

Abstract
Heteronucleus-detected dipolar based correlation spectroscopy is established for assignments of 1H, 13C, and 15N resonances and structural analysis in fully protonated proteins. We demonstrate that 13C detected 3D experiments are highly efficient and permit assignments of the majority of backbone resonances, as shown in an 89-residue dynein light chain 8, LC8 protein. With these experiments, we have resolved many ambiguities that were persistent in our previous studies using moderate MAS frequencies and lacking the 1H dimension. The availability of 1H isotropic chemical shifts measured with the heteronucleus-detected fast-MAS experiments presented here is essential for the accurate determination of the 1H CSA tensors, which provide very useful structural probe. Finally, our results indicate that 13C detection in fast-MAS HETCOR experiments may be advantageous compared with 1H detection as it yields datasets of significantly higher resolution in the 13C dimension than the 1H detected HETCOR versions.
Keywords: magic angle spinning, fast MAS, dynein light chain 8, resonance assignments, secondary structure, proton chemical shift, heteronuclear detection
INTRODUCTION
Magic angle spinning NMR spectroscopy (MAS NMR) is a powerful technique to study, at atomic resolution, three-dimensional structure and molecular motions of large biological molecules, including proteins, nucleic acids and their assemblies (McDermott 2009; Yan et al. 2013). Despite recent impressive advances in the field, sensitivity and resolution still remain a major concern for the MAS NMR studies of large biomolecules. To overcome these limitations, fast MAS conditions (rotation frequencies of 40 kHz and higher) are increasingly employed (Agarwal et al. 2013; Ernst et al. 2001; Webber et al. 2012; Wickramasinghe and Ishii 2006; Wickramasinghe et al. 2009; Yan et al. 2013; Zhou et al. 2007).
Resonance assignment is an essential step in the protocol for structural and dynamics analysis by MAS NMR. By correlating the resonance frequencies of backbone or side-chain atoms in a series of 2D and 3D spectra, site-specific chemical shifts are obtained for a subsequent structure determination using distance restraints and/or residue-specific dynamics characterization. Proton-detected sequences generally produce excellent sensitivity and are the preferred route for structural studies in solution NMR (Clore and Gronenborn 1994). Unlike in solution NMR, the network of 1H–1H dipolar coupling in the solid-state is so strong that the proton linewidths are poorly resolved in 1H-detected SSNMR experiments, at the MAS frequencies below 60 kHz that have been conventionally available to an experimentalist. To overcome this challenge, proton dilution by partial or full deuteration has been demonstrated to be one of the approaches to largely improve 1H resolution in MAS NMR under moderate MAS frequencies (10–20 kHz) (Akbey et al. 2010; Chevelkov et al. 2006; Linser et al. 2011; Reif et al. 2001). Different deuteration schemes are successfully applied to amyloid fibrils, membrane proteins and protein-RNA complex for precise assignments, structural restraints and interface identification (Asami et al. 2013; Asami and Reif 2013; Linser et al. 2011; Zhou et al. 2012). However, the dilution of proton bath compromises to a great extent the potentially high sensitivity associated with proton detection. Furthermore, the expression protocols required for production of deuterated biomolecules as well as the back-exchange steps needed for reprotonation make this technique not applicable to many proteins and biomolecular assemblies.
In this context, proton detection under fast magic angle spinning conditions is a potential approach that can be widely used to achieve narrow 1H lindewidths. As demonstrated by Rienstra and coworkers, fast MAS substantially improves the sensitivity and resolution if combined with full protonation (Zhou et al. 2007). In that study, high-resolution spectra have been acquired on fully protonated proteins with proton detection at MAS frequencies ~40 kHz and high magnetic field. Using fast MAS frequency of 60 kHz, Pintacuda and colleagues have demonstrated that the resolved 1H dimension can be incorporated to help removing assignment ambiguities so that the resonance assignments of backbone 1H, 15N, 13C nuclei of fully protonated protein can be made in an efficient and precise way (Marchetti et al. 2012). In addition to the 1H chemical shifts that provide rich information for predicting secondary structure, the 1H–1H distance restraints for structural determination can potentially be measured by 1H detection experiments in fully protonated proteins (Reif et al. 2001).
Even though 1H detection offers large sensitivity enhancements in various applications discussed above, heteronucleus (13C) detection is advantageous in the cases where the resolution is inadequate for 1H-detected experiments. For instance, in paramagnetic proteins the 1H linewidths are often paramagnetically broadened beyond the detectable limit (Bermel et al. 2003; Bermel et al. 2006a; Bertini et al. 2005). Another example is natively unfolded proteins, which have very small chemical shift dispersion in the proton dimension, making 1H detection impractical for resonance assignments of many unfolded proteins (Bermel et al. 2006b). Finally, the 13C chemical shifts in diamagnetic proteins span ca. 200 ppm, and covering the entire chemical shift range with adequate digital resolution is time consuming if 13C is sampled in an indirect dimension when full spectral widths need to be covered. The use of nonuniform sampling may alleviate this challenge to some extent; however, in the case of 13C experiments, the number of points that need to be sampled to retain the intensity information is still rather large (Suiter et al. 2014). Therefore, 13C detected experiments may be preferred for the acquisition of high-resolution spectra in the above situations. Indeed, 13C-detected scalar coupling based experiments have been reported to have high efficiency and resolution under fast MAS conditions and enable nearly complete backbone resonance assignments of fully protonated dimeric 153-residue protein in its diamagnetic and paramagnetic states (Barbet-Massin et al. 2013). However, heteronucleus detection in the context of dipolar based fast-MAS 3D experiments have not been yet demonstrated.
