

Abstract
There is increasing evidence implicating astrocytes in multiple forms of chronic pain, as well as in the specific context of chemotherapy-induced peripheral neuropathy (CIPN). However, it is still unclear what the exact role of astrocytes may be in the context of CIPN. Findings in oxaliplatin and paclitaxel models have displayed altered expression of astrocytic gap junctions and glutamate transporters as means by which astrocytes may contribute to observed behavioral changes. The current study investigated whether these changes were also generalizable to the bortezomib CIPN. Changes in mechanical sensitivity were verified in bortezomib-treated animals, and these changes were prevented by co-treatment with a glial activation inhibitor (minocycline), a gap junction decoupler (carbenoxolone), and by a glutamate transporter upregulator (ceftriaxone). Immunohistochemistry data at day 30 in bortezomib-treated animals showed increases in expression of GFAP and connexin 43 but decrease in GLAST expression. These changes were prevented by co-treatment with minocycline. Follow-up Western blotting data showed a shift in connexin 43 from a non-phosphorylated state to a phosphorylated state, indicating increased trafficking of expressed connexin 43 to the cell membrane. These data suggest that increases in behavioral sensitivity to cutaneous stimuli may be tied to persistent synaptic glutamate resulting from increased calcium flow between spinal astrocytes.
Keywords: bortezomib, minocycline, GFAP, connexin 43, GLT-1, GLAST
3.0 Introduction
Bortezomib is a first-generation proteasome inhibitor chemotherapy drug used primarily for the treatment of multiple myeloma. Among its multiple side effects, bortezomib is known to produce chemotherapy-induced peripheral neuropathy (CIPN) that is often dose-limiting (Argyriou et al, 2012; Argyriou et al, 2014; Ferrier et al, 2013; Miltenburg and Boogerd, 2014). Bortezomib CIPN may extend long after the cessation of treatment, and current treatment options to manage CIPN symptoms are few and limited in their efficacy.
Among other forms of neuropathic pain, there is emerging evidence that spinal astrocytes and microglia may contribute to the phenotype’s development and maintenance. However, it is becoming apparent that astrocytes are activated in the apparent absence of microglia in CIPN models. Our own lab has now demonstrated this in paclitaxel, oxaliplatin, and bortezomib models of CIPN (Zhang et al, 2012; Robinson et al, 2014b). However, there is currently limited evidence demonstrating potential mechanism by which astrocytes may contribute to CIPN. Downregulation of glutamate transporters in paclitaxel CIPN (Zhang et al, 2012) and the upregulation of astrocytic gap junctions in oxaliplatin CIPN suggest these also may contribute to bortezomib CIPN (Yoon et al, 2013).
The investigation of glutamate transporter dysfunction as a possible mechanism for CIPN may be justified by observations of behavior and neuronal activity in the spinal dorsal horn. Downregulation of glutamate transporters leads to persistent synaptic glutamate, which is sufficient to potentiate action potentials, drive persistent after-discharges, and promote spontaneous ectopic activity (Matute et al, 2006; Yi and Hazell, 2006; López-Bayghen and Ortega, 2011). Downregulation of glutamate transporters through pharmacological means is sufficient to drive spontaneous nociceptive behaviors (Nakagawa and Kaneko, 2010). Previous work from our lab has demonstrated altered activity in spinal wide dynamic range neurons and downregulation of glutamate transporters in support of such a model in animals treated with vincristine or paclitaxel (Weng et al, 2003; Cata et al, 2006). Work within the bortezomib model has demonstrated behaviors indicating persistent sensation and in vivo recordings showing potentiated responses and persistent after-discharges in spinal wide dynamic range neurons (Robinson et al, 2014a). Taken together, it is possible that the changes observed in glutamate transporters in paclitaxel may also carry over to the bortezomib model, as well.
The activity of connexin 43 is another point of interest in CIPN. Connexin 43 is the protein in astrocytes that composes gap junction hemichannels that connect astrocytes together in a functional syncytium (Giaume and Liu, 2012; Theis and Giaume, 2012). The openings that are formed by astrocytic gap junctions are small, allowing only ions and molecules smaller than 1.2 kDa to pass. This is too small for the majority of signaling molecules, but astrocytic gap junctions permit the flow of such messengers as calcium ions and glutamate between cells (Tabernero et al, 2006; Theis and Giaume, 2012; Pannasch and Rouach, 2013). Hemichannel-forming proteins are upregulated in conjunction with astrocyte activation in multiple models of insult or injury, suggesting an increase in intercellular communication in these reactive astrocytes (Homkajorn et al, 2010; Chen et al, 2014; Chen et al, 2012). The potential downstream effects of this increased hemichannel expression are of great interest in CIPN. Increases in intracellular calcium in astrocytes may lead to decreased glutamate uptake or even direct release of glutamate from astrocytes (Malarkey and Parpura, 2008; Devinsky et al, 2013; Hansen and Malcangio, 2013). Thus, increases in connexin 43 potentially indicate a parallel means by which astrocytes decrease glutamate uptake at the tripartite synapse.
The focus of the present study is to assess the activity of the astrocytic glutamate transporters GLT-1 and GLAST and the activity of connexin 43 in bortezomib-treated animals. Data in support of previous findings in other models would explain behaviors and electrophysiological data seen in bortezomib-treated animals. The present work also includes minocycline in treatment groups, since this has been shown to prevent behavioral changes in bortezomib-treated animals. Therefore a direct role for connexins and glutamate transporters in bortezomib-induced peripheral neuropathy may only be established if (1) these proteins are altered in accordance with an observed change of behavior, and (2) if prevention of changes to behavior also prevent changes in these proteins. Carbenoxolone was included in additional treatment groups for behavioral data as a gap junction decoupler (Juszczak and Swiergiel, 2009; Yoon et al, 2013), and ceftriaxone was included as an upregulator of glutamate transporter expression (Lee et al, 2008; Kim et al, 2011). The inclusion of these treatment groups was to establish whether pharmacological strategies to directly counteract possible changes would also prevent the development of behavioral changes. Positive data would indicate that changes to these proteins are sufficient to drive the bortezomib phenotype of CIPN in the animal model.
