2012/397103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barallobre-Barreiro J, Didangelos A, Yin X, Domenech N, Mayr M. A sequential extraction methodology for cardiac extracellular matrix prior to proteomics analysis. Methods Mol. Biol. 2013;1005:215–223. doi: 10.1007/978-1-62703-386-2_17. [DOI] [PubMed] [Google Scholar]
- Morgan TH. Localization of the hereditary material in the germ cells. Proc. Natl. Acad. Sci. USA. 1915;1:420–429. doi: 10.1073/pnas.1.7.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annu. Rev. Genomics Hum. Genet. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- Cammarato A, Dambacher CM, Knowles AF, Kronert WA. Myosin transducer mutations differentially affect motor function, myofibril structure, and the performance of skeletal and cardiac muscles. Mol. Biol. Cell. 2008;19:553–562. doi: 10.1091/mbc.E07-09-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonberger J, Wang L, Shin JT, Kim SD. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat. Genet. 2005;37:418–422. doi: 10.1038/ng1527. [DOI] [PubMed] [Google Scholar]
- Taghli-Lamallem O, Akasaka T, Hogg G, Nudel U. Dystrophin deficiency in Drosophila reduces lifespan and causes a dilated cardiomyopathy phenotype. Aging Cell. 2008;7:237–249. doi: 10.1111/j.1474-9726.2008.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Meiler SE, Zhong TP, Mohideen M. Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nat. Genet. 2002;30:205–209. doi: 10.1038/ng816. [DOI] [PubMed] [Google Scholar]
- Yu L, Daniels J, Glaser AE, Wolf MJ. Raf-mediated cardiac hypertrophy in adult Drosophila. Dis. Model. Mech. 2013;6:964–976. doi: 10.1242/dmm.011361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozhimuttam Viswanathan M, Kaushik G, Engler AJ, Lehman WJ, Cammarato A. A Drosophila melanogaster model of diastolic dysfunction and cardiomyopathy based on impaired troponin-T function. Circ. Res. 2014;114:e6–e17. doi: 10.1161/CIRCRESAHA.114.302028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink M, Callol-Massot C, Chu A, Ruiz-Lozano P. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. Biotechniques. 2009;46:101–113. doi: 10.2144/000113078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MJ, Amrein H, Izatt JA, Choma MA. Drosophila as a model for the identification of genes causing adult human heart disease. Proc. Natl. Acad. Sci. USA. 2006;103:1394–1399. doi: 10.1073/pnas.0507359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CG, Milan DJ, Grande EJ, Rottbauer W. High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat. Chem. Biol. 2005;1:263–264. doi: 10.1038/nchembio732. [DOI] [PubMed] [Google Scholar]
- Tsang M. Zebrafish: a tool for chemical screens. Birth Defects Res. C. Embryo. Today. 2010;90:185–192. doi: 10.1002/bdrc.20183. [DOI] [PubMed] [Google Scholar]
- Dulcis D, Levine RB. Glutamatergic innervation of the heart initiates retrograde contractions in adult Drosophila melanogaster. J. Neurosci. 2005;25:271–280. doi: 10.1523/JNEUROSCI.2906-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin N, Badie N, Yu L, Abraham D. A method to measure myocardial calcium handling in adult Drosophila. Circ. Res. 2011;108:1306–1315. doi: 10.1161/CIRCRESAHA.110.238105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserthal LT. Drosophila flies combine periodic heartbeat reversal with a circulation in the anterior body mediated by a newly discovered anterior pair of ostial valves and ‘venous’ channels. J. Exp. Biol. 2007;210:3707–3719. doi: 10.1242/jeb.007864. [DOI] [PubMed] [Google Scholar]
- Bodmer R. Heart development in Drosophila and its relationship to vertebrates. Trends Cardiovasc. Med. 1995;5:21–28. doi: 10.1016/1050-1738(94)00032-Q. [DOI] [PubMed] [Google Scholar]
- Bodmer R, Venkatesh TV. Heart development in Drosophila and vertebrates: conservation of molecular mechanisms. Dev. Genet. 1998;22:181–186. doi: 10.1002/(SICI)1520-6408(1998)22:3<181::AID-DVG1>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Cammarato A, Ahrens CH, Alayari NN, Qeli E. A mighty small heart: the cardiac proteome of adult Drosophila melanogaster. PLoS One. 2011;6:e18497. doi: 10.1371/journal.pone.0018497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cripps RM, Olson EN. Control of cardiac development by an evolutionarily conserved transcriptional network. Dev. Biol. 2002;246:14–28. doi: 10.1006/dbio.2002.0666. [DOI] [PubMed] [Google Scholar]
- Schoenebeck JJ, Yelon D. Illuminating cardiac development: advances in imaging add new dimensions to the utility of zebrafish genetics. Semin. Cell Dev. Biol. 2007;18:27–35. doi: 10.1016/j.semcdb.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Buszczak M, Paterno S, Lighthouse D, Bachman J. The carnegie protein trap library: a versatile tool for Drosophila developmental studies. Genetics. 2007;175:1505–1531. doi: 10.1534/genetics.106.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin X, Daneman R, Zavortink M, Chia W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc. Natl. Acad. Sci. USA. 2001;98:15050–15055. doi: 10.1073/pnas.261408198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner E, Ahrens CH, Mohanty S, Baetschmann H. A high-quality catalog of the Drosophila melanogaster proteome. Nat. Biotechnol. 2007;25:576–583. doi: 10.1038/nbt1300. [DOI] [PubMed] [Google Scholar]