In this work, we present heteronucleus-detected dipolar-based 3D correlation spectroscopy for assignments of 1H, 13C, and 15N resonances and structural analysis in fully protonated proteins. We establish this approach on an 89-residue microtubule-associated protein, dynein light chain 8 (LC8), whose primary sequence and secondary structure are shown in Figure 1. LC8 is an integral subunit of cytoplasmic dynein and plays important roles in both dynein-dependent and dynein-independent cellular functions. It has been implicated in viral infections and cancer progression (Barbar 2008; Vadlamudi et al. 2004). Previously, we have employed paramagnetically doped LC8 for the development of fast MAS NMR methods (40 kHz) for sensitivity and resolution enhancement needed for structural analysis of large protein assemblies (Sun et al. 2012). Here, we demonstrate that high-resolution 13C-detected 3D HNCA, HNCO, and HNCOCX spectra can be acquired in fully protonated U-13C,15N-LC8, in a time-efficient way, at the magnetic field of 19.9 T and MAS frequency of 62 kHz. The resolution in the 1H dimension is excellent and allows for unambiguous assignments of most backbone 1H resonances of LC8. With this set of 3D experiments, we have accomplished backbone resonance assignments for the majority of the residues in LC8 and resolved many ambiguities that were persistent in our previous studies using moderate MAS frequencies and experiments lacking the 1H dimension (Sun et al. 2011). The secondary structure predicted based on the 1H, 13C, and 15N chemical shifts agrees very well with the X-ray crystal structure 3DVT (PDB ID) (Lightcap et al. 2008). Finally, we compare heteronucleus and 1H detection under fast MAS conditions using 2D HETCOR experiments and demonstrate that heteronucleus detection is advantageous from the resolution and time-saving standpoints.
Figure 1.
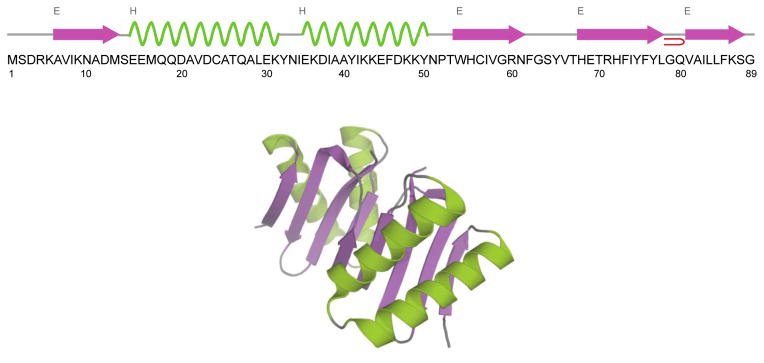
Amino acid sequence, secondary structure (top) and 3D X-ray structure (bottom) of Drosophila dynein light chain LC8. The structure is generated from PDB entry file 3DVT (Lightcap et al. 2008). LC8 is shown as homodimer. The α-helices are shown in green and β-sheets are in purple.
MATERIALS AND METHODS
Sample Preparation
The expression and purification of U–13C,15N-LC8 was reported by us previously (Lightcap et al. 2008). 15N–NH4Cl and U–13C6–glucose were used for isotopic enrichment. Purified U–13C,15N LC8 was dialyzed against 10 mM MES buffer (10 mM MgCl2, pH = 6.0) and then concentrated to 30 mg/ml. To make paramagnetically doped sample, Cu(II)-EDTA solution was added to both concentrated LC8 solution and PEG-3350 solution (32%, w/v) to the final concentration of 5 mM. The PEG-3350 solution was gradually added into the LC8 solution following the controlled precipitation protocol (Marulanda et al. 2004) to get protein precipitates. The detailed preparation procedures of Cu(II)-EDTA doped LC8 have been described previously (Sun et al. 2012). Finally 3.1 mg of U–13C,15N LC8 solid sample containing 5 mM Cu(II)-EDTA was centrifuged into 1.3 mm Bruker MAS rotor for subsequent solid-state NMR experiments.
MAS NMR Spectroscopy
The MAS NMR experiments were carried out on Bruker AVIII 850 MHz spectrometer at the magnetic field of 19.9 T using a 1.3 mm 1H/13C/15N triple-resonance probe. All 3D spectra were recorded at the MAS frequency of 62 kHz with apparent temperature controlled at −33±1 °C (sample temperature was 2 °C). The 1H, 13C and 15N chemical shifts were referenced with respect to DSS, admantane and ammonium chloride used as external referencing standards. The pulse sequences for three-dimensional HNCA, HNCO and HN(CO)CX experiments in U–13C,15N LC8 doped with Cu(II)-EDTA are shown in Figure 2 A–B. For HNCA and HNCO experiments, 1H–15N Hartmann–Hahn cross-polarization (CP) was set after 1H evolution and followed by 15N–13C specific cross-polarization. For HN(CO)CX experiment, to establish 13C–13C correlations, RFDR was used with a mixing time of 2.6 ms after the double cross-polarization (DCP) (Schaefer et al. 1979). Low-power TPPM decoupling was used during the acquisition and indirect-dimension evolution periods (Bennett et al. 1995). All the 3D experiments were acquired with 2064 complex points in direct 13C dimension (t3), 48 and 24 complex points in indirect 15N (t2) and 1H dimension (t1) respectively. The experimental time were 3.5 days for HNCA, 1.5 days for HNCO and 7 days for HNCOCX. It has been demonstrated that with the paramagnetic dopant the 1H longitudinal relaxation time T1 of LC8 is significantly shortened and the 15N transverse relaxation time T2 is slightly affected and close to that of neat samples. However, most chemical shifts of LC8 resonances are unperturbed and only a few resonances exhibiting small chemical shift perturbations in the 5mM Cu(II)-EDTA doped sample (Sun et al. 2012).
Figure 2.