4.0 Experimental Procedures
4.1 Animals
All procedures were reviewed and approved by the M.D. Anderson Institutional Animal Care and Use Committee and were in accordance with the guidelines established by the NIH and the International Association for the Study of Pain. Ninety-four (94) Male Sprague-Dawley rats between 60–75 days of age upon beginning treatment (300–350 g) were used for all experiments. Of these rats, all 94 were used for behavioral testing, but 21 of these were used for immunohistochemistry. Another 14 of these were used for Western blotting. Rats were housed in a facility with a 12h light/dark cycle and were given food and water ad libitum. All efforts were taken at each stage of the experiments to limit the numbers of animals used and any discomfort to which they might be exposed.
4.2 Drugs
Saline, minocycline, and bortezomib were administered by intraperitoneal injection, and volumes were calculated based on body mass to approximate a volume of 0.5 ml. The gap junction decoupler ceftriaxone (Sigma Aldrich) and the glutamate transporter upregulator carbenoxolone (Sigma Aldrich) were administered intrathecally in the space between spinal segments L5 and L6 at a volume of 10μL. Animals were divided into eight treatment groups: saline alone (n=11), saline + minocycline (n=11), saline + carbenoxolone (n=12), saline + ceftriaxone (n=12), bortezomib alone (n=12), bortezomib + minocycline (n=12), bortezomib + carbenoxolone (n=12), and bortezomib + ceftriaxone (n=12). Bortezomib (Millennium Pharmaceuticals) was administered in saline vehicle at a dose of 0.15 mg/kg on days 1, 3, 5, and 7 of experimentation for a cumulative dose of 0.60 mg/kg. Equivolume saline was administered to rats not treated with bortezomib. Animals treated with minocycline hydrochloride (Sigma Aldrich) were injected daily with 25.0 mg/kg minocycline in saline vehicle beginning at day 0 and continuing daily through day 8 (one day past chemotherapy treatment) of experimentation for a cumulative dose of 225mg/kg. Carbenoxolone (25μg/day) and ceftriaxone (150μg/day) were administered on the same schedule as minocycline for cumulative intrathecal doses of 225μg and 1350 μg, respectively. Animals not injected with carbenoxolone or ceftriaxone were administered an equivalent volume of saline intrathecally on the same dosing schedule.
4.3 Behavior Testing
Von Frey filament testing was used to assess mechanical sensitivity over time as previously described (Boyette-Davis et al, 2011; Boyette-Davis and Dougherty, 2011; Robinson et al, 2014b). Briefly, filaments calibrated to a bending force equal to 4g, 10g, 15g, and 26g were applied 6 times each to the mid-plantar surface of each hindpaw. Animals were allowed a half-hour to habituate to the testing apparatus prior to application of filaments. Testing began with the lowest (4g) filament and escalated in filament size until a withdrawal threshold was reached. 5 to 10 minutes was allowed between filaments in order to minimize responses due to anxiety or sensitization. Filaments were applied with steady force until bending of the filament was observed and held for approximately 1second. Responses were classified as a rapid withdrawal of the paw immediately followed by a return of the paw to the mesh. The threshold for sensitivity to mechanical stimuli was recorded as the bending force of the filament for which at least 50% of applications elicited a response. The mean was reported for each treatment group at each time point. Error was reported as standard error of the mean (SEM). Significant differences between groups were tested using a one-way ANOVA, followed by post-hoc Tukey’s test. Significance was further tested at each time point using Mann-Whitney tests.
4.4 Tissue Collection
On day 30, following the final time point of behavioral testing, animals were sacrificed for tissue collection. Rats were overdosed with sodium pentobarbital (150mg), and then perfused intracardially with room temperature heparinized saline, followed by cold 4% paraformaldehyde in 0.1M phosphate buffer. After tissue fixation was verified by rigidity of the rat’s extremities, spinal cords were removed and post-fixed in 4% paraformaldehyde at 4°C overnight. Spinal cords were then moved to 15% sucrose the following day, and then moved after another 24h to 30% sucrose for a minimum of 48h. The lumbar enlargement was mounted and the L5 segment cut in a cryostat at a thickness of 30μm.
For Western blotting, fresh tissue was collected from a different group of animals. Animals were overdosed with sodium pentobarbital (150mg). Each animal was then sacrificed via decapitation, and the spinal cord was quickly removed. The dorsal horns of the spinal cord were taken and frozen rapidly using liquid nitrogen. All samples were then kept at -80°C until tissue could be processed. Following this, tissue was homogenized in lysis buffer and centrifuged. The supernatant was taken and protein concentration quantified via Lowry assay. Protein concentration was then adjusted to 1μg/μL for all samples and heated at 70°C for 10 minutes.
4.5 Immunohistochemistry
Spinal cord slices were washed in phosphate buffered saline (PBS) for 6 washes lasting 15 minutes each and then blocked in normal donkey serum (5% NDS and 0.2% triton X in PBS). Slices were incubated overnight at 4°C with primary antibodies against GFAP (mouse anti-rat 1:1000, Cell Signaling Technology), connexin 43 (rabbit anti-rat 1:1000, Invitrogen), GLT-1 (guinea pig anti-rat 1:2000, Chemicon), or GLAST (rabbit anti-rat 1:250, Abcam). Slices were washed the following day in PBS for 6 washes lasting 15 minutes each, and then incubated with secondary antibodies. For one set of tissue, slices were incubated with FITC-conjugated donkey anti-mouse secondary antibody for GFAP detection (1:500, Jackson Immunoresearch) and Cy-3-conjugated donkey anti-rabbit secondary antibody for Cx43 detection (1:500, Jackson Immunoresearch). A second set of tissue was incubated with FITC-conjugated donkey anti-guinea pig secondary antibody for GLT-1 detection (1:500, Jackson Immunoresearch) and Cy-3-conjugated donkey anti-rabbit secondary antibody for GLAST detection (1:500, Jackson Immunoresearch). All tissue was incubated with secondary antibodies for 2 hours at room temperature. Slices were washed for a final course of 6 washes lasting 15 minutes each, then mounted onto glass slides using Vectamount medium.
4.6 Quantification of Immunohistochemistry
Slices were viewed and images captured using fluorescent illumination at 20X magnification using a Nikon Eclipse E600 microscope. Captured images were analyzed using NIS Elements software (Nikon, USA). A region of interest was drawn within the dorsal horn containing lamina I and II for quantification purposes. A region outside of the tissue slice was selected as background, and its signal was subtracted from the entire image. The signal within the dorsal horn region of interest was then expressed as intensity in pixels/μm2. For each treatment group, the mean of these values was calculated and expressed as a percent versus the mean values of the saline-treated group. Error was expressed as the combined standard error of the mean (SEM) for the corresponding treatment and saline-treated groups to account for variability within controls. Significance was determined via t-test (P = 0.05, 0.01).
