

Abstract
Ross River virus (RRV), chikungunya virus (CHIKV), and related alphaviruses cause debilitating polyarthralgia and myalgia. Mouse models of RRV and CHIKV have demonstrated a role for the adaptive immune response in the control of these infections. However, questions remain regarding the role for T cells in viral control, including the magnitude, location, and dynamics of CD8+ T cell responses. To address these questions, we generated a recombinant RRV expressing the H-2b-restricted gp33 determinant derived from the glycoprotein (gp) of lymphocytic choriomeningitis virus (LCMV) (“RRV-LCMV”). Utilizing tetramers, we tracked gp33-specific CD8+ T cells during RRV-LCMV infection. We found that acute RRV infection induces activation of CD8+ T cell responses in lymphoid and musculoskeletal tissues that peak from 10 to 14 days post-inoculation (dpi), suggesting that CD8+ T cells contribute to control of acute RRV infection. Mice genetically deficient for CD8+ T cells or wild-type mice depleted of CD8+ T cells had elevated RRV loads in skeletal muscle tissue, but not joint-associated tissues, at 14 dpi, suggesting that the ability of CD8+ T cells to control RRV infection is tissue-dependent. Finally, adoptively transferred T cells were capable of reducing RRV loads in skeletal muscle tissue of Rag1−/− mice, indicating that T cells can contribute to the control of RRV infection in the absence of B cells and antibody. Collectively, these data demonstrate a role for T cells in the control of RRV infection and suggest that the antiviral capacity of T cells is controlled in a tissue-specific manner.
INTRODUCTION
Ross River virus (RRV), chikungunya virus (CHIKV), and several related viruses are mosquito-transmitted, positive-sense, single-stranded RNA viruses in the Alphavirus genus of the Togaviridae family that cause a debilitating musculoskeletal inflammatory disease in humans (1). These globally distributed viruses cause endemic disease and, occasionally, large epidemics. In 1979–1980, RRV spread from Australia to multiple islands in the Pacific Region, including Fiji, the Cook Islands, and America Samoa, resulting in more than 60,000 cases (2–5). After several decades of relative absence, CHIKV re-emerged in both Africa and Asia, causing large outbreaks and becoming a substantial global public health concern (6). Due to international travel, imported CHIKV cases have been reported in nearly 40 countries including the USA, Japan, and several European countries. In addition, local transmission of CHIKV has been documented for the first time in several locations, including Italy, France, New Caledonia, Papua New Guinea, and Yemen (7–11). Most recently, autochthonous transmission of CHIKV has occurred for the first time in the W. Hemisphere, with over 750,000 confirmed and suspected cases reported from a number of Caribbean islands as well as a few countries in Central America and in South America (12, 13).
The hallmark clinical manifestation following infection with arthritogenic alphaviruses is severe polyarthralgia that mainly affects the peripheral small joints (1, 14–16). Other classical symptoms include the sudden onset of fever, myalgias, an impaired ability to ambulate, and sometimes rash (1, 17). Surveys conducted for RRV-infected patients have shown that symptoms for most patients progressively resolve over 3–6 months (16). Up to 60% of persons infected with CHIKV complain of musculoskeletal pain for months to years, although the cause of these long-lasting symptoms is unclear (1, 17). Atypical outcomes of CHIKV infection occur and include neurologic manifestations, myocarditis, and death; these outcomes are associated with age and underlying medical conditions (18). There are currently no licensed antivirals or vaccines for any of the arthritogenic alphaviruses; treatment is limited to supportive care with analgesics and anti-inflammatory drugs (19).
A number of studies have identified the importance of the host innate immune responses, particularly the type I IFN response, for controlling arthritogenic alphavirus infections (20–28). In addition, studies in humans and animal models have demonstrated that antibodies are important mediators of protection. In humans, the early appearance of anti-CHIKV IgG3 antibodies was associated with efficient virus clearance from the serum (29). Similarly, CHIKV-infected Rag1−/−, Rag2−/−, and Ighm−/− mice, but not Cd4−/− or Cd8−/− mice, have increased and sustained viremia, further supporting an important role for antibodies in clearance of virus from the serum (30–32). In addition, prophylaxis via passive transfer of human or murine immune serum, virus-specific IgG, or virus-specific monoclonal antibodies to highly susceptible Ifnar1−/− mice or WT mice can protect against RRV or CHIKV-induced mortality and musculoskeletal disease (31, 33–40). The importance of antibodies during alphavirus infection was also demonstrated in studies in mice with neurotropic Sindbis virus (SINV), which showed that antibody is important for the clearance of infectious virus (41), although replication competent RNA remained for months after antibody treatment in SINV-infected immunodeficient SCID mice (42).
In contrast to the role of antibodies, the kinetics of T cell activation and the role of T cells in the control of alphavirus infection in musculoskeletal tissues are poorly defined. During acute CHIKV infection in humans and macaques, an increase in activated peripheral T cells has been reported (43–45). Additionally, it was recently shown that CD4+ and CD8+ T cells from CHIKV-infected patients produce IFN-γ in response to several CHIKV antigens, with responses against E2 being the highest in magnitude (46). T cells are also an important component of the inflammatory infiltrate: T cells have been found in synovial fluid as well as synovial and muscle tissue biopsies collected from RRV- and CHIKV-infected patients (44, 47, 48). Consistent with these findings, studies in RRV- and CHIKV-infected mice have shown that CD4+ and CD8+ T cells infiltrate infected muscle and joint tissue (49–52). In mice, CD4+ T cells were shown to contribute to CHIKV-induced foot swelling and musculoskeletal tissue injury (32); however, the extent to which either CD4+ or CD8+ T cells contribute to the control and clearance of alphavirus infections in musculoskeletal tissues has not been investigated. T cells are critical for the clearance of viral RNA from central nervous system tissues of mice infected with neuroadapted SINV (53–55), and RRV-specific CD8+ T cells derived from previously infected mice mediated clearance of RRV from persistently-infected macrophages in vitro (56), suggesting that T cells may contribute to controlling arthritogenic alphavirus infections in vivo.
In this study, utilizing a relevant mouse model of arthritogenic alphavirus-induced rheumatological disease (49), we sought to characterize the kinetics of the CD8+ T cell response and investigate the role of CD8+ T lymphocytes in the control of arthritogenic alphavirus infection in musculoskeletal tissues. To investigate antigen-specific CD8+ T cell responses, we generated a recombinant RRV expressing the CD8 (gp33-41) immunodominant epitope of the well-studied lymphocytic choriomeningitis virus (LCMV). Utilizing this system, we found that RRV infection induces virus antigen-specific CD8+ T cell responses, characterized by expansion, modulated expression of cell surface proteins associated with activation, and cytokine production, in both lymphoid and musculoskeletal tissues. Furthermore, we found that CD8α−/− mice and mice depleted of CD8+ T cells had significantly increased viral RNA levels in muscle tissue compared to control mice. Remarkably, the levels of viral RNA in joint-associated tissues was unaffected by genetic deletion or antibody-mediated depletion of CD8+ T cells. Consistent with these data, Rag1−/− mice reconstituted with CD8+ and CD4+ T cells had significantly decreased viral loads in muscle tissue but not joint-associated tissue, suggesting that T cells can contribute to viral control in the absence of B cells and antibody. These studies provide important new insights into the CD8+ T cell response during arthritogenic alphavirus infection.