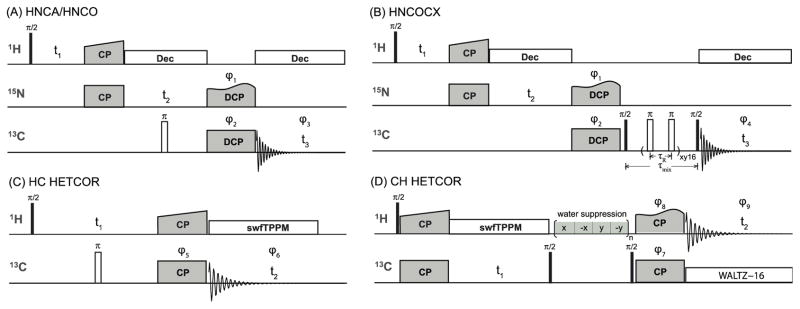
Schematic representations of pulse sequences for 3D 1H-based heteronucleus-detected experiments and 2D HETCOR conducted at fast MAS conditions. Solid and open bars represent π/2 and π pulse. A) The pulse sequence of HNCA and HNCO 3D experiments. The 13C carrier frequency was set to 55.0 ppm for HNCA and 170.0 ppm for HNCO. B) The pulse scheme of HN(CO)CX 3D experiment. A rotor synchronized RFDR mixing sequence was used to establish the 13C–13C correlations, with a mixing time of 2.6 ms. No proton decoupling was applied during the RFDR period at fast MAS frequency. C) HC HETCOR with carbon detection. D) CH HETCOR with proton detection. Phase cycling was applied to suppress water signal during the 13C–13C mixing period. The phases of the RF pulses are as follows: φ1 = x, x, –x, –x; φ2 = x, x, y, y, –x, –x, –y, –y; φ3 = x, –x, –y, y, –x, x, y, –y; φ4 = y, –y, –y, y, x, –x, –x, x, –y, y, y, –y, –x, x, x, –x; φ5 = x, x, –x, –x, y, y, –y, –y; φ6 = x, –x, –x, x, y, –y, –y, y; φ7 = x, x, –x,– x; φ8 = x, x, x, x, –x, –x, –x, –x, y, y, y, y, –y, –y, –y, –y; φ9 = x, –x, –x, x, –x, x, x, –x, y, –y, –y, y, –y, y, y, –y.
To examine the resolution benefits of carbon-detected spectra, we conducted 2D heteronuclear (HETCOR) experiments with both 13C detection and 1H detection at the MAS frequency of 60 kHz. The 2D HETCOR experiments exploited coherence transfers through dipolar couplings; the pulse schemes are displayed in Figure 2 C–D (Maudsley and Ernst 1977). For 1H–13C carbon-detected HETCOR, swept-frequency TPPM decoupling (swfTPPM) was employed during the 13C FID acquisition (Thakur et al. 2006) following the Hartmann–Hahn polarization transfer. In the 13C–1H proton-detected HETCOR, the WALTZ-16 broadband decoupling was executed during the FID acquisition, after the magnetization was transferred back to 1H. Phase cycling was applied to suppress the water signal during the 13C–13C spin diffusion. In both spectra, the total experiment time was 4.5 hours. The HC HETCOR was collected with 128 scans for each t1 point, while 32 scans were required for the CH HETCOR. The spectra were processed in NMRpipe (Delaglio et al. 1995) and analyzed with CCPNMR analysis (Stevens et al. 2011). Detailed information on the acquisition and processing parameters is given in Table S1.
RESULTS AND DISCUSSION
13C-Detected Fast MAS NMR Spectroscopy: Resonance Assignments of Dynein Light Chain 8
For resonance assignments of LC8, a set of 13C-detected dipolar-based 3D HNCA, HNCO and HN(CO)CX spectra was acquired. As expected, these spectra are of high quality, and the resolution is excellent with most of the cross peaks well separated. From these spectra, the resonance assignments could be readily derived as discussed below.
Several 2D H–C planes extracted from the 3D HNCA and HN(CO)CX spectra at different 15N chemical shifts are presented in Figure 3. The correlations between the amide proton and Cα of the same residue (i) or Cα of the previous residue (i-1) clearly reveal the connectivity of adjacent amino acids (shown as dashed line in Figure 3 A–C). From all slices it is easily seen that most cross peaks are resolved and distinguishable. Even though the resolution of the carbonyl region of HN(CO)CX spectrum is generally slightly worse than that of the aliphatic region of the same spectrum, most the peaks in the carbonyl region can be assigned unambiguously when the data set is combined with the HNCO spectrum. Notably, the experimental times for these high-quality 3D heteronuclear-detected data sets are relatively short: 3.5 days for HNCA, 1.5 days for HNCO and 7 days for HNCOCX. Therefore, 13C detection under fast MAS is a time efficient method yielding high sensitivity and resolution and enabling facile resonance assignments in fully protonated proteins.
Figure 3.

Representative 2D H–C planes of the 13C-detected 3D HNCOCX (purple) and HNCA (green) spectrum of U–13C,15N-LC8 at (A–C) δ15N = 128.3 ppm; (D–F) δ15N = 120.8 ppm; (G–I) δ15N = 118.1 ppm. The correlations between amide proton and Cα of the same residue (i) or Cα of the previous residue (i-1) clearly reveal the connectivity of adjacent amino acids (shown as dashed line in Figure 3. A–C). All the spectra were acquired at the magnetic field of 19.9 T (850 MHz 1H frequency) and MAS frequency of 62 kHz. The experimental times are 3.5 days for HNCA, 1.5 days for HNCO and 7 days for HNCOCX.