4.7 Western Blotting
For each Western blot, 20μL of each sample (20μg protein) was separated via gel electrophoresis and transferred to polyvynilidene (PVDF) membranes. After transferring, membranes were washed three times for 10 minutes each in Tris-buffered saline with Tween-20 (TBST). Membranes were then blocked in a solution of 5% powdered nonfat milk in TBST for 1 hour at room temperature. Primary antibodies were added, and membranes were incubated overnight at 4°C. The membranes were then washed three times for 10 minutes each in TBST. These were then incubated with secondary antibodies conjugated against horseradish protein (HRP) in 5% nonfat milk/TBST solution for 30 minutes at room temperature and washed a final three times at 10 minutes each in TBST. Target proteins on membranes were then visualized using an enhanced chemiluminescence system (Amersham Pharmacia Biotech, Little Chalfont, UK). Primary antibodies were selected in order to verify changes observed in immunohistochemistry. The primary antibodies used for Western blotting were the same as those used for immunohistochemistry, incubated at a 1:1000 concentration. Mouse anti-rat beta-actin (1:10,000, Sigma) was also included as a control for basal protein expression. Secondary antibodies were either goat anti-rabbit or goat anti-mouse HRP-conjugated antibodies (1:10,000, Calbiochem).
Band intensity was quantified using ImageJ software, available from the NIH website. A region of interest containing the full band was taken, and background was subtracted. The expression of a protein was then quantified as the ratio of the expression of that protein over the expression of beta-actin in the same lane. The average of these values was taken for saline treated controls. The other treatment groups were then normalized as a percentage of this value. Error bars were calculated as the SEM, and significance was determined via t-test (p = 0.05, 0.01). For the connexin 43 antibody, which detects three bands for different phosphorylation states, this calculation was performed for total amount of connexin 43 and for each phosphorylation state.
5.0 Results
5.1 Mechanical Withdrawal Thresholds
Mean withdrawal thresholds across groups were not significantly different at baseline (Fig. 1). Animals treated with saline alone or saline with minocycline, carbenoxolone, or ceftriaxone (not shown) did not change over time versus baseline withdrawal threshold and did not differ significantly from one another according to ANOVA data. Tukey’s test data following ANOVA indicated that the bortezomib-treated group differed significantly in behavior from saline-treated controls (p<0.05) and other treatment groups (p<0.01). Bortezomib-treated animals first showed a significant difference in withdrawal thresholds versus saline-treated controls at day 14 (19.5 ± 1.9g saline/11.2 ± 0.9g bortezomib). Thresholds continued to decrease and showed significant differences at day 21 (18.1 ± 2.4g saline/6.5 ± 0.9g bortezomib) and day 28 (20.6 ± 2.1g saline/7.1 ± 1.2g bortezomib). Co-treatment with minocycline, ceftriaxone, or carbenoxolone prevented the development of mechanical hypersensitivity and did not differ significantly from saline-treated controls at any time point that was tested.
Figure 1.
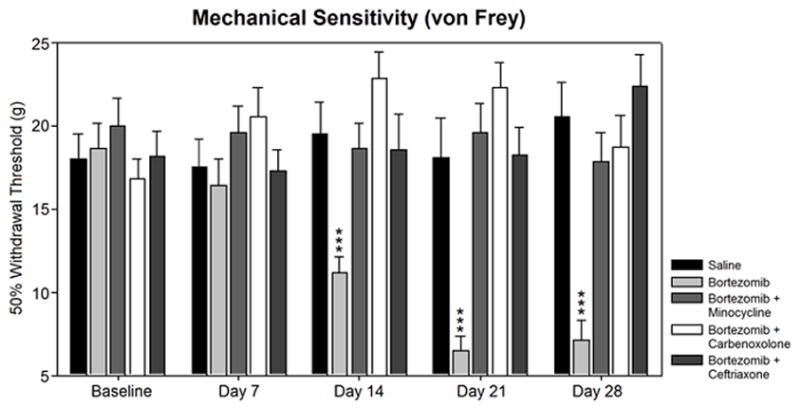
Mechanical sensitivity was assesed in bortezomib-treated animals versus preventative treatment groups. Treatment of animals with bortezomib (n=12) induced mechanical hypersensitivity versus saline-treated controls (n=11) that was prevented by co-treatment with minocycline (n=12), carbenoxolone (n=12), or ceftriaxone (n=12). Rats treated with minocycline (n=11), carbenoxolone (n=12), or ceftriaxone (n=12) alone did not develop mechanical hypersensitivity (not shown).
5.2 Astrocyte Activation
GFAP staining for astrocytes showed no difference in minocycline-treated rats versus saline-treated controls (104.9 ± 13.6% of control) (Fig. 2). Astrocytes from these groups showed thin arbors and uniform distribution throughout the dorsal horn gray matter (top-left and bottom-left panels). Bortezomib-treated rats showed significantly higher GFAP activation versus controls (132.1 ± 16.9% of control). Astrocytes from this group showed brighter staining, hypertrophy, and increased arborization (top-right panel). Distribution of astrocytes was uniform throughout the dorsal horn in this group, as well. Minocycline co-treatment prevented this activation, and these animals were not significantly different from controls (113.1 ± 16.2% of control). Qualitative observations of cell size, brightness, arborization, and distribution indicated no apparent difference in the astrocytes in this tissue versus saline-treated controls (bottom-right panel). These findings are in line with previous data from our lab showing the same profile of activation (Robinson et al, 2014b).
Figure 2.