MATERIALS AND METHODS
Ethics Statement
All mouse studies were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All mouse studies were performed at the University of Colorado Anschutz Medical Campus (Animal Welfare Assurance #A 3269-01) using protocols approved by the University of Colorado Institutional Animal Care and Use Committee. All studies were performed in a manner designed to minimize pain and suffering in infected animals.
Viruses
The T48 stain of RRV was isolated from Aedes vigilax mosquitoes in Queensland, Australia (57). Prior to cDNA cloning, the virus was passaged 10 times in suckling mice, followed by two passages on Vero cells (58, 59). Lymphocytic choriomeningitis virus (LCMV) Armstrong (clone 53b) was originally obtained from Dr. M. Oldstone, and stocks were prepared by a single passage on BHK-21 cells. Plaque assays for determination of LCMV titers were performed as previously described (60).
The recombinant “RRV-LCMV” was generated by inserting a tandem sequence, similar in design to a sequence inserted in the influenza virus genome that encodes the LCMV CD8 T cell receptor epitope gp33-41 (KAVYNFATC) and CD4 T cell receptor epitope gp61–80 (GLKGPDIYKGVYQFKSVEFD) (61) in-frame with the RRV structural polyprotein. The LCMV peptides were linked to 19 amino acids of the 2A protease of foot-and-mouth disease virus (FMDV), and this entire sequence was synthesized by Genewiz. A plasmid encoding the full-length T48 cDNA clone along with green fluorescent protein in-frame (pGH1.2) (kindly provided by Mark T. Heise, University of North Carolina) was mutagenized at RRV nucleotide 6476 to remove an ApaI restriction enzyme site. The pGH1.2ΔApa16476 plasmid was digested with AgeI to release a 1426 base pair (bp) AgeI-AgeI fragment. The vector lacking this fragment was religated (pGH1.2ΔApaI6476.1), which created a vector with a single ApaI and single AgeI site. The pGH1.2ΔApaI6476.1 plasmid was digested with AgeI and ApaI and ligated with the AgeI-ApaI–digested Genewiz construct. The AgeI-AgeI fragment was re-inserted and screened for directionality, resulting in the LCMV and FMDV 2A protease sequences inserted in-frame between the capsid and E3 genes of RRV. During replication, the LCMV-2A fusion protein is synthesized as a component of the viral structural polyprotein that is then released by intramolecular cleavages mediated by the RRV capsid and FMDV 2A proteases. Stocks of infectious RRV-LCMV and RRV were generated from cDNAs and titered by direct plaque assay on BHK-21 cells as previously described (62).
Mice
C57BL/6 wild-type (stock # 000664), Rag1−/− (stock # 002216), and CD8α−/− (stock # 002665) mice were obtained from The Jackson Laboratory and bred in house. Animal husbandry and experiments were performed in accordance with all University of Colorado School of Medicine Institutional Animal Care and Use Committee guidelines. All mouse studies were performed in an animal biosafety level 3 laboratory. For LCMV infections, six-to-ten week-old mice were inoculated with 2 × 105 PFU via the intraperitoneal (i.p.) route and monitored daily. For RRV infection, three-to-four week old mice were used. In contrast to older mice in which RRV replication and dissemination is restricted, three-to-four week old mice develop a disseminated infection and musculoskeletal disease signs similar to RRV disease in humans (63). Mice were inoculated in the left rear footpad with 103 PFU of wild-type RRV or RRV-LCMV in diluent (PBS/1% bovine calf serum) in a 10-μl volume. Mock-infected animals received diluent alone. Mice were monitored for disease signs and weighed at 24-h intervals. Disease scores were determined by assessing grip strength, hind limb weakness, and altered gait, as previously described (49). On the termination day of each experiment, mice were sacrificed by exsanguination, blood was collected, and mice were perfused by intracardial injection of 1x PBS. PBS-perfused tissues were removed by dissection and homogenized in TRIzol Reagent (Life Technologies) for RNA analysis with a MagNA Lyser (Roche). Alternatively, quadriceps muscles were dissected, minced, and incubated for 1.5 h with vigorous shaking at 37°C in digestion buffer (RPMI 1640, 10% FBS, 15 mM HEPES, 2.5 mg/ml Collagenase Type 1 [Worthington Biochemical], 1.7 mg/ml DNase I [Roche], 1x gentamicin [Life Technologies], 1% penicillin/streptomycin). Following digestion, cells were passed through a 100-μm cell strainer (BD Falcon) and banded on Lympholyte-M (Cedarlane Laboratories) to isolate infiltrating leukocytes. Additionally, spleens and draining popliteal lymph nodes were dissected from mice and passed through a 100-μm cell strainer. Following red blood cell lysis (spleens only), cells were washed in wash buffer (1x PBS, 15 mM HEPES, 1x gentamicin, 1% penicillin/streptomycin), and total viable cells were determined by trypan blue exclusion. Sera samples were titered by direct plaque assay on BHK-21 cells.
Flow cytometry and adoptive transfer
Leukocytes isolated from enzymatically digested tissues were incubated with anti-mouse FcγRII/III (2.4G2; BD Pharmingen) for 20 min on ice to block nonspecific Ab binding and then stained in FACS staining buffer (1x PBS, 2% FBS) with the following Abs: anti-CD3-fluorescein isothiocyanate (FITC, clone 145-2C11), anti-CD3-allophycocyanin (APC, 145-2C11), anti-CD4-Pacific Blue (RM4-5), anti-CD8α-phycoerythrin (PE, 53-6.7), anti-CD44-PE-Cy7 (IM7), anti-CD62L-PE-Cy7 (MEL-14), anti-TNF-α-FITC (MP6-XT22), anti-IFN-γ-PE-Cy7 (XMG1.2), anti-IL-2-APC (JES6-5H4), anti-B220-PE-Cy7 (RA3-6B2), and anti-CD19-FITC (6D5) (all from BioLegend); anti-CD8β-PE (H35-17.2), anti-CD11b-APC (M1/70), anti-F4/80-FITC (BM8), and anti-Gr-1-PE-Cy7 (RB6-8C5) (all from eBioscience). gp33-APC H-2Db KAVYNFATM tetramer was kindly provided by the National Institutes of Health Tetramer Core Facility. Cells were fixed overnight in 1% paraformaldehyde and analyzed on an LSR II using FACSDiva software (Becton Dickinson). Further analysis was done using FlowJo Software (Tree Star). Doublets were excluded using forward-scatter height and width parameters.
For intracellular cytokine analysis, spleen cells from Rag1−/− mice were used as antigen presenting cells (APCs) and pulsed with or without 1 μg of gp33 peptide per 5 × 106 APCs for 1 h at 37°C in RPMI containing 7% FBS. Muscle-infiltrating leukocytes from RRV-LCMV-infected mice were isolated at 10 dpi. 0.5 × 106 APCs (pulsed with or without gp33 peptide) per well were added to a 96 well round-bottom plate in 100 μl RPMI containing 7% FBS and Brefeldin A followed by addition of muscle-infiltrating leukocytes in 100 μl RPMI containing 7% FBS. Cells were incubated together for 5 h in the presence of Brefeldin A to prevent cytokine release. Cells were harvested, and incubated with anti-mouse FcγRII/III (2.4G2) for 20 min on ice followed by surface marker staining in FACS buffer for an additional 20 min on ice. After one wash step, cells were fixed and permeabilized in a 1% paraformaldehyde and saponin solution for 15 min at room temperature. Cells were washed with PBS containing saponin and then stained for intracellular cytokines for 45 min on ice in PBS with saponin. Finally, cells were washed 1X with PBS containing saponin, 1X with FACS buffer, and then fixed overnight in 1% paraformaldehyde. Muscle-infiltrating leukocytes from WT RRV-infected mice and splenocytes from a LCMV Armstrong-infected mouse were used as controls for the specificity of restimulation.