Using the above approach, we have successfully extracted assignments for 82 out of 89 residues of LC8. The representative sequential backbone walk for the A21–A28 segment is shown in Figure 4. Correlations in different spectra are found by matching the same amide 1H and amide 15N chemical shifts. The incorporation of 1H dimension helps remove many ambiguities that have previously impeded our resonance assignments of LC8 from experiments executed at the MAS frequency of 14 kHz (Sun et al. 2011). The residues that could not be assigned because the corresponding cross peaks were missing in the spectra include M1, S2, D3, R4, K5 (at the N-terminus), H68 (in a loop region) and G89 (at the C-terminus). As these residues are located in dynamic regions they may be either undergoing motions on intermediate timescales or are conformationally heterogeneous, either of which would lead to broader or undetectable peaks. For several other residues only backbone resonances are partially assigned. The amide 1H and 15N resonances are not obtained for P52 and E69. P52 does not have an amide 1H. E69 is in a loop region and may be undergoing conformational exchange on the experimental timescales. The backbone carbonyl 13C resonances are missing for A6, E16, L29, E30, K31, Y51, T67, T70, F86 and K87. Most of these residues are adjacent to mobile residues, and therefore we speculate that dynamics interferes with polarization transfer. The solid-state chemical shifts of LC8 obtained from 3D HNCA, HNCO, and HN(CO)CX spectra are listed in Table 1. Compared to our previous resonance assignments, under the fast MAS conditions we have detected and assigned 31 new resonances and also assigned the overwhelming majority of the backbone 1H chemical shifts. In addition to the sequential assignments, we have detected and assigned a number of additional side chain resonances in HN(CO)CX spectra, which were not present in the NCOCX and NCACX experiments performed by us previously at the MAS frequencies of 10–20 kHz. On the basis of the HN(CO)CX spectra alone, we have assigned Cβ resonances for 29 residues (Table 1) and Cγ resonances for 5 residues (see Table S2). Furthermore, we have detected the aromatic side chain resonances of F86 as well (Table S2).
Figure 4.

Sequential backbone walk for residues A21–A28 of LC8 using 3D fast MAS experiments: HNCA (green), HNCO (black) and HNCOCX (purple). The solid-state NMR experiments were carried out on Bruker Ascend 850 MHz spectrometer at the magnetic field of 19.9 T. All 3D spectra were recorded at the MAS frequency of 62 kHz with apparent temperature controlled at −33±1 °C (sample temperature was 2 °C).
Table 1.
Chemical shifts of U-13C,15N-LC8 assigned based on 3D MAS NMR HNCA, HNCO and HNCOCX spectra.
| Residue | H (ppm) | N (ppm) | C′ (ppm) | Cα (ppm) | Cβ (ppm) |
|---|---|---|---|---|---|
| K5 | 176.1 | ||||
| A6 | 9.3 | 134.9 | 51.6 | ||
| V7 | 9.1 | 123.4 | 175.4 | 61.7 | 34.2 |
| I8 | 9.1 | 129.0 | 176.0 | 61.4 | |
| K9 | 8.3 | 129.0 | 176.9 | 56.2 | |
| N10 | 8.2 | 114.4 | 172.5 | 53.6 | |
| A11 | 8.8 | 124.7 | 175.2 | 52.0 | 22.4 |
| D12 | 8.9 | 122.5 | 173.7 | 53.5 | |
| M13 | 8.1 | 120.7 | 174.9 | 55.9 | |
| S14 | 9.4 | 123.9 | 175.1 | 58.5 | 64.2 |
| E15 | 9.3 | 123.6 | 179.1 | 60.5 | |
| E16 | 9.1 | 118.7 | 60.0 | ||
| M17 | 7.5 | 120.8 | 177.5 | 59.6 | |
| Q18 | 8.5 | 119.0 | 178.1 | 59.4 | 28.9 |
| Q19 | 7.7 | 117.0 | 178.3 | 58.2 | |
| D20 | 7.8 | 119.4 | 178.7 | 57.4 | |
| A21 | 8.7 | 125.1 | 178.7 | 56.0 | 17.6 |
| V22 | 7.8 | 118.7 | 179.8 | 66.8 | 31.7 |
| D23 | 8.9 | 124.0 | 178.6 | 58.0 | |
| C24 | 8.9 | 120.7 | 177.1 | 63.3 | 27.7 |
| A25 | 8.6 | 120.1 | 178.3 | 55.0 | 19.8 |
| T26 | 8.6 | 115.5 | 176.6 | 68.0 | |
| Q27 | 7.7 | 120.1 | 178.7 | 58.7 | 28.4 |
| A28 | 8.3 | 122.1 | 175.9 | 55.1 | 20.9 |
| L29 | 8.2 | 116.6 | 56.9 | 42.2 | |
| E30 | 7.3 | 117.5 | 57.8 | ||
| K31 | 7.1 | 116.9 | 58.3 | 35.5 | |
| Y32 | 8.6 | 118.5 | 173.5 | 56.5 | 33.6 |
| N33 | 6.9 | 111.2 | 175.8 | 53.2 | 41.8 |
| I34 | 9.6 | 124.2 | 178.1 | 61.5 | |
| E35 | 10.2 | 130.1 | 178.6 | 62.6 | 28.4 |
| K36 | 8.5 | 116.7 | 177.1 | 59.9 | |
| D37 | 6.8 | 118.9 | 178.5 | 56.7 | |
| I38 | 7.7 | 122.2 | 177.0 | 65.4 | 38.4 |
| A39 | 8.4 | 119.1 | 178.6 | 54.8 | 17.9 |
| A40 | 7.9 | 119.3 | 178.3 | 55.0 | 18.6 |
| Y41 | 7.7 | 117.5 | 177.5 | 61.9 | 38.3 |
| I42 | 7.8 | 117.6 | 177.4 | 65.0 | 38.4 |
| K43 | 8.4 | 118.8 | 178.7 | 61.1 | 35.4 |
| K44 | 8.4 | 117.4 | 180.5 | 59.8 | |
| E45 | 8.0 | 119.4 | 178.9 | 59.0 | |
| F46 | 8.5 | 123.1 | 177.9 | 63.5 | |
| D47 | 8.6 | 119.9 | 178.7 | 57.3 | 40.2 |
| K48 | 7.3 | 117.2 | 177.7 | 59.0 | |
| K49 | 7.9 | 117.6 | 177.9 | 59.2 | |
| Y50 | 8.6 | 114.3 | 176.8 | 58.1 | |
| N51 | 7.0 | 113.1 | 55.2 | ||