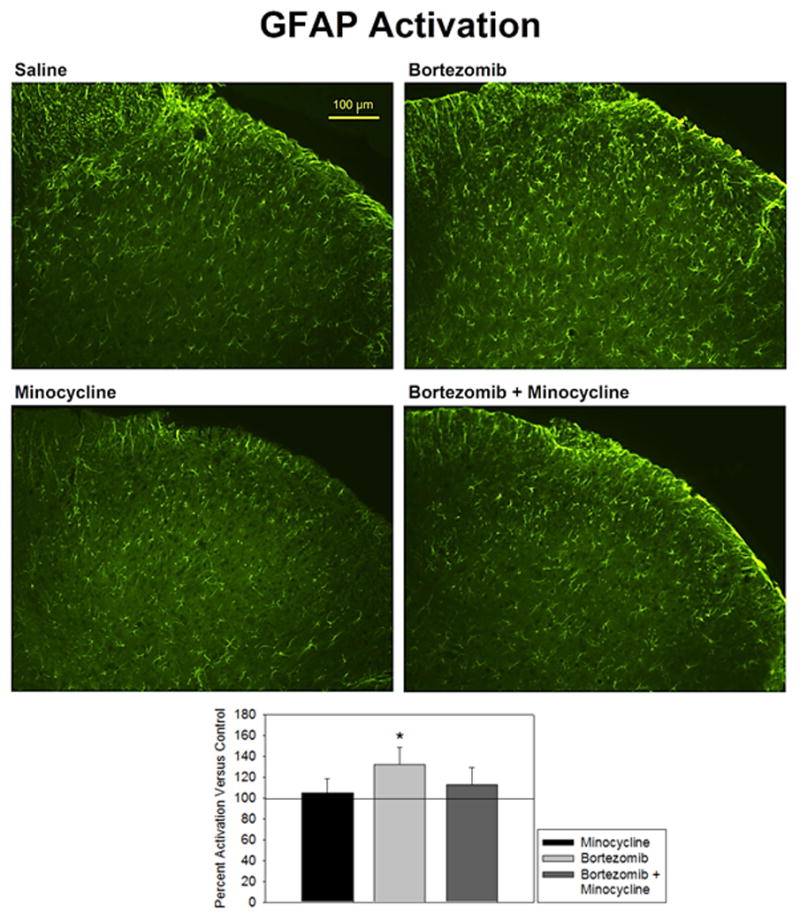
Activation of GFAP was expressed as percent fluorescence intensity versus mean fluorescence intensity of control. Treatment of animals with bortezomib (n=5) induced a significantly increased expression of GFAP in astrocytes versus saline-treated controls (n=6) that was prevented by co-treatment with minocycline (n=5). Minocyline alone (n=5) did not affect GFAP activity.
5.3 Connexin 43 Expression
Staining for connexin 43 activation was equivalent in minocycline-treated rats versus saline-treated controls (97.3 ± 17.6% of control) (Fig. 3). Connexin 43 staining was distributed evenly throughout the dorsal horn (top-left and bottom-left panels). The staining was granular in appearance, indicating staining of gap junctions apart from background levels of staining. Bortezomib-treated rats showed a significant upregulation of connexin 43 expression (131.3 ± 19.3% of control). The dorsal horn staining appeared brighter in this tissue than in saline-treated controls (top-right panel). This could indicate either increases in the brightness of individual gap junctions or an increase in the number of gap junctions. This activation was prevented in rats that were also treated with minocycline (111.7 ± 19.9% of control). Levels of connexin 43 in these animals were higher than in saline-treated controls, but the difference was not significant (bottom-right panel). Staining was slightly brighter in this tissue than in saline-treated controls, but it maintained an even distribution and granular appearance. This would indicate a significant abrogation by minocycline of the connexin increases induced by bortezomib.
Figure 3.
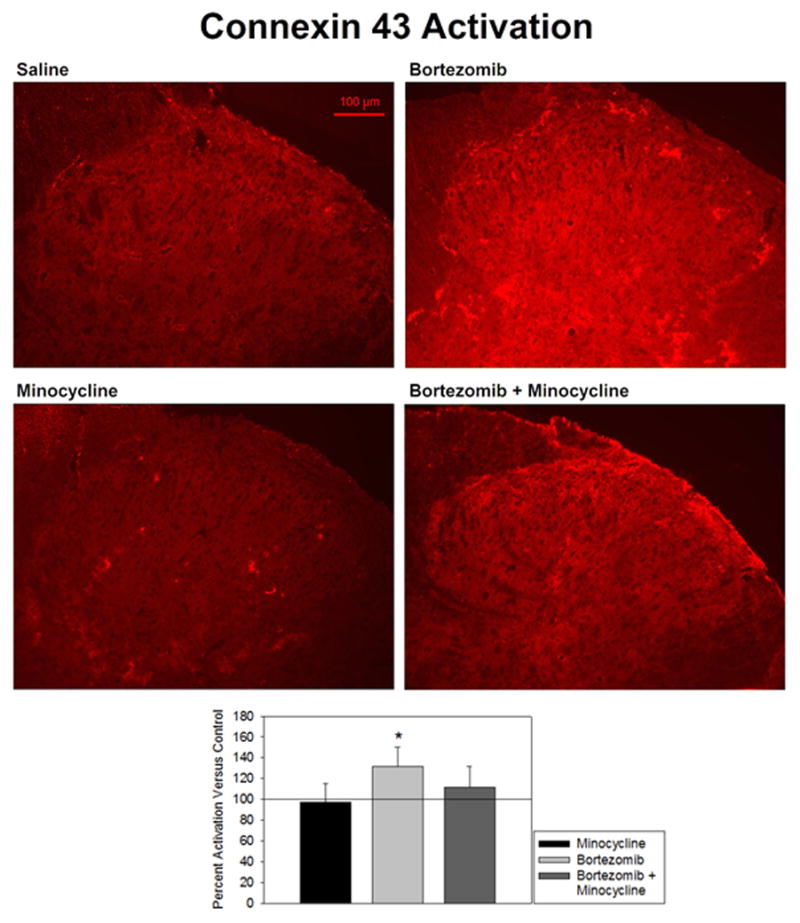
Activation of connexin 43 was expressed as percent fluorescence intensity versus mean fluorescence intensity of control. Treatment of animals with bortezomib (n=5) induced a significantly increased expression of connexin 43 in the spinal dorsal horn versus saline-treated controls (n=6) that was prevented by co-treatment with minocycline (n=5). Minocyline alone (n=5) did not affect connexin 43 activity.