For T cell adoptive transfer experiments, CD4 plus CD8 T cells or CD8 T cells alone were isolated from the spleens of naïve wild-type C57BL/6 mice via negative selection using a pan-T cell isolation kit or CD8 T cell isolation kit, respectively (Miltenyi Biotec). Following isolation, cells were counted and 2.5 × 106 CD4 plus CD8 T cells or 1 × 106 CD8 T cells were resuspended in RPMI containing 2% FBS in a total volume of 200 μl for i.p. injection into Rag1−/− mice one day prior to RRV infection. Control Rag1−/− mice received 200 μl of media alone. Of the transferred T cells, ≥ 95% of the cells were CD3+. In the pan T cell groups, ~60% of the CD3+ T cells were CD4+ and ~35% were CD8+; in the CD8 T cell groups, ≥ 95% of the CD3+ T cells were also CD8+. Additionally, ≤ 1% of CD19+ B cells remained in the injected cell preparations. T cell transfer was confirmed by flow cytometric analysis of spleen and muscle tissue at 14 dpi as described above for the presence of cells staining negatively for B220 (RA3-6B2) and Gr-1 (RB6-8C5) and positively for CD3 (145-2C11) and CD4 (RM4-5) or CD8α (53–6.7).
Quantitative RT-PCR
RNA was isolated using a PureLink RNA Mini Kit (Life Technologies). Absolute quantification of viral RNA was performed as previously described (62). Briefly, a sequence-tagged (small caps) RRV-specific RT primer (4415-5'-ggcagtatcgtgaattcgatgcAACACTCCCGTCGACAACAGA-3') was used for reverse transcription. A tag sequence-specific reverse primer (5'-GGCAGTATCGTGAATTCGATGC-3') was used with an RRV sequence-specific forward primer (4346 5'-CCGTGGCGGGTATTATCAAT-3') and an internal TaqMan probe (4375 5'-ATTAAGAGTGTAGCCATCC-3') during qPCR to enhance specificity. To create standard curves, 10-fold dilutions, from 108 to 100 copies of RRV genomic RNAs, synthesized in vitro, were spiked into RNA from BHK-21 cells, and reverse transcription and qPCR were performed in an identical manner.
Histological analysis
At 10 dpi, mice were sacrificed and perfused by intracardial injection of 1X PBS, and the left gastrocnemius muscle was dissected and fixed in 4% paraformaldehyde (pH 7.3). Tissues were embedded in paraffin, and 5-μm sections were prepared. To assess the degree of tissue inflammation, tissue sections were stained with H&E and evaluated by light microscopy by a trained pathologist. Each section was scored in a blinded manner using the following scoring system: 0, absent; 1, minimal, <10% of tissue affected; 2, mild, 10–24% of tissue affected; 3, moderate, 25–39% of tissue affected; 4, marked, 40–59% of tissue affected; 5, severe, >60% of tissue affected.
Creatine Kinase Activity Assay
Creatine kinase activity in the serum of mock- and RRV-inoculated mice was determined by utilizing a colorimetric assay; the manufacturer's instructions were followed (Stanbio Laboratory).
Statistical Analysis
All data were analyzed using GraphPad Prism 5 software. Data were evaluated for statistically significant differences using either a two-tailed, unpaired t-test with or without Welch's correction, a one-way analysis of variance (ANOVA) test followed by Tukey's multiple comparison test, or a two-way ANOVA followed by a Bonferroni multiple comparison test. A P-value < 0.05 was considered statistically significant. All differences not specifically indicated to be significant were not significant (P > 0.05).
RESULTS
RRV-infected Rag1−/− mice have increased viral loads in musculoskeletal tissues
Previous studies in mice suggested that the adaptive response is dispensable for the development of acute RRV-induced musculoskeletal clinical disease signs (49, 64) and for control of acute RRV infection (65). To evaluate a role for the adaptive immune response at later times post-infection, we quantified viral RNA loads in the quadriceps muscle and ankle tissue at 7 and 14 dpi of RRV-inoculated WT and Rag1−/− mice which lack functional T and B lymphocytes (66). RRV-inoculated Rag1−/− mice had significantly higher viral loads in both muscle tissue (13.8-fold increase, P < 0.001) and joint tissue (6-fold increase, P < 0.001) at 7 dpi and at 14 dpi (quadriceps: 31.7-fold increase, P < 0.001; ankle: 34.5-fold increase, P < 0.001) (Figure 1A and B). These data suggest that the adaptive immune response is important for the control of RRV infection.
Figure 1. Rag1−/− mice have increased RRV loads compared to WT mice in musculoskeletal tissue at 7 and 14 dpi.
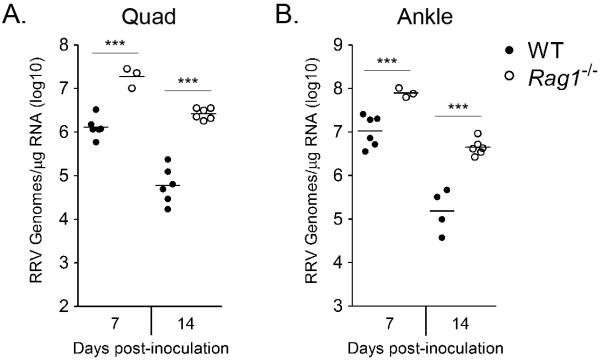
Three-to-four week-old C57BL/6J WT (7 dpi, n = 6; 14 dpi, n = 6) and Rag1−/− (7 dpi, n = 3; 14 dpi, n = 6) mice were inoculated with 103 pfu by injection into the left rear footpad. At 7 and 14 dpi, mice were sacrificed, perfused by intracardial injection with 1x PBS, and total RNA was isolated from the (A) right quadriceps muscle and (B) right ankle/foot. RRV genomes were quantified by absolute RT-qPCR as described in the Materials and Methods. Horizontal bars indicate the mean. *** P < 0.001, as determined by one-way ANOVA followed by Tukey's multiple comparison test.