| P52 | 178.3 | 60.4 | |||
| T53 | 7.9 | 121.6 | 172.0 | 64.8 | |
| W54 | 9.7 | 128.3 | 174.3 | 55.9 | |
| H55 | 8.5 | 118.5 | 174.8 | 55.1 | 34.5 |
| C56 | 8.3 | 120.9 | 171.4 | 56.7 | 30.9 |
| I57 | 9.4 | 132.4 | 173.6 | 59.8 | |
| V58 | 8.4 | 125.0 | 175.8 | 59.6 | 36.3 |
| G59 | 9.7 | 112.6 | 172.7 | 46.8 | |
| R60 | 8.6 | 118.5 | 178.2 | 56.7 | |
| N61 | 7.7 | 117.3 | 172.6 | 54.9 | |
| F62 | 8.0 | 119.4 | 173.4 | 57.5 | |
| G63 | 9.6 | 107.8 | 172.0 | 43.8 | |
| S64 | 8.2 | 111.8 | 172.4 | 57.2 | |
| Y65 | 9.0 | 122.6 | 173.2 | 61.5 | |
| V66 | 8.0 | 119.9 | 177.3 | 57.2 | |
| T67 | 9.0 | 122.6 | 61.6 | ||
| H68 | |||||
| E69 | 179.5 | 60.7 | |||
| T70 | 8.1 | 123.4 | 64.9 | ||
| R71 | 9.0 | 123.3 | 176.6 | 58.7 | |
| H72 | 8.6 | 116.2 | 173.3 | 56.7 | |
| F73 | 8.1 | 120.8 | 172.6 | 58.3 | |
| I74 | 8.6 | 129.0 | 169.1 | 61.8 | |
| Y75 | 8.5 | 127.3 | 174.8 | 54.8 | 41.1 |
| F76 | 9.4 | 125.3 | 170.1 | 54.8 | |
| Y77 | 9.3 | 118.2 | 176.5 | 55.6 | |
| L78 | 8.9 | 123.1 | 176.6 | 54.1 | |
| G79 | 8.9 | 114.3 | 175.1 | 47.0 | |
| Q80 | 8.9 | 124.4 | 174.5 | 56.3 | |
| V81 | 8.8 | 121.2 | 172.5 | 62.7 | |
| A82 | 8.5 | 128.8 | 176.2 | 50.3 | 20.7 |
| I83 | 8.8 | 120.2 | 173.3 | 60.5 | 39.8 |
| L84 | 9.3 | 128.0 | 173.9 | 53.1 | 46.2 |
| L85 | 8.9 | 130.2 | 174.1 | 53.9 | |
| F86 | 9.0 | 121.2 | 56.1 | ||
| K87 | 9.1 | 116.3 | 54.6 | ||
| S88 | 7.4 | 117.1 | |||
| G89 |
We also note that the availability of 1H isotropic chemical shifts measured directly under MAS conditions is essential for the determination of the 1H CSA tensors. We have recently developed an experimental approach for deriving 1H CSA tensors in proteins and other biological molecules, in a residue-specific way (Hou et al. 2014; Hou et al. 2013), and corroborated that the principal components are strongly correlated with hydrogen bonding environments, providing very useful structural probe. At the same time, we could not record 1H isotropic chemical shifts under moderate MAS conditions and had to rely on solution values, an approach, which is likely to be error-prone due to the different environments of the solid-like and solution states. The heteronucleus-detected fast-MAS experiments presented here overcome this limitation.
Secondary Structure Prediction in LC8
To predict the secondary structure from the resonance assignments of LC8, we used TALOS+ (Shen et al. 2009) to derive the backbone torsion angles ϕ and ψ of each residue. Overall, the secondary structure predicted from the fast-MAS NMR chemical shifts is generally consistent with the X-ray structure of LC8 (PDB file 3DVT) and with our previous results (Sun et al. 2011).
The plots of derived torsion angles and cartoon of predicted secondary structure are shown in Figure 5. The α-helices are shown in green and β-sheets are in purple. Since the chemical shifts of residues M1–R4 are unavailable, the predicted results are presented from K5. Our prediction is identical to the secondary structure derived from the X-ray data in the two α-helices segments and the β-sheet structure at the end (V81–S88). In other segments slight discrepancies exist. For instance, G59–N61 and T70–H72 are predicted to have no secondary structure and belong to loop regions, instead of being at the termini of two β-sheets in the X-ray structure 3DVT. In our case, this is not surprising since the resonances of these two segments are missing in most of our previous heteronuclear spectra (Sun et al. 2011; Sun et al. 2012). Even though we were able to detect the resonances corresponding to these residues in the 3D experiments performed with fast MAS at 850 MHz reported here, the peak intensities of these residues are extremely weak compared to those of other peaks. These results suggest that the G59–N61 and T70–H72 segments are mobile on the experimental timescales, and therefore whether these residues belong to the beginning and end of a loop connecting the two β sheets or to the corresponding β-sheet termini regions is likely dependent on the sample conditions. To this end, we note that the X-ray structure was determined under cryogenic conditions at temperature of 100 K and the termini of secondary structure elements appear rigid, unlike the MAS NMR experiments conducted under the temperatures close to physiological conditions, T = 2 °C.
Figure 5.

Backbone torsion angles Φ (top) and φ (bottom) derived by TALOS+ (Shen et al. 2009) based on the isotropic chemical shifts of backbone 1H, 13C and 15N atoms in LC8. The chemical shifts (see Table 1) were recorded by 3D fast MAS experiments. The amino acid sequence and predicted secondary structure of LC8 by TALOS+ are shown on top. The α-helices are shown in green and β-sheets are in purple.