5.4 GLT-1 Expression
Staining for GLT-1 activation did not differ significantly between minocycline-treated and saline-treated groups (111.7 ± 8.1% of control) (Fig. 4). There was also no change detected in rats treated with bortezomib (102.9 ± 10.5% of control) or bortezomib and minocycline (98.4 ± 12.3% of control). GLT-1 staining was not localized to any one region of the spinal cord gray matter. There was minimal staining within the white matter, which reflects the paucity of synapses in these regions. Furthermore, staining was granular in appearance, but difficult to distinguish in appearance from background staining. The difficulty in distinguishing background from signal could potentially mask smaller changes, but the current data did not indicate any difference between treatment groups with this staining.
Figure 4.
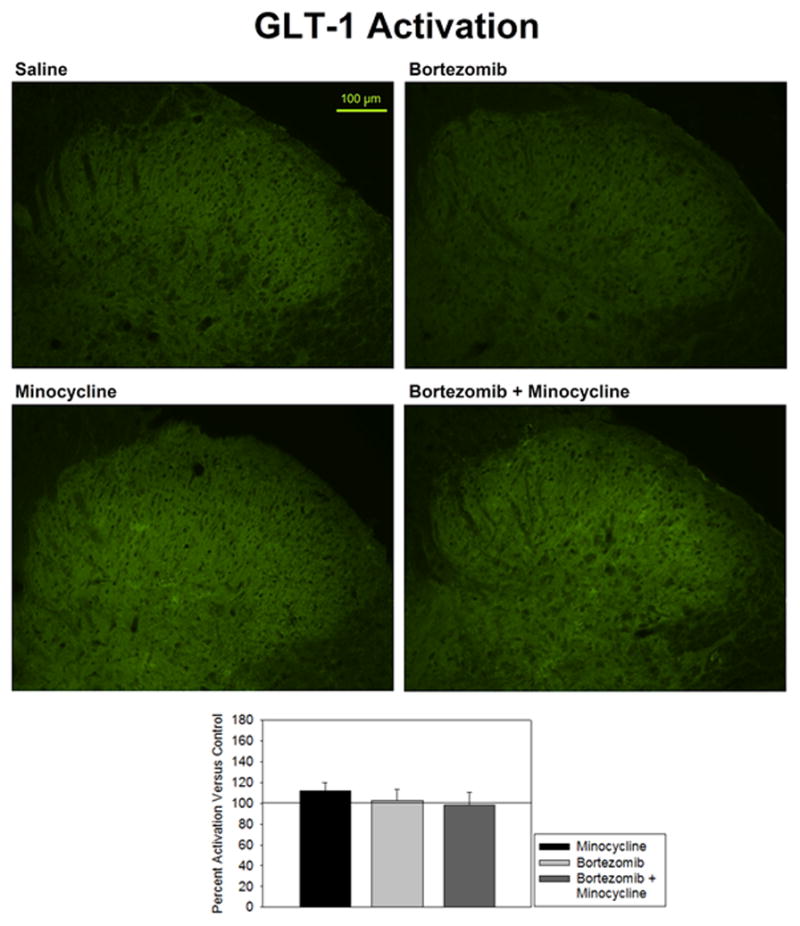
Activation of GLT-1 was expressed as percent fluorescence intensity versus mean fluorescence intensity of control. Treatment of animals with bortezomib (n=5), minocycline alone (n=5), or bortezomib + minocycline (n=5) showed no change in GLT-1 expression in the spinal dorsal horn versus saline-treated controls (n=6).
5.5 GLAST Expression
Expression of GLAST was not significantly different in minocycline-treated rats versus saline-treated rats (106.7 ± 10.5% of control) (Fig. 5). Staining was most densely localized to the first two laminae of the dorsal horn, which is where nociceptive neurons synapse (top-left and bottom-left panels). Staining was granular in appearance. Bortezomib-treated rats showed significantly lower expression of GLAST (81.5 ± 8.5% of control), which was only partially prevented by co-treatment with minocycline (top-right and bottom-right panels). However, there did not appear to be analogous differences in staining in regions outside of the quantified region. It is difficult to say whether this is truly the case, considering that the far greater brightness in the first two laminae could overshadow lesser changes. Animals treated with both bortezomib and minocycline still showed significantly lower expression of GLAST versus saline-treated animals (87.0 ± 7.9% of control), although this change was not as significant as in animals treated with bortezomib alone.
Figure 5.
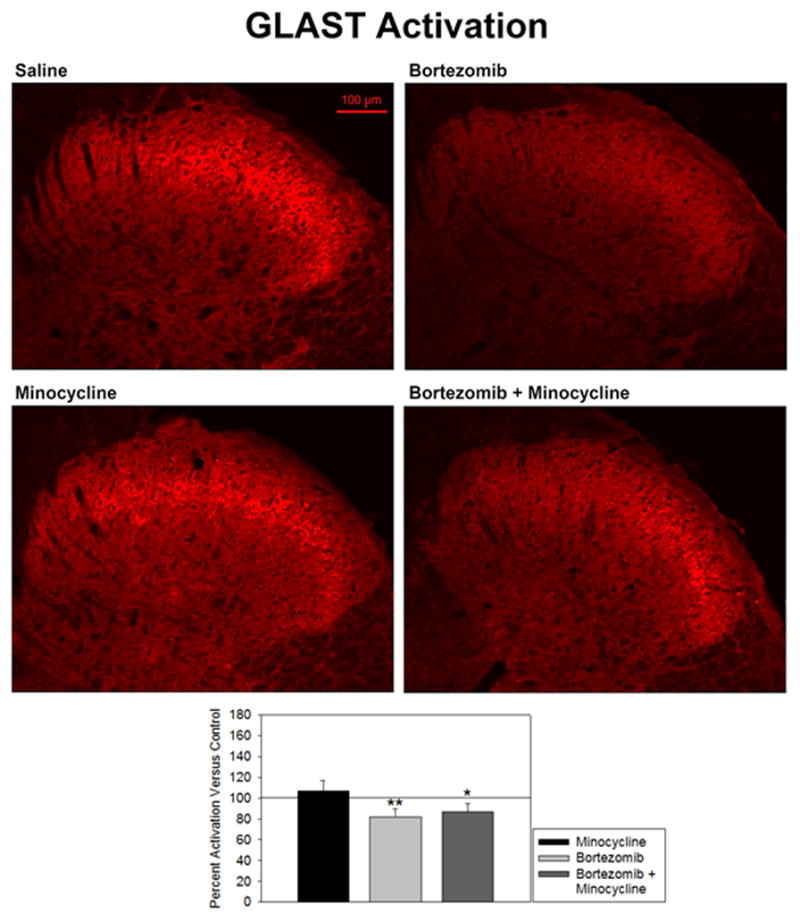
Activation of GLAST was expressed as percent fluorescence intensity versus mean fluorescence intensity of control. Treatment of animals with bortezomib (n=5) significantly decreased expression of GLAST in the spinal dorsal horn versus saline-treated controls (n=6). This was partially prevented by co-treatment with minocycline (n=5), but decrease of GLAST was still significant in these animals. Minocyline alone (n=5) did not affect GLAST activity.