- Link V, Shevchenko A, Heisenberg CP. Proteomics of early zebrafish embryos. BMC. Dev. Biol. 2006;6:1. doi: 10.1186/1471-213X-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrimpf SP, Weiss M, Reiter L, Ahrens CH. Comparative functional analysis of the Caenorhabditis elegans and Drosophila melanogaster proteomes. PLoS Biol. 2009;7:e48. doi: 10.1371/journal.pbio.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay TL, Lin Q, Seow TK, Tan KH. Proteomic analysis of protein profiles during early development of the zebrafish, Danio rerio. Proteomics. 2006;6:3176–3188. doi: 10.1002/pmic.200600030. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lanham KA, Heideman W, Peterson RE, Li L. Characterization of zebrafish cardiac proteome using online pH gradient SCXRP HPLCMSMS platform. Methods Mol. Biol. 2013;1005:119–127. doi: 10.1007/978-1-62703-386-2_10. [DOI] [PubMed] [Google Scholar]
- Waterborg JH. Evolution of histone H3: emergence of variants and conservation of post-translational modification sites. Biochem. Cell Biol. 2012;90:79–95. doi: 10.1139/o11-036. [DOI] [PubMed] [Google Scholar]
- Hake SB, Garcia BA, Duncan EM, Kauer M. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J. Biol. Chem. 2006;281:559–568. doi: 10.1074/jbc.M509266200. [DOI] [PubMed] [Google Scholar]
- Huang Y, Niu B, Gao Y, Fu L, Li W. CDHIT suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic. Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling N, Walsh RA, Song G, Estridge T. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- Kooij V, Holewinski RJ, Murphy AM, Van Eyk JE. Characterization of the cardiac myosin binding protein-C phosphoproteome in healthy and failing human hearts. J. Mol. Cell Cardiol. 2013;60:116–120. doi: 10.1016/j.yjmcc.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J. Mol. Cell Cardiol. 2007;42:247–259. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Cole HA, Perry SV. The phosphorylation of troponin I from cardiac muscle. Biochem. J. 1975;149:525–533. doi: 10.1042/bj1490525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeacocke SA, England PJ. Phosphorylation of a myofibrillar protein of Mr 150 000 in perfused rat heart, and the tentative indentification of this as C-protein. FEBS Lett. 1980;122:129–132. doi: 10.1016/0014-5793(80)80418-2. [DOI] [PubMed] [Google Scholar]
- Jeacocke SA, England PJ. Phosphorylation of myosin light chains in perfused rat heart. Effect of adrenaline and increased cytoplasmic calcium ions. Biochem. J. 1980;188:763–768. doi: 10.1042/bj1880763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh N, Wise BC, Kuo JF. Phosphorylation of cardiac troponin inhibitory subunit (troponin I) and tropomyosin-binding subunit (troponin T) by cardiac phospholipid-sensitive Ca2+-dependent protein kinase. Biochem. J. 1983;209:189–195. doi: 10.1042/bj2090189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaro RJ, Moir AJ, Perry SV. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- Zabrouskov V, Ge Y, Schwartz J, Walker JW. Unraveling molecular complexity of phosphorylated human cardiac troponin I by top down electron capture dissociation/electron transfer dissociation mass spectrometry. Mol. Cell Proteomics. 2008;7:1838–1849. doi: 10.1074/mcp.M700524-MCP200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dong X, Hacker TA, Ge Y. Deciphering modifications in swine cardiac troponin I by top-down high-resolution tandem mass spectrometry. J. Am. Soc. Mass. Spectrom. 2010;21:940–948. doi: 10.1016/j.jasms.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk JA, Zhang P, Murphy AM, Van Eyk JE. Troponin I alterations detected by multiple-reaction monitoring: how might this impact the study of heart failure. Expert. Rev. Proteomics. 2013;10:5–8. doi: 10.1586/epr.12.77. [DOI] [PubMed] [Google Scholar]
- Zhang J, Guy MJ, Norman HS, Chen YC. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J. Proteome Res. 2011;10:4054–4065. doi: 10.1021/pr200258m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LJ, Kuhn M, Stark M, Chaffron S. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic. Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Divergence and convergence across the seven species (human, pig, dog rat, mouse, zebrafish and fruit fly) at varying levels of amino acid sequence homology. A. The number of proteins with a 100%, ≥90% or ≥80% amino acid sequence homology are depicted in the bar graph. The proteins unique to a species at 100%, 90% or 80% are represented as line graphs. B. The functional analyses that were generated through the use of IPA (Ingenuity® Systems, www.ingenuity.com) for those with 80% homology showed a distribution of proteins involved in cardiovascular disease, cardiovascular development and function, or other. C. Top 5 functional categories, assigned by IPA, associated with the 82 proteins (Table 1). The number proteins assigned to the 5 categories is presented in the bar graph.