Comparison of Heteronucleus Detection and 1H Detection
As discussed above, heteronucleus-detected 3D fast-MAS experiments are very efficient for resonance assignments. In certain instances, 13C-detection is anticipated to be advantageous vis-à-vis 1H detection at fast MAS in terms of spectral resolution. One such example is H-C HETCOR experiments.
The HC (carbon-detected) and CH (proton-detected) HETCOR spectra acquired with the same total experiment time are shown in Figure 6 A and D, and the magnified representations of 1HN-13Cα regions are given in B and C. The number of scans for CH HETCOR was set to be four times as small as the number for HC HETCOR, in order to keep the total time of the CH experiment reasonable. We note that this does not affect the resolution comparison between the two experiments. 1D slices of 1H and 13C dimensions extracted from 2D HETCOR are shown in Figure 6. The 1H and 13C linewidths of the cross peaks in the 1HN-13Cα region are listed in Table S3. For the majority of the cross peaks, the linewidths of the 13C dimensions in the HC HETCOR are smaller than those in the CH HETCOR, indicating distinct resolution improvement in the 13C dimension in the carbon detection experiment. The resolution in 1H dimension is very similar in both experiments: half of the cross peaks have somewhat smaller 1H linewidths in the 13C-detected experiment while for the other half of the peaks, the linewidths in 1H dimension are somewhat larger. One such example is shown in Figure 6 E, where the 1H linewidth of the cross peak at 5.9 ppm is 269 Hz for carbon-detected HETCOR (red curve) and 322 Hz for proton-detected HETCOR (black curve). The 1H-detected HETCOR thus does not exhibit obvious advantages in terms of the 1H resolution either. Our results generally suggest that 1H detection may not offer resolution benefits in the cases where indirect-dimension spectral width is large, such as 13C experiments. In these cases, experiment times of 1H-detected measurements are excessively long to attain sufficient resolution for unambiguous assignments, if full spectral width in the 13C dimension needs to be covered.
Figure 6.

2D H–C 13C-detected HETCOR spectrum (A) and 2D C–H 1H-detected HETCOR spectrum (D) of U–13C,15N-LC8 acquired at 60 kHz MAS and 850 MHz. Both spectra were processed with 30-degree sine bell apodization. The lowest contour level is set at 10 × σ RMSD with multiplier of 1.2. The zoom-in representations of the 1HN-13Cα region of each HETCOR spectrum are displayed in B and C. The experimental time was the same in both cases (4.5 hours) while different numbers of scans were used: 128 scans for HC HETCOR and 32 scans for CH HETCOR. Note that several cross peaks that appear in the 1H-detected HETCOR are not visible in 13C-detected experiment, because of the lower sensitivity of the latter. The peaks in the 13C-detected HETCOR that appear around δ13C = 20–45 ppm/δ1H = 5.2 ppm (1H) are most likely artifacts. 1D slices were extracted from 2D HETCOR spectra processed with 60-degree sine bell at the position of crosshairs shown in B and C: (E) 1H slices of HC HETCOR (red) and CH HETCOR (black) extracted at δ13C = 61.5 ppm. The 1H linewidth of the peak at δ1H = 5.9 ppm is 269 Hz for 13C-detected HETCOR and 322 Hz for 1H-detected HETCOR. F) 13C slices of HC HETCOR (red) and CH HETCOR (black) extracted at δ1H = 4.25 ppm.
While better resolution is an advantage of 13C-detected HETCOR, sensitivity is considerably higher in the 1H-detected version, consistent with prior reports by other investigators (Marchetti et al. 2012; Zhou et al. 2007). In the spectra reported here, the average signal-to-noise ratio (SNR) for the cross peaks is 69 and 208 for the 13C- and 1H-detected HETCOR, respectively. In both cases, the experiment time was 4.5 hours. The HC HETCOR was collected with 128 scans, and the SNR is ca. 6 per scan for the 13C-detection. The CH HETCOR was acquired with 32 scans, and the SNR is 37 per scan for the 1H-detection. Curiously, several cross peaks that appear in the 1H-detected experiment vanished in the 13C-detected spectrum, e. g., the peaks in the region of δ13C = 20–35 ppm/δ1H = 4–5 ppm, δ13C = 59 ppm/δ1H = 2.8 ppm, and δ13C = 69 ppm/δ1H = 2.9 ppm. This is likely due to the lower sensitivity of the 13C detection.
CONCLUSIONS
13C detected 3D heteronuclear correlation spectroscopy under fast MAS conditions is highly efficient for resonance assignments and structural analysis of fully protonated proteins. The sensitivity and the resolution of 3D HNCA, HNCO, and HNCOCX experiments are excellent, and with these three datasets we have assigned the majority of the backbone and a number of sidechain 1H/13C/15N resonances in dynein’s LC8. With these experiments, we have resolved many ambiguities that were persistent in our previous studies using moderate MAS frequencies and lacking the 1H dimension. Finally, we demonstrate that 13C detection in the fast-MAS HETCOR experiments may be advantageous vis-à-vis 1H detection as it yields datasets of significantly higher 13C resolution.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH Grants R01GM085306, 8P30GM103519-03 from NIGMS, and 5P30RR031160-03 from NCRR). We acknowledge the support of the National Science Foundation (NSF Grant CHE0959496) for the acquisition of the 850 MHz NMR spectrometer at the University of Delaware. We thank Dr. Si Yan for preparing and packing the Cu-EDTA doped LC8 protein sample.
Footnotes
Acquisition and processing parameters for the 3D heteronuclear-detected and 2D HETCOR MAS NMR spectra (Table S1); additional chemical shifts of 5mM Cu(II)-EDTA doped U-13C,15N-LC8 assigned based on 3D 13C-detected MAS NMR HNCA, HNCO and HNCOCX spectra (Table S2); 1H and 13C linewidths of cross peaks in the 1HN-13Cα regions of 2D 13C-detected HETCOR and 1H-detected HETCOR spectra (Table S3).