5.6 Western Blotting
Western blotting data for the GLAST antibody yielded no significant differences between any of the treatment groups (Fig. 6). Tissue from animals treated with minocycline alone showed slightly lower staining than saline-treated animals, but these differences were not significant. However, connexin 43 expression showed significant changes in the expression of its different phosphorylation states (Fig. 7). The different bands detected by the connexin 43 antibody corresponded to non-phosphorylated (NP), phosphorylated (P1), and phosphorylated (P2) forms of connexin 43. There was no significant difference between groups in the NP bands or the P2 bands. There was, however, a significantly higher expression of phosphorylated (P1) connexin 43 in both the groups treated with bortezomib alone (494.7 ± 103.7% of control) and those treated with both bortezomib and minocycline (308.0 ± 85.3% of control). The antibody used was for the detection of overall connexin 43 expression, rather than phosphorylated connexin 43 selectively. It is therefore unlikely that the observed effects are a result of preferential staining of one state over another. The tissue corresponding to bortezomib-treated animals also appeared to have greater overall staining, but this was not compared quantitatively.
Figure 6.
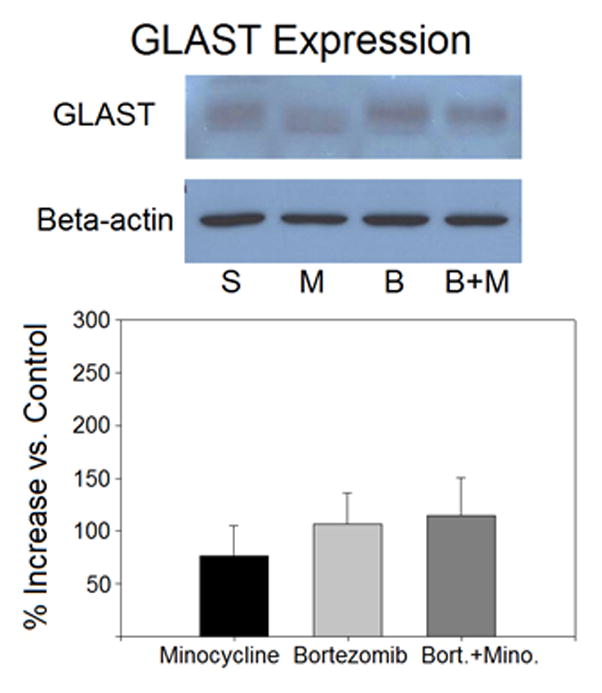
Western blotting data for GLAST did not indicate significant differences between treatment groups. Treatment groups included saline (n=4), minocycline (n=4), bortezomib (n=4), and bortezomib + minocycline (n=4). Ratios for each band were calculated as GLAST intensity over beta-actin intensity within the same lane. Band intensity was expressed as a percent increase of the average ratio for a treatment group versus the average ratio of saline-treated controls.
Figure 7.
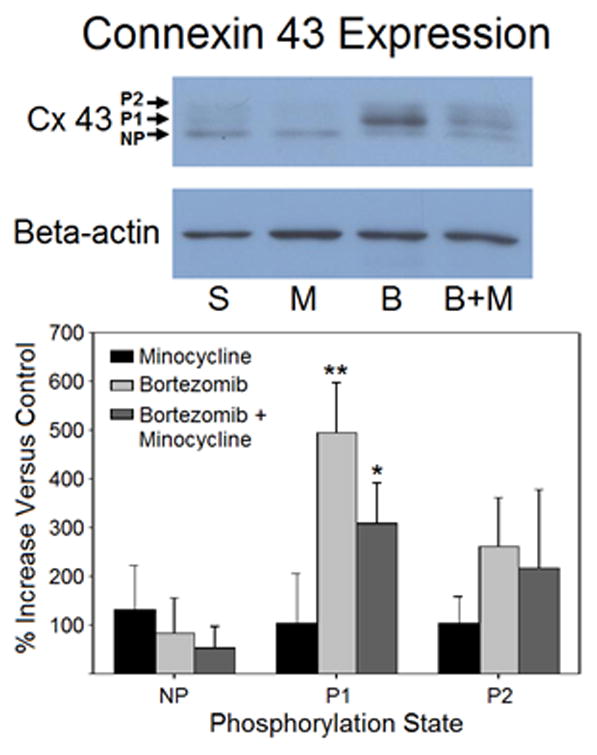
Western blotting data for connexin 43 indicated significant differences between treatment groups. Treatment groups included saline (n=3), minocycline (n=4), bortezomib (n=3), and bortezomib + minocycline (n=4). Bands for individual phosphorylation states indicated an increase in the P1 phosphorylation state of connexin 43 in animals treated with bortezomib or bortezomib + minocycline. Ratios for each band were calculated as connexin 43 intensity over beta-actin intensity within the same lane. Band intensity was expressed as a percent increase of the average ratio for a treatment group versus the average ratio of saline-treated controls.
6.0 Discussion
The data in the present study show alterations in astrocytic connexins and glutamate transporters in animals treated with bortezomib. Previous work in our lab has shown a clear involvement of astrocytes in various forms of CIPN, and these changes provide a possible means by which astrocytes may contribute to the phenotype. First, GFAP staining and von Frey mechanical sensitivity were used to verify the bortezomib-induced phenotype. Development of mechanical hypersensitivity and astrocyte activation were in line with previous data from our lab (Robinson et al, 2014b; Robinson et al, 2014a). The use of ceftriaxone, carbenoxolone, and minocycline were each successful in preventing the behavioral changes. However, it is important to note that the use of either intraperitoneal or intrathecal drug administration could impact activity in the dorsal root ganglia. It is therefore possible that the aforementioned effects of drugs in this study could occur by means of affecting this activity, rather than the dorsal horn itself. The fact that bortezomib does not cross the blood-brain barrier indicates that its primary site of activity in CIPN is indeed more likely at the dorsal root ganglia. The possibility therefore must not be ruled out that preventative drugs used in the present study may act upstream of the dorsal horn.