Recombinant RRV-LCMV behaves similarly to WT virus in vitro and in vivo
Our experiments in Rag1−/− mice did not distinguish the role of T versus B lymphocytes in controlling RRV infection. Several studies have evaluated the role of antibodies in control of arthritogenic alphavirus infection; however, the role of T cells in control of these infections is less clear. Due to the availability of reagents such as tetramers and TCR transgenic mice that recognize LCMV-specific T cells and epitopes, respectively, previous groups have generated recombinant viruses that express the immunodominant CD8 (gp33) and/or CD4 (gp61) epitopes of LCMV to investigate antigen-specific T cell responses during infection (61, 67, 68). To take advantage of these immunological tools, we generated a recombinant Ross River virus that contains a tandem sequence encoding the gp3333-41 and gp6161–80 peptides (61) in-frame with the RRV structural polyprotein, termed “RRV-LCMV” (Figure 2A). Specifically, the LCMV peptide sequences were linked to 19 amino acids of the 2A protease of foot-and-mouth disease virus (FMDV) and then inserted in-frame between the capsid and E3 genes of RRV. During replication, the LCMV-2A fusion protein is synthesized as a component of the viral structural polyprotein that is then released by intramolecular cleavages mediated by the RRV capsid and FMDV 2A proteases. This design was selected based on previous studies indicating that insertion of foreign sequences into alphavirus genomes in this manner results in minimal attenuation and enhanced genome stability (69, 70). An in vitro multistep growth curve was performed to compare WT RRV and RRV-LCMV growth in BHK-21 cells. Importantly, yields of infectious RRV-LCMV were similar to WT RRV at all time points (Figure 2B). We next compared the outcome of infection in vivo. Three-to-four week old C57BL/6 mice inoculated with RRV-LCMV gained significantly more weight than WT RRV-inoculated mice on days 7–10 pi, indicating some degree of attenuation of the recombinant virus (Figure 2C), as expected from previous studies utilizing this design (69, 70). However, musculoskeletal disease scores for RRV-LCMV-inoculated mice during the acute stage were not significantly different from WT RRV-inoculated mice (Figure 2D). To further compare RRV-induced disease, viral loads from mice inoculated with WT RRV and the recombinant RRV-LCMV were quantified 5 and 10 dpi in muscle (Figure 2E) and joint-associated tissues (Figure 2F). Viral loads in muscle tissue were similar at 5 dpi, although RRV-LCMV-infected mice had significantly reduced viral loads at 10 dpi compared to WT RRV-infected mice (8.9-fold decrease, P = 0.02) (Figure 2E), suggesting that the recombinant RRV-LCMV is cleared more efficiently. However, viral loads in ankle tissue of RRV-LCMV-inoculated mice were similar to WT RRV-inoculated mice at 10 dpi and slightly lower, albeit statistically significant, at 5 dpi, (Figure 2F). Consistent with these data, WT RRV and RRV-LCMV induced similar muscle inflammation and tissue damage during the acute phase of disease as seen in representative images of H&E-stained tissue sections derived from gastrocnemius muscle tissue at 10 dpi (Figure 2G), which were scored for the level of inflammation by a blinded pathologist (Figure 2H). Additionally, similar levels of creatine kinase activity, which is a marker of muscle tissue damage, were measured in sera from mice inoculated with WT RRV and RRV-LCMV at 7 and 10 dpi, the peak of acute disease (Figure 2I). Overall, these data demonstrate that the recombinant RRV-LCMV virus behaves similarly to WT RRV in cell culture and in mice.
Figure 2. Recombinant RRV-LCMV is similar to WT RRV in vitro and in vivo.

(A) Schematic of recombinant RRV-LCMV virus encoding the CD8 (gp33) and CD4 (gp61) immunodominant epitopes of the LCMV glycoprotein. (B) BHK-21 cells were inoculated with a MOI of 0.01 PFU/cell of WT RRV or RRV-LCMV, and infectious virus in the supernatant was titered at 0 (inoculum), 1, 3, 6, 12, and 24 hpi by direct plaque assay on BHK-21 cells. Three-to-four week-old C57BL/6J WT mice were mock-inoculated (PBS) (n = 3) or inoculated with 103 PFU of WT RRV (n = 7) or RRV-LCMV (n = 15) and assessed for (C) weight gain and (D) musculoskeletal disease signs including loss of gripping ability and altered gait at 24-hour intervals. Each data point represents the arithmetic mean ± SEM. * P < 0.05 for days 7−10 pi between WT RRV and RRV-LCMV groups as determined by two-way ANOVA followed by a Bonferroni multiple comparison test. (E, F) Three-to-four week-old C57BL/6J WT mice were or inoculated with 103 PFU of WT RRV or RRV-LCMV, and RRV genome levels in the right quadriceps muscle (E) and right ankle/foot (F) were analyzed by absolute RT-qPCR at 5 dpi (n = 4) and 10 dpi (n = 5). Horizontal bar represents the mean. * P = 0.02, ** P = 0.009, as determined by two-way, unpaired t-tests. (G) Five-μm paraffin-embedded sections were generated from the gastrocnemius muscles of mock (n = 2), WT RRV (n = 4), and RRV-LCMV (n = 5) infected mice at 10 dpi and H&E stained. (H) H&E-stained sections from 10 dpi were scored in a blinded manner for the degree of inflammation based on the following scale for the percentage of tissue affected: 0, absent (0%); 1, minimal (<10%); 2, mild (11–25%); 3, moderate (26–40%); 4, marked (41–60%); and 5, severe (>60%). Graphs represent the arithmetic mean ± SEM. (I) Three-to-four week-old C57BL/6J WT mice were mock-inoculated (n = 6) or inoculated with 103 PFU of WT RRV (7 dpi, n =7; 10 dpi, n = 11) or RRV-LCMV (7 dpi, n = 11; 10 dpi, n = 12). At 7 and 10 dpi, blood was collected for analysis of creatine kinase activity in the serum. Graphs represent the arithmetic mean ± SEM.
RRV infection induces virus antigen-specific T cells responses in lymphoid and musculoskeletal tissues
One value of the recombinant RRV-LCMV is that it allows the kinetics of virus-specific CD8+ T cell responses to be investigated via gp33 tetramer staining of isolated cell populations. Mice were inoculated with 103 PFU of RRV-LCMV and spleens, draining (left) popliteal lymph nodes, and quadriceps muscle tissues were taken at 5, 7, 10, and 14 dpi for analysis of gp33-specific CD8+ T cells. WT RRV-infected mice (10 dpi) and LCMV-Arm-infected mice (7 dpi) were used as negative and positive controls for gp33 tetramer staining, respectively. After gating on lymphocytes and excluding doublets (Fig. S1A), the gp33-specific CD8+ T cell population was quantified by gating on CD8+CD4− T cells followed by gating on gp33 tetramer-positive cells (Figure 3A and Fig. S1B). This gp33 tetramer-positive gate was set such that there were ≤ 0.1% of gp33+ cells from the CD8+CD4− population from mock or WT RRV-infected control samples for each tissue. Utilizing this gating scheme, we identified a small number of gp33-specific CD8+ T cells in lymphoid and muscle tissues of RRV-LCMV-infected mice starting at day 5 pi (Figure 3B and C and Fig. S1B). These virus-specific CD8+ T cells expanded, and a distinct population was readily detectable in both the spleen and muscle tissue at 7 and 10 dpi. By 14 dpi, abundant gp33-specific CD8+ T cells were still present but in diminished numbers in all tissues analyzed. These data demonstrate that RRV infection in mice induces a virus-specific CD8+ T cell response and that these cells traffic to the sites of infection. Of note, a significantly increased percentage of CD8+ T cells was identified in the spleen and muscle tissue, but not the pLN, of RRV-LCMV-infected mice compared to WT RRV-infected mice (spleen, P = 0.008; muscle, P = 0.003); however, the total number of CD8+ T cells were not significantly different in either tissue (Figure 3B). These data suggest that expression of the immunodominant gp33 epitope stimulated an increased CD8+ T cell response in mice infected with the recombinant RRV-LCMV compared to mice infected with WT RRV, although this did not affect the total CD8+ T cell numbers in the tissues.