Contributor Information
Changmiao Guo, Email: cmguo@udel.edu.
Guangjin Hou, Email: hou@udel.edu.
Xingyu Lu, Email: luxingyu@udel.edu.
Bernie O’Hare, Email: Bernie.O’Hare@bruker-biospin.com.
Jochem Struppe, Email: Jochem.Struppe@bruker-biospin.com.
Tatyana Polenova, Email: tpolenov@udel.edu.
References
- Agarwal V, Sardo M, Scholz I, Bockmann A, Ernst M, Meier BH. PAIN with and without PAR: variants for third-spin assisted heteronuclear polarization transfer. J Biomol NMR. 2013;56(4):365–377. doi: 10.1007/s10858-013-9756-4. [DOI] [PubMed] [Google Scholar]
- Akbey U, Lange S, Franks WT, Linser R, Rehbein K, Diehl A, van Rossum BJ, Reif B, Oschkinat H. Optimum levels of exchangeable protons in perdeuterated proteins for proton detection in MAS solid-state NMR spectroscopy. J Biomol NMR. 2010;46(1):67–73. doi: 10.1007/s10858-009-9369-0. [DOI] [PubMed] [Google Scholar]
- Asami S, Rakwalska-Bange M, Carlomagno T, Reif B. Protein-RNA Interfaces probed by 1H-detected MAS solid-state NMR spectroscopy. Angew Chem Int Ed. 2013;52(8):2345–2349. doi: 10.1002/anie.201208024. [DOI] [PubMed] [Google Scholar]
- Asami S, Reif B. Proton-detected solid-state NMR spectroscopy at aliphatic sites: application to crystalline systems. Acc Chem Res. 2013;46(9):2089–2097. doi: 10.1021/ar400063y. [DOI] [PubMed] [Google Scholar]
- Barbar E. Dynein light chain LC8 is a dimerization hub essential in diverse protein networks. Biochemistry. 2008;47(2):503–508. doi: 10.1021/bi701995m. [DOI] [PubMed] [Google Scholar]
- Barbet-Massin E, Pell AJ, Knight MJ, Webber AL, Felli IC, Pierattelli R, Emsley L, Lesage A, Pintacuda G. 13C-detected through-bond correlation experiments for protein resonance assignment by ultra-fast MAS solid-state NMR. Chem Phys Chem. 2013;14(13):3131–3137. doi: 10.1002/cphc.201201097. [DOI] [PubMed] [Google Scholar]
- Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J Chem Phys. 1995;103(16):6951–6958. [Google Scholar]
- Bermel W, Bertini I, Felli IC, Kummerle R, Pierattelli R. 13C direct detection experiments on the paramagnetic oxidized monomeric copper, zinc superoxide dismutase. J Am Chem Soc. 2003;125(52):16423–16429. doi: 10.1021/ja037676p. [DOI] [PubMed] [Google Scholar]
- Bermel W, Bertini I, Felli IC, Kummerle R, Pierattelli R. Novel 13C direct detection experiments, including extension to the third dimension, to perform the complete assignment of proteins. J Magn Reson. 2006a;178(1):56–64. doi: 10.1016/j.jmr.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Bermel W, Bertini I, Felli IC, Lee YM, Luchinat C, Pierattelli R. Protonless NMR experiments for sequence-specific assignment of backbone nuclei in unfolded proteins. J Am Chem Soc. 2006b;128(12):3918–3919. doi: 10.1021/ja0582206. [DOI] [PubMed] [Google Scholar]
- Bertini I, Luchinat C, Parigi G, Pierattelli R. NMR spectroscopy of paramagnetic metalloproteins. Chem Bio Chem. 2005;6(9):1536–1549. doi: 10.1002/cbic.200500124. [DOI] [PubMed] [Google Scholar]
- Chevelkov V, Rehbein K, Diehl A, Reif B. Ultrahigh resolution in proton solid-state NMR spectroscopy at high levels of deuteration. Angew Chem Int Ed. 2006;45(23):3878–3881. doi: 10.1002/anie.200600328. [DOI] [PubMed] [Google Scholar]
- Clore GM, Gronenborn AM. Multidimensional heteronuclear nuclear magnetic resonance of proteins. Methods Enzymol. 1994;239:349–363. doi: 10.1016/s0076-6879(94)39013-4. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRpipe: a multidimensional spectral processing system based on Unix pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Ernst M, Samoson A, Meier BH. Low-power decoupling in fast magic-angle spinning NMR. Chem Phys Lett. 2001;348(3–4):293–302. [Google Scholar]
- Hou GJ, Gupta R, Polenova T, Vega AJ. A magic-angle-spinning NMR Spectroscopy method for the site-specific measurement of proton chemical-shift anisotropy in biological and organic solids. Isr J Chem. 2014;54(1–2):171–183. doi: 10.1002/ijch.201300099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou GJ, Paramasivam S, Yan S, Polenova T, Vega AJ. Multidimensional magic angle spinning NMR spectroscopy for site-resolved measurement of proton chemical shift anisotropy in biological solids. J Am Chem Soc. 2013;135(4):1358–1368. doi: 10.1021/ja3084972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightcap CM, Sun S, Lear JD, Rodeck U, Polenova T, Williams JC. Biochemical and structural characterization of the Pak1-LC8 interaction. J Biol Chem. 2008;283(40):27314–27324. doi: 10.1074/jbc.M800758200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linser R, Dasari M, Hiller M, Higman V, Fink U, Lopez del Amo JM, Markovic S, Handel L, Kessler B, Schmieder P, Oesterhelt D, Oschkinat H, Reif B. Proton-detected solid-state NMR spectroscopy of fibrillar and membrane proteins. Angew Chem Int Ed. 2011;50(19):4508–4512. doi: 10.1002/anie.201008244. [DOI] [PubMed] [Google Scholar]