For immunohistochemistry, the 30-day time point in this model was selected because it corresponds to peak behavioral sensitivity. In addition to the activation of GFAP, increased expression of connexin 43 was observed. The changes in the levels of these two markers were nearly identical. This increase in connexin 43 expression suggests possible increased flow of calcium ions between astrocytes that may result in glutamate release. A possible point of interest for future experimentation would be to verify whether this glutamate release occurs in vitro. Alternatively, in vivo electrophysiology in rats treated with gap junction decouplers like carbenoxolone could be employed to see whether this inhibition of connexin 43 prevents potentiated responses to stimuli. However, it is possible that the connexins are responsible for the transport of other intercellular signal molecules. What exactly they may contribute in the context of this model is currently unknown, but the data in the present study show a clear correlation between peak hyperalgesia and connexin activity.
The involvement of glutamate transporters in the present study was less clear-cut than that of connexin 43. While GLAST was downregulated in the immunohistochemistry data in this model, no change was observed in GLT-1 activity. It is unknown why one form of astrocytic glutamate transporter would be depressed without another. However, the decreased levels of at least one form of glutamate transporter at the peak of the behavioral phenotype suggests a mechanism for persistent sensations and persistent after-discharges observed in other studies from our lab (Robinson et al, 2014b; Robinson et al, 2014a). One possibility for the disparity between GLT-1 and GLAST is that GLT-1 activity may be involved at a different time point. The possibility of early and late forms of glutamate transporter downregulation has not been explored to the knowledge of the author, but another study from our lab has shown GLT-1 and GLAST downregulation at earlier time points that returns to baseline at later time points (Zhang et al, 2012). The successful use of ceftriaxone, which is generally considered to be specific to GLT-1 upregulation (Lee et al, 2008), would suggest that early GLT-1 changes are a strong possibility. It is therefore possible that a follow-up study examining earlier time points in bortezomib-treated rats would yield a similar result of early downregulation of GLT-1. The early downregulation of glutamate transporters would then suggest the induction of central sensitization that persists long after the changed expression resolves.
The Western blotting data both validated and contradicted the changes observed in immunohistochemistry. The lack of any change observed in GLAST is difficult to explain, but it is possible that the means of processing the tissue for Western blotting washed out any changes that may have occurred. Considering that the change in expression observed in immunohistochemistry was less than 20% and surveyed only in the first two laminae of the dorsal horn within the gray matter, it is possible that inclusion of the entire dorsal half of the spinal cord for Western blotting diluted any changes that may have occurred in a much smaller region. The changes observed in connexin 43 Western blotting, on the other hand, were of particular interest. There was a clear shift in phosphorylation of connexin 43 in bortezomib-treated animals. The P1 phosphorylation state of connexin 43 corresponds to increased trafficking of connexin 43 from the cytoplasm to the membrane (Solan and Lampe, 2007). This would indicate that there is not only an apparent increase in overall expression of the protein, but that it is also being transported for use within a greater number of gap junctions, as well.
The activity of minocycline in the present study showed an overall prevention of the observed phenotype. There was a complete abrogation of the changes in behavior, GFAP, and connexin 43, as well as a partial abrogation of the changes observed in GLAST. Furthermore, the phosphorylation states of connexin 43 in animals treated with both bortezomib and minocycline indicate that minocycline exerted its effects on connexin 43 activity by a partial prevention of the increase in expression, rather than by altering its post-translational modification. This suggests that minocycline acts on a target upstream of the regulation of connexin 43. The behavioral data obtained from animals co-treated with carbenoxolone or ceftriaxone showed prevention of the phenotype in these animals, as well. Further studies using these three agents may provide important information on the mechanisms of CIPN as well as potential therapeutic strategies for its prevention.
Previous studies from our lab have clearly demonstrated a correlation between astrocyte activation and the development of the CIPN behavioral phenotype. The changes to connexin and glutamate transporter expression observed now in bortezomib-treated animals provide mechanistic support for direct astrocytic involvement. In conjunction with similar studies in oxaliplatin and paclitaxel, there is increasing evidence for a unified pathway for the development of CIPN. However, upstream mechanisms for astrocyte activation are not well understood. A recent study from our lab has demonstrated increases in TLR4 and MyD88 in paclitaxel (Li et al., 2014). This suggests that damage-associated molecular patterns from outside of the spinal cord may activate astrocytes. It also strongly suggests increases in proinflammatory cytokines released from astrocytes. This could have a direct impact on glutamate transporter expression, and it could further sensitize surrounding neurons to glutamate signaling directly. Furthermore, TLR4 activation is capable of upregulating connexin 43 (Aguirre et al, 2013), which strongly suggests a possible single site for all of the activity observed in gap junction, glutamate transporter, and cytokine regulation. However, whether or not TLR4 is activated by chemotherapy treatment as a whole has yet to be verified outside of the paclitaxel model. Future studies should be conducted to verify whether this occurs in the bortezomib model, as well. The activity of astrocytes provides a very promising point for studies to elaborate on an overall pathway for CIPN, as well as for the development of therapeutic strategies for its prevention.
2.0 Highlights.
Mechanical sensitivity in bortezomib-treated rats was prevented by co-treatment with minocycline, carbenoxolone, or ceftriaxone.
Connexin 43 expression was increased, and GLAST expression was decreased alongside increased expression of GFAP.
Acknowledgments
This work was supported by NCI grant CA124787 and NIH grant NS046606 and the H.E.B. Professorship in Cancer Research.