Figure 3. Virus-specific CD8+ T cells expand in RRV-LCMV-infected mice.

Three-to-four week-old C57BL/6J mice were mock-inoculated (n = 3) or inoculated with 103 PFU of WT RRV (n = 4) or RRV-LCMV (5 dpi, n = 6; 7 dpi n = 9; 10 dpi, n = 9; 14 dpi, n = 6). At the indicated day post-inoculation, leukocytes were isolated from spleens, draining popliteal lymph nodes (pLN), and quadriceps muscles (following enzymatic digestion) for FACS analysis. Spleen cells from a mouse inoculated with LCMV-Armstrong i.p. and harvested on day 7 pi was used as a control for gp33 tetramer staining. (A) Representative flow plots from 10 dpi indicating the gating strategy to identify CD8+CD4−gp33+ T cells, after gating on lymphocytes and excluding doublets. (B) Frequency (top panel) and total number (bottom panel) of CD8+CD4− T cells and CD8+CD4−gp33+ T cells in the spleen, pLN, and muscle tissues. Data are combined from 2–3 independent experiments. Graphs represent the arithmetic mean ± SEM. ** P < 0.01, as determined by two-way, unpaired t-tests.
To determine the activation status of the virus-specific CD8+ T cells, we analyzed cell surface expression of T cell activation markers on gp33+CD8+ T cells from the spleen and muscle tissue during the peak of acute disease at 10 dpi, compared to mock splenocytes or splenocytes from a LCMV-Arm-infected mouse (7 dpi) (Figure 4A). CD44 is an adhesion molecule that is upregulated on antigen-experienced T cells (71). As expected, nearly 100% of the gp33+CD8+ T cells in the spleen of RRV-LCMV-infected mice were CD44+ at 10 dpi (Figure 4B). In contrast, on average, 16.5% of the muscle-infiltrating gp33+CD8+ T cells were negative for CD44. We also analyzed cell surface expression of CD62L, which is a lymph node homing receptor that is downregulated following antigen stimulation (71). As expected, very few of the spleen or muscle-infiltrating gp33+CD8+ T cells stained for CD62L (Figure 4B). Due to the lack of CD44 staining on a percentage of muscle-infiltrating virus antigen-specific CD8+ T cells, we also investigated the CD44 staining on all CD8+ T cells in the muscle and spleen of both WT RRV-and RRV-LCMV-infected mice at 10 dpi. Here, we found about 24% of total CD8+ T cells in the muscle tissue did not stain for CD44, either when mice were infected with WT RRV or RRV-LCMV (Figure 4C and D). In contrast, significantly more total CD8+ T cells in the spleens of RRV-LCMV-infected mice were CD44+ than CD8+ T cells in the spleens of WT RRV-infected mice (67% versus 57%, P = 0.002) (Figure 4C and D), suggesting slightly increased CD8+ T cell activation in RRV-LCMV-infected mice, potentially due to expression of the immunodominant gp33 epitope. In addition, we quantified CD62L expression on total CD8+ T cells in the spleen and muscle tissues of WT RRV- and RRV-LCMV-infected mice; in this case, no significant differences were identified (Figure 4C and D). Overall, these data indicate that the CD8+ T lymphocyte response to the recombinant RRV-LCMV is very similar to WT RRV.
Figure 4. Virus-specific CD8+ T cells from the spleen and muscle tissues of RRV-LCMV-infected mice express different levels of activation markers and produce cytokines following ex vivo restimulation.

(A and B) Three-to-four week-old C57BL/6J mice were mock-inoculated (n = 3) or inoculated with 103 PFU of RRV-LCMV (n = 9). At 10 dpi, leukocytes were isolated from spleens and quadriceps muscles (following enzymatic digestion) for FACS analysis. Spleen cells from a mock-inoculated mouse and a mouse inoculated with LCMV-Armstrong i.p. and harvested on day 7 pi was used as a control for gp33 tetramer staining. (A) Representative histogram plots showing CD44 and CD62L staining on T cells. (B) Frequency of activation marker-positive T cells in spleen compared to muscle tissue from RRV-LCMV-inoculated mice. Data are combined from two independent experiments. Graphs represent the arithmetic mean ± SEM. * P > 0.05, ** P < 0.01, as determined by two-tailed, unpaired t-test with or without Welch's correction. (C and D) Three-to-four week-old C57BL/6J mice were or inoculated with 103 PFU of WT RRV (n = 4) or RRV-LCMV (n = 9). At 10 dpi, leukocytes were isolated from spleens and quadriceps muscles (following enzymatic digestion) for FACS analysis. (C) Representative histogram plots showing CD44 and CD62L staining on total CD8+ T cells from WT RRV- or RRV-LCMV-inoculated mice compared to unstained splenocytes. (D) Frequency of activation marker-positive T cells from the spleen or muscle tissue of WT RRV- or RRV-LCMV-inoculated mice. Data are combined from two independent experiments. Graphs represent the arithmetic mean ± SEM. ** P = 0.002, as determined by a two-way, unpaired t-test. (E–G) Three-to-four week-old C57BL/6J mice were inoculated with 103 PFU of WT RRV (n = 2) or RRV-LCMV (n = 5). At 10 dpi, infiltrating leukocytes were isolated from the quadriceps muscles (following enzymatic digestion) and restimulated ex vivo for 5 hours with gp33 peptide followed by intracellular cytokine staining. Spleen cells from a mouse inoculated with LCMV-Arm i.p. and harvested on day 7 pi was used as a control for gp33 peptide restimulation. (E) Representative flow plots showing IFN-γ, TNF-α, and IL-2 production by CD8+CD4− T cells following stimulation with antigen presenting cells (APCs) alone (top panel) or APCs with peptide (middle and bottom panels). (F) Graph showing the distribution of zero, one, two, or three-cytokine-producing CD8+CD4− T cells isolated from quadriceps muscle tissue of RRV-LCMV-inoculated mice at 10 dpi. (G) Total number of one, two, or three-cytokine-producing CD8+CD4− T cells. Graphs represent the arithmetic mean ± SEM.
In addition to T cell activation markers, we investigated the functionality of the virus-specific CD8+ T cells by determining expression levels of multiple cytokines in response to peptide stimulation. At 10 dpi, muscle-infiltrating leukocytes were isolated and restimulated ex vivo with or without gp33 peptide for 5 hours in the presence of Brefeldin A, followed by surface staining for CD4 and CD8 and intracellular cytokine staining for IFN-γ, TNF-α, and IL-2. Muscle-infiltrating leukocytes from WT RRV-infected mice and splenocytes from a LCMV-Arm-infected mouse were used as controls for the specificity of peptide restimulation. As shown in Figure 4E, cells incubated with APCs only (no peptide) showed minimal cytokine production (≤ 1% in any cytokine group). Similarly, muscle-infiltrating CD8+ T cells from WT RRV-infected mice did not produce cytokines after gp33 peptide restimulation. In contrast, about one third of the muscle-infiltrating CD8+ T cells from RRV-LCMV-infected mice produced TNF-α and/or IFN-γ after gp33 restimulation, with the majority of cytokine-producing cells secreting both TNF-α and IFN-γ (Figure 4E–G). These double-producing cells were also analyzed for IL-2 production. Figure 4F shows a pie chart illustrating the division of muscle-infiltrating, cytokine-expressing CD8+ T cells into single-positive, double-positive, and triple-positive groups. The average percentages were as follows: TNF-α+ (4.7%), IFN-γ+ (8.4%), TNF-α+IFN-γ+ (20.8%), and TNF-α+IFN-γ+IL-2+ (2.4%). These data demonstrate that cytokine-producing, virus-specific CD8+ T cells are present in skeletal muscle tissue, a major site of infection and injury during infection with arthritogenic alphaviruses.
CD8+ T cells contribute to control of acute RRV infection
Based on our observations that muscle-infiltrating, virus-specific CD8+ T cells were activated and produced cytokines following ex vivo analysis, we hypothesized that CD8+ T cells contribute to the control of RRV infection. To investigate this hypothesis, WT and CD8α−/− mice were inoculated with 103 PFU of RRV, and skeletal muscle and ankle joint tissues were harvested at 7 and 14 dpi for analysis of RRV RNA levels via absolute RT-qPCR. CD8α−/− mice had similar viral loads to WT mice at 7 dpi in muscle tissue (Figure 5A). CD8α−/− mice had slightly lower viral loads, albeit significant, than WT mice at 7 dpi in joint tissue (2.7-fold decrease, P < 0.05) (Figure 5B). In contrast, CD8α−/− mice had significantly increased viral loads in quadriceps muscle tissue at 14 dpi (9.8-fold increase, P < 0.001) and 21 dpi (5.4-fold increase, P < 0.01) (Figure 5A). CD8α−/− mice had similar viral loads to WT mice in the ankle joint tissue at 14 dpi but slightly greater viral loads at 21 dpi in this tissue (2.7-fold increase, P < 0.05) (Figure 5B). These data suggest that CD8+ T cells more effectively control RRV infection in muscle tissue compared to joint-associated tissues. WT and CD8α−/− mice had similar serum viremia levels at 3 dpi (data not shown). Additionally, RRV-infected CD8α−/− mice had similar weight gain and disease scores as infected WT mice (Figure 5C and D), suggesting that the loss of CD8+ T cells results in a similar acute disease course.
Figure 5. CD8α−/− mice have increased RRV loads in muscle but not joint tissue at 14 and 21 dpi.
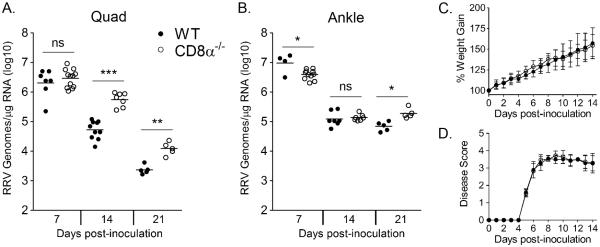
Three-to-four week-old C57BL/6J WT (7 dpi, n = 7; 14 dpi, n = 10; 21 dpi, n = 5) and CD8α−/− (7 dpi, n = 12; 14 dpi, n = 7; 21 dpi, n = 5) mice were inoculated with 103 PFU of RRV. At 7, 14, and 21 dpi mice were sacrificed, perfused by intracardial injection with 1× PBS, and total RNA was isolated from the (A) right quadriceps muscle and (B) right ankle/foot. RRV genome levels were quantified by absolute RT-qPCR. Horizontal bars indicate the mean. * P < 0.05, ** P < 0.01, *** P < 0.001, as determined by one-way ANOVA followed by Tukey's multiple comparison test. (C and D) Three-to-four week-old C57BL/6J WT (n = 21) and CD8α−/− (n = 28) mice were inoculated with 103 PFU of RRV and assessed for (C) weight gain and (D) musculoskeletal disease signs including loss of gripping ability and altered gait at 24-hour intervals. Each data point represents the arithmetic mean ± SD.
The findings in CD8α−/− mice were confirmed by antibody-mediated depletion studies. RRV-inoculated WT mice were treated with an anti-CD8α antibody or an isotype control antibody at 7 and 12 dpi, and muscle and joint tissues were harvested at 14 dpi for quantification of RRV RNA levels. The anti-CD8α antibody treatments effectively depleted CD8+ T cells as determined by flow cytometric analysis of splenocytes from control antibody- or anti-CD8α antibody-treated mice at 14 dpi for the surface markers CD3 and CD8β (Figure 6A and B). As with CD8α−/− mice, anti-CD8α antibody-treated mice had significantly increased viral loads in quadriceps muscle (17.3-fold increase, P = 0.0014) but not ankle joint tissue at 14 dpi (Figure 6C and D). These findings suggest that while CD8+ T cells contribute to viral control in muscle tissue they play a limited role in joint tissue of WT mice following RRV infection.
Figure 6. CD8 T cell depletion significantly increases RRV loads in muscle but not joint tissue at 14.

Three-to-four week-old C57BL/6J WT mice were inoculated with 103 PFU of RRV and treated with an anti-CD8α antibody (n = 4) or a control antibody (n = 4) on days 7 and 12 pi. At 14 dpi mice were sacrificed and perfused by intracardial injection with 1× PBS. Spleens were harvested for FACS analysis; the right quadriceps and right ankle/foot were dissected for RNA analysis. Spleen cells were stained for CD3, CD4, and CD8β to confirm efficient depletion. (A) Representative flow plots demonstrating depletion of CD8α+ T cells. (B) Frequency of CD3+CD8β+ T cells in spleens. Total RNA was isolated from the (C) right quadriceps muscle and (D) right ankle/foot, and RRV genome levels were quantified by absolute RT-qPCR. Horizontal bars indicate the mean. P-value was determined by a two-tailed, unpaired t-test. Data are combined from two independent experiments.
Adoptive transfer of CD8+ and CD4+ T cells reduces RRV infection in Rag1−/− mice
To test whether T cells are sufficient to control RRV infection in the absence of B cells or antibody, we performed adoptive transfer experiments of T cells into Rag1−/− mice. Pan T cells were isolated via negative selection from the spleens of adult WT C57BL/6 mice. Rag1−/− mice received 2.5 × 106 T cells i.p. and were inoculated with RRV 24 hours later. Muscle and joint tissues were harvested for RRV RNA analysis at 14 dpi. Additionally, T cell transfer was confirmed by flow cytometric analysis of spleen and quadriceps muscle tissue at 14 dpi for the presence of cells staining positively for CD3, CD4, and CD8 and negatively for B220 and Gr-1 (Figure 7A–C). T cell engraftment varied between the mice, but T cells could be identified in the spleen and muscle tissue of all mice (Figure 7B and C). RRV RNA levels were significantly lower in muscle tissue of mice that received T cells compared to mice that received media (5.3-fold decrease, P < 0.001) (Figure 7D). Additionally, T cell transfer decreased viremia in most of the mice although the difference did not reach statistical significance (P > 0.05) (Figure 7E). Moreover, the mice that had the best T cell engraftment also had the lowest RNA levels; that is, viral loads correlated significantly with the percent of CD3+ T lymphocytes found in the spleen (r2 = 0.79, P < 0.0001) (Figure 7F) and quadriceps muscle tissue (r2 = 0.70, P = 0.0002) (Figure 7G) as well as the total number of T cells found in both tissues (spleen: r2 = 0.44, P = 0.0095; muscle: r2 = 0.50, P = 0.0045) (data not shown). Consistent with our results in CD8α−/− mice and CD8-depleted WT mice, adoptively transferred T cells did not reduce viral loads in the ankle tissue of Rag1−/− mice (data not shown). To determine if CD8+ T cells could decrease RRV RNA levels independent of CD4+ T cell help, we adoptively transferred 1 × 106 naïve CD8+ T cells into Rag1−/− mice one day prior to RRV inoculation. We found that naïve CD8+ T cells had no effect on RRV RNA levels at 14 dpi (Figure 7D), suggesting that naïve CD8+ T cells require CD4+ T cell help to control RRV infection. Further studies are needed to determine if RRV-immune CD8+ T cells can control RRV infection in the absence of CD4+ T cell help.
Figure 7. T cells contribute to the control of RRV infection in the absence of B cells and Ab.

Pan T cells were negatively selected from the spleen of WT mice, and 2.5 × 106 cells were injected i.p. into Rag1−/− mice (n = 8) one day prior to RRV inoculation. Alternatively, CD8+ T cells were negatively selected from the spleen of WT mice, and 1 × 106 cells were injected i.p. into Rag1−/− mice (n = 4) one day prior to RRV inoculation. As a negative control, 6 Rag1−/− mice received media alone on day −1. Spleens and left quadriceps were harvested at 14 dpi for flow cytometric analysis of the presence of T cells, as shown in (A). Cells were gated on Gr-1−B220−, then CD3+, then divided into CD4+ or CD8+. The percent of CD3+CD8+ T cells (squares) and CD3+CD4+ T cells (circles) in the spleen (closed shapes) and muscle (open shapes) are graphed in (B), and the total number of CD3+CD8+ T cells and CD3+CD4+ T cells in the spleen and muscle are graphed in (C). Right quadriceps (D) and right ankle/foot (not shown) were harvested for analysis of RRV RNA. Horizontal bar indicates the mean. ** P < 0.01, *** P < 0.001, as determined by one-way ANOVA followed by Tukey's multiple comparison test. (E) Serum viremia levels measured by direct plaque assay on BHK-21 cells. Dashed line indicates the limit of detection. (F) The percent of CD3+ T cells (gated on Gr-1−B220− cells) in the spleen plotted against the corresponding RRV RNA level in the quadriceps muscle at 14 dpi. Linear regression analysis was performed to determine the r2 values and P-values. Data are combined from four independent experiments.
DISCUSSION
Although type I IFN is critical for controlling alphavirus infection (1, 23, 72), epidemiological evidence suggests that arthritogenic alphavirus infection results in lifelong protective immunity (73), indicating a role for the adaptive immune response. Indeed, antibodies are considered important mediators of protection against arthritogenic alphaviruses (33, 74, 75). Consistent with this, we recently demonstrated that CHIKV-infected Rag1−/− mice have significantly increased viral RNA levels in inflamed musculoskeletal tissues for weeks after inoculation (31). Similarly, here we show that RRV-infected Rag1−/− mice have significantly higher viral loads in both muscle and joint tissue at 7 and 14 dpi (Figure 1). In this study, we sought to elucidate the role for T cells, specifically CD8+ T cells, in the control of arthritogenic alphavirus infection in musculoskeletal tissues.
Previous publications have demonstrated that both CD4+ and CD8+ T cells are a major component of the muscle-infiltrating leukocytes starting at day 5 post-RRV infection (49, 50, 76). Additionally, an early report on RRV infection in mice showed virus-specific T cell proliferation when exposed to virus ex vivo (77), and a recent report on CHIKV infection in mice demonstrated that spleen and footpad-infiltrating T cells could produce IFN-γ after ex vivo stimulation with virus (32). Additionally, several studies have shown virus-specific T cell responses following immunization with different CHIKV vaccines in mouse and macaque models (36, 78), indicating an expansion and activation of virus-specific T cells. However, the kinetics of virus-specific T cell responses in mice and the role of T cells in the control of infection in musculoskeletal tissues have not been evaluated. To identify virus antigen-specific T cells in C57BL/6 mice, we generated a recombinant Ross River virus that encodes the major MHC-I and MHC-II-restricted LCMV determinants, gp33 and gp61, in tandem (Figure 2), similar to a system recently utilized to study the virus-specific T cell response to influenza virus in mice (61). Using the recombinant RRV-LCMV we detected virus-specific CD8+ T cells in lymphoid tissues as well as inflamed musculoskeletal tissues of RRV-infected mice (Figure 3). Small numbers of virus-specific CD8+ T cells were detectable at 5 dpi in lymphoid and musculoskeletal tissues which expanded at 7 and 10 dpi and began to contract by 14 dpi. These data are consistent with previous reports on the kinetics of total T cell activation in CHIKV-infected mice and macaques (32, 45). Subsequent work will analyze CD4+ T cell immunity utilizing the recombinant RRV-LCMV.
As expected, nearly all of the virus-specific CD8+ T cells in the spleen and muscle uniformly down-regulated the lymph node-homing molecule CD62L to aid in peripheral homing (Figure 4). Also anticipated was that nearly all of the virus-specific CD8+ T cells in the spleen at 10 dpi were CD44+ since it is a marker of antigen-experienced T cells (71). However, approximately 15–20% of the virus-specific CD8+ T cells in the muscle tissue at 10 dpi were CD44 low or negative. We previously reported that macrophages in the sites of infection and inflammation have immunosuppressive activity (62). The lack of CD44 staining on a portion of the virus-specific CD8+ T cells suggests that the inflamed, immunosuppressive microenvironment within the muscle tissue may inhibit some aspect of T cell activation. We further found that a subset of total CD8+ T cells in the muscle tissue of WT RRV- and RRV-LCMV-infected mice did not stain for CD44, indicating that this differential CD44 expression was not particular to gp33-specific CD8+ T cells. The absence of CD44 staining could play an important role in T cell-mediated control of RRV infection since CD44 expression on T cells is important for trafficking to and within sites of inflammation (79). Additionally, downregulation of CD44 on virus-specific CD8+ T cells has been observed under conditions of chronic LCMV infection (80); this may also be occurring following infection with arthritogenic alphaviruses, which cause persistent infections in humans and animals (1, 16, 17, 31).
Following ex vivo peptide restimulation we found that a small portion of the virus-specific CD8+ T cells produced IFN-γ, TNF-α, and IL-2 (Figure 5). The majority of cytokine-producing cells secreted both IFN-γ and TNF-α, followed by single IFN-γ-producers, then single TNF-α-producers. IL-2 production, although present, was minimal, with only 2–3% of all gp33-specific cells producing IL-2 (10% of IFN-γ+TNF-α+ cells). These findings are similar to previous analyses of cytokine-producing T cells in the CNS following infection with the neurotropic alphaviruses VEEV and SINV (81, 82). Irani and colleagues (82) found that IL-2 production by brain-infiltrating T cells was particularly deficient when compared with the levels made by stimulated spleen cells, suggesting that the local microenvironment regulates T cells that enter the brain. We did not evaluate cytokine production by spleen T cells from RRV-LCMV-infected mice in these studies; however, given the differences in activation marker staining between T cells in the spleen and muscle tissue of RRV-LCMV-infected mice, we hypothesize that cytokine production by the muscle-infiltrating T cells would be lower than spleen T cells due to the immunosuppressive environment within the inflamed muscle tissue (62).
Given that activated, virus-specific CD8+ T cells are present in infected musculoskeletal tissues of RRV-infected mice, we next sought to determine the contribution of CD8+ T cells in controlling RRV infection. A number of studies have shown that T cells can directly contribute to the control of alphavirus infection. Adoptive transfer of primed T cells but not antibody prevented lethal VEEV-induced encephalitis in TCRαβ−/− mice (83, 84). Furthermore, recovery from VEEV infection in μMT mice has been shown to be T cell-dependent, with both CD8+ and CD4+ T cells contributing to control of VEEV replication systemically and in the CNS (81). Similarly, clearance of SINV RNA from the CNS of infected mice required both CD4+ and CD8+ T cells (54, 55). Consistent with these data, we found that mice depleted of CD8+ T cells as well as CD8α−/− mice had ~10-fold more RRV RNA in the muscle tissue than WT control mice at 14 dpi (Figures 5 and 6). Experiments in CD8α−/− mice infected with West Nile virus (WNV) demonstrated a clear role for CD8+ T cells in control of that positive-strand RNA virus infection. However, WNV-infected CD8α−/− mice had 1,000-fold more virus in the brain at 10 dpi than WT mice (85), which contrasts with the ~10-fold difference we detected following RRV infection of CD8α−/− mice versus WT mice at 14 dpi. These data suggest that although CD8+ T cells contribute to the control of RRV infection, their antiviral activities may be blunted in the inflamed, immunosuppressive microenvironment of the infected muscle tissue (62).
Tomov and colleagues (86) demonstrated that adoptive transfer of murine norovirus-specific CD8+ T cells into persistently infected Rag1−/− mice led to a significant reduction in viral loads that was dependent on the number of T cells transferred. Similarly, we found that T cells transferred into Rag1−/− mice one day prior to RRV infection reduced RRV RNA levels in muscle tissue by 14 dpi when compared to loads in Rag1−/− mice that received media, and the reduction in viral loads correlated with the frequency and total number of T cells present in the spleen and muscle tissue on the day of harvest (Figure 7). These data demonstrate that T cells control arthritogenic alphavirus infection in a relevant tissue – muscle tissue – in the absence of B cells and antibody. We further showed that adoptively transferred CD8+ T cells alone did not reduce RRV levels in muscle tissue of Rag1−/− mice (Figure 7). This occurred despite a similar number of CD8+ T cells in spleen and muscle tissue of mice that received CD8+ T cells alone compared to mice that received both CD4+ and CD8+ T cells. These data suggest that CD4 T cell-help is required for CD8+ T cell-mediated control of RRV infection.
Interestingly, CD8α−/− mice, CD8+ T cell-depleted mice, and Rag1−/− mice that received adoptively transferred T cells had similar RRV RNA levels as control mice at 14 dpi in the ankle joint tissue (Figures 5 and 6, and data not shown), suggesting that T cells have minimal effects on virus control in joint tissue. This could be a result of i) inefficient migration into tissues, ii) inopportune location of target cells, iii) suppression of T cell antiviral functions in the joint, and/or a variety of other mechanism(s). Consistent with these data, Teo and colleagues (32) found that CD8−/− mice had similar levels of CHIKV RNA in the serum through day 12 pi, when the virus levels became undetectable, as well as similar joint swelling as CHIKV-infected WT mice. Additionally, mice depleted of CD8+ T cells had similar viral loads in joint tissue as determined by luciferase intensity following infection with a recombinant Firefly luciferase-expressing CHIKV (32). These data, together with our findings, suggest that CD8+ T cells are ineffective at controlling arthritogenic alphavirus infection in joint tissues. Although T cell transfer resulted in a modest decrease in viremia for most mice (Figure 7E), this may not have reached statistical significance because of continued virus production from joint tissues. Overall, these data demonstrate an important but limited and tissue-specific role for CD8+ T lymphocytes in the control of pathogenic RRV infection.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge Bennett Davenport and Phil Pratt for assistance with flow cytometry. We thank Kelsey Haist for assistance with RT-qPCR.
This research was supported by NIH-NIAID research grant R01 AI108725-01A1 (T.E.M). K.S.B. was supported by NIH-NIAID training grant T32 AI052066.
REFERENCES
- 1.Suhrbier A, Jaffar-Bandjee MC, Gasque P. Arthritogenic alphaviruses--an overview. Nat Rev Rheumatol. 2012;8:420–429. doi: 10.1038/nrrheum.2012.64. [DOI] [PubMed] [Google Scholar]
- 2.Aaskov JG, Mataika JU, Lawrence GW, Rabukawaqa V, Tucker MM, Miles JA, Dalglish DA. An epidemic of Ross River virus infection in Fiji, 1979. Am J Trop Med Hyg. 1981;30:1053–1059. doi: 10.4269/ajtmh.1981.30.1053. [DOI] [PubMed] [Google Scholar]
- 3.Rosen L, Gubler DJ, Bennett PH. Epidemic polyarthritis (Ross River) virus infection in the Cook Islands. Am J Trop Med Hyg. 1981;30:1294–1302. doi: 10.4269/ajtmh.1981.30.1294. [DOI] [PubMed] [Google Scholar]
- 4.Tesh RB, McLean RG, Shroyer DA, Calisher CH, Rosen L. Ross River virus (Togaviridae: Alphavirus) infection (epidemic polyarthritis) in American Samoa. Trans R Soc Trop Med Hyg. 1981;75:426–431. doi: 10.1016/0035-9203(81)90112-7. [DOI] [PubMed] [Google Scholar]
- 5.Fauran P, Donaldson M, Harper J, Oseni RA, Aaskov JG. Characterization of Ross River viruses isolated from patients with polyarthritis in New Caledonia and Wallis and Futuna Islands. Am J Trop Med Hyg. 1984;33:1228–1231. doi: 10.4269/ajtmh.1984.33.1228. [DOI] [PubMed] [Google Scholar]
- 6.Powers AM, Logue CH. Changing patterns of chikungunya virus: re-emergence of a zoonotic arbovirus. J Gen Virol. 2007;88:2363–2377. doi: 10.1099/vir.0.82858-0. [DOI] [PubMed] [Google Scholar]
- 7.Rezza G, Nicoletti L, Angelini R, Romi R, Finarelli AC, Panning M, Cordioli P, Fortuna C, Boros S, Magurano F, Silvi G, Angelini P, Dottori M, Ciufolini MG, Majori GC, Cassone A. Infection with chikungunya virus in Italy: an outbreak in a temperate region. Lancet. 2007;370:1840–1846. doi: 10.1016/S0140-6736(07)61779-6. [DOI] [PubMed] [Google Scholar]
- 8.Grandadam M, Caro V, Plumet S, Thiberge JM, Souares Y, Failloux AB, Tolou HJ, Budelot M, Cosserat D, Leparc-Goffart I, Despres P. Chikungunya virus, southeastern France. Emerg Infect Dis. 2011;17:910–913. doi: 10.3201/eid1705.101873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zayed A, Awash AA, Esmail MA, Al-Mohamadi HA, Al-Salwai M, Al-Jasari A, Medhat I, Morales-Betoulle ME, Mnzava