- Marchetti A, Jehle S, Felletti M, Knight MJ, Wang Y, Xu ZQ, Park AY, Otting G, Lesage A, Emsley L, Dixon NE, Pintacuda G. Backbone assignment of fully protonated solid proteins by 1H detection and ultrafast magic-angle-spinning NMR spectroscopy. Angew Chem Int Ed. 2012;51(43):10756–10759. doi: 10.1002/anie.201203124. [DOI] [PubMed] [Google Scholar]
- Marulanda D, Tasayco ML, McDermott A, Cataldi M, Arriaran V, Polenova T. Magic angle spinning solid-state NMR spectroscopy for structural studies of protein interfaces. Resonance assignments of differentially enriched Escherichia coli thioredoxin reassembled by fragment complementation. J Am Chem Soc. 2004;126(50):16608–16620. doi: 10.1021/ja0464589. [DOI] [PubMed] [Google Scholar]
- Maudsley AA, Ernst RR. Indirect detection of magnetic-resonance by heteronuclear 2-dimensional spectroscopy. Chem Phys Lett. 1977;50(3):368–372. [Google Scholar]
- McDermott A. Structure and dynamics of membrane proteins by magic angle spinning solid-state NMR. Annu Rev Biophys. 2009;38:385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- Reif B, Jaroniec CP, Rienstra CM, Hohwy M, Griffin RG. 1H-1H MAS correlation spectroscopy and distance measurements in a deuterated peptide. J Magn Reson. 2001;151(2):320–327. doi: 10.1006/jmre.2001.2354. [DOI] [PubMed] [Google Scholar]
- Schaefer J, Mckay RA, Stejskal EO. Double-cross-polarization NMR of solids. J Magn Reson. 1979;34(2):443–447. [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS plus: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44(4):213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens TJ, Fogh RH, Boucher W, Higman VA, Eisenmenger F, Bardiaux B, van Rossum BJ, Oschkinat H, Laue ED. A software framework for analysing solid-state MAS NMR data. J Biomol NMR. 2011;51(4):437–447. doi: 10.1007/s10858-011-9569-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suiter CL, Paramasivam S, Hou G, Sun S, Rice D, Hoch JC, Rovnyak D, Polenova T. Sensitivity gains, linearity, and spectral reproducibility in nonuniformly sampled multidimensional MAS NMR spectra of high dynamic range. J Biomol NMR. 2014;59(2):57–73. doi: 10.1007/s10858-014-9824-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SJ, Butterworth AH, Paramasivam S, Yan S, Lightcap CM, Williams JC, Polenova T. Resonance assignments and secondary structure analysis of dynein light chain 8 by magic-angle spinning NMR spectroscopy. Can J Chem. 2011;89(7):909–918. doi: 10.1139/v11-030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SJ, Yan S, Guo CM, Li MY, Hoch JC, Williams JC, Polenova T. A time-saving strategy for MAS NMR spectroscopy by combining nonuniform sampling and paramagnetic relaxation assisted condensed data collection. J Phys Chem B. 2012;116(46):13585–13596. doi: 10.1021/jp3005794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur RS, Kurur ND, Madhu PK. Swept-frequency two-pulse phase modulation for heteronuclear dipolar decoupling in solid-state NMR. Chem Phys Lett. 2006;426(4–6):459–463. [Google Scholar]
- Vadlamudi RK, Bagheri-Yarmand R, Yang Z, Balasenthil S, Nguyen D, Sahin AA, den Hollander P, Kumar R. Dynein light chain 1, a p21-activated kinase 1-interacting substrate, promotes cancerous phenotypes. Cancer Cell. 2004;5(6):575–585. doi: 10.1016/j.ccr.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Webber AL, Pell AJ, Barbet-Massin E, Knight MJ, Bertini I, Felli IC, Pierattelli R, Emsley L, Lesage A, Pintacuda G. Combination of DQ and ZQ coherences for sensitive through-bond NMR correlation experiments in biosolids under ultra-fast MAS. Chem Phys Chem. 2012;13(9):2405–2411. doi: 10.1002/cphc.201200099. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe NP, Ishii Y. Sensitivity enhancement, assignment, and distance measurement in 13C solid-state NMR spectroscopy for paramagnetic systems under fast magic angle spinning. J Magn Reson. 2006;181(2):233–243. doi: 10.1016/j.jmr.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe NP, Parthasarathy S, Jones CR, Bhardwaj C, Long F, Kotecha M, Mehboob S, Fung LW, Past J, Samoson A, Ishii Y. Nanomole-scale protein solid-state NMR by breaking intrinsic 1H T1 boundaries. Nat Methods. 2009;6(3):215–218. doi: 10.1038/nmeth.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Suiter CL, Hou GJ, Zhang HL, Polenova T. Probing structure and dynamics of protein assemblies by magic angle spinning NMR spectroscopy. Acc Chem Res. 2013;46(9):2047–2058. doi: 10.1021/ar300309s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou DH, Nieuwkoop AJ, Berthold DA, Comellas G, Sperling LJ, Tang M, Shah GJ, Brea EJ, Lemkau LR, Rienstra CM. Solid-state NMR analysis of membrane proteins and protein aggregates by proton detected spectroscopy. J Biomol NMR. 2012;54(3):291–305. doi: 10.1007/s10858-012-9672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou DH, Shah G, Cormos M, Mullen C, Sandoz D, Rienstra CM. Proton-detected solid-state NMR spectroscopy of fully protonated proteins at 40 kHz magic-angle spinning. J Am Chem Soc. 2007;129(38):11791–11801. doi: 10.1021/ja073462m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.