Footnotes
There are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
8.0 Reference List
- 1.Aguirre A, Maturana CJ, Harcha PA, Sáez JC. Possible involvement of TLRs and hemichannels in stress-induced CNS dysfunction via mastocytes, and glia activation. Mediators Inflam. 2013;2013:893521. doi: 10.1155/2013/893521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Argyriou AA, Bruna J, Marmiroli P, Cavaletti G. Chemotherapy-induced peripheral neurotoxicity (CIPN): an update. Crit Rev Oncol Hematol. 2012;82:51–77. doi: 10.1016/j.critrevonc.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 3.Argyriou AA, Kyritsis AP, Makatsoris T, Kalofonos HP. Chemotherapy-induced peripheral neuropathy in adults: a comprehensive update of the literature. Cancer Manag Res. 2014;6:135–147. doi: 10.2147/CMAR.S44261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyette-Davis J, Dougherty PM. Protection against oxaliplatin-induced mechanical hyperalgesia and intraepidermal nerve fiber loss by minocycline. Exp Neurol. 2011;229:353–357. doi: 10.1016/j.expneurol.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyette-Davis J, Xin W, Zhang H, Dougherty PM. Intraepidermal nerve fiber loss corresponds to the development of Taxol-induced hyperalgesia and can be prevented by treatment with minocycline. Pain. 2011;152:308–313. doi: 10.1016/j.pain.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cata JP, Weng HR, Chen JH, Dougherty PM. Altered discharges of spinal wide dynamic range neurons and down-regulation of glutamate transporter expression in rats with paclitaxel-induced hyperalgesia. Neuroscience. 2006;138:329–338. doi: 10.1016/j.neuroscience.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain following spinal cord injury. Brain. 2014;137:2193–2209. doi: 10.1093/brain/awu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen MJ, Kress B, Han X, Moll K, Peng W, Ji RR, Nedergaard M. Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. GLIA. 2012;60:1660–1670. doi: 10.1002/glia.22384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Devinsky O, Vessani A, Najjar S, de Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36:174–184. doi: 10.1016/j.tins.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 10.Ferrier J, Pereira V, Busserolles J, Authier N, Balayssac D. Emerging trends in understanding chemotherapy-induced peripheral neuropathy. Curr Pain Headache Rep. 2013;17:364. doi: 10.1007/s11916-013-0364-5. [DOI] [PubMed] [Google Scholar]
- 11.Giaume C, Liu X. From a glial syncytium to a more restricted and specific glial networking. J Physiol Paris. 2012;106:34–39. doi: 10.1016/j.jphysparis.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Hansen RR, Malcangio M. Astrocytes - multitaskers in chronic pain. Eur J Pharmacol. 2013;716:120–128. doi: 10.1016/j.ejphar.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 13.Homkajorn B, Sims NR, Muyderman H. Connexin 43 regulates astrocytic migration and proliferation in response to injury. Neurosci Lett. 2010;486:197–201. doi: 10.1016/j.neulet.2010.09.051. [DOI] [PubMed] [Google Scholar]
- 14.Juszczak GR, Swiergiel AH. Properties of gap junction blockers and their behavioral, cognitive and electrophysiological effects: animal and human studies. Prog Neuropsychopharmacol Biol Psychiat. 2009;33:181–198. doi: 10.1016/j.pnpbp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 15.Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK, Dash R, Dasgupta S, Barral PM, Hedvat M, Diaz P, Reed JC, Stebbins JL. Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol. 2011;226:2484–2493. doi: 10.1002/jcp.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SG, Su ZZ, Embad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB. Mechanisms of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem. 2008;283:13116–13123. doi: 10.1074/jbc.M707697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.López-Bayghen E, Ortega A. Glial glutamate transporters: new actors in brain signaling. IUBMB Life. 2011;63:816–823. doi: 10.1002/iub.536. [DOI] [PubMed] [Google Scholar]
- 18.Malarkey EB, Parpura V. Mechanisms of glutamate release from astrocytes. Neurochem Int. 2008;52:142–154. doi: 10.1016/j.neuint.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matute C, Domercq M, Sanchez-Gomez MV. Glutamate-mediated glial injury: mechanisms and clinical importance. GLIA. 2006;53:212–224. doi: 10.1002/glia.20275. [DOI] [PubMed] [Google Scholar]
- 20.Miltenburg NC, Boogerd W. Chemotherapy-induced neuropathy: a comprehensive survey. Cancer Treat Rev. 2014;40:872–882. doi: 10.1016/j.ctrv.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 21.Nakagawa T, Kaneko S. Spinal astrocytes as therapeutic targets for pathological pain. J Pharmacol Sci. 2010;114:347–353. doi: 10.1254/jphs.10r04cp. [DOI] [PubMed] [Google Scholar]
- 22.Pannasch U, Rouach N. Emerging role for astroglial networks in information processing: from synapse to behavior. Trends Neurosci. 2013;36:405–417. doi: 10.1016/j.tins.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 23.Robinson CR, Zhang H, Dougherty PM. Altered discharges of spinal wide dynamic range neurons parallel the behavioral phenotype shown by rats with bortezomib related chemotherapy induced peripheral neuropathy. Brain Res. 2014a;1574:6–13. doi: 10.1016/j.brainres.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson CR, Zhang H, Dougherty PM. Astrocytes, but not microglia, are activated in oxaliplatin and bortezomib-induced peripheral neuropathy. Neuroscience. 2014b;274:308–317. doi: 10.1016/j.neuroscience.2014.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solan JL, Lampe PD. Key connexin43 phosphorylation events regulate the gap junction life cycle. J Membr Biol. 2007;217:35–41. doi: 10.1007/s00232-007-9035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tabernero A, Medina JM, Giaume C. Glucose metabolism and proliferation in glia: role of astrocytic gap junctions. J Neurochem. 2006;99:1049–1061. doi: 10.1111/j.1471-4159.2006.04088.x. [DOI] [PubMed] [Google Scholar]
- 27.Theis M, Giaume C. Connexin-based intercellular communication and astrocyte heterogeneity. Brain Res. 2012;1487:88–98. doi: 10.1016/j.brainres.2012.06.045. [DOI] [PubMed] [Google Scholar]
- 28.Weng HR, Cordella JV, Dougherty PM. Changes in sensory processing in the spinal dorsal horn accompany vincristine-induced hyperalgesia and allodynia. Pain. 2003;103:131–138. doi: 10.1016/s0304-3959(02)00445-1. [DOI] [PubMed] [Google Scholar]
- 29.Yi JH, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int. 2006;48:394–403. doi: 10.1016/j.neuint.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Yoon SY, Robinson CR, Zhang H, Dougherty PM. Gap junction protein connexin 43 is involved in the induction of oxaliplatin-related neuropathic pain. J Pain. 2013;14:205–214. doi: 10.1016/j.jpain.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H, Yoon SY, Zhang H, Dougherty PM. Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of paclitaxel-induced painful neuropathy. J Pain. 2012;13:293–303. doi: 10.1016/j.jpain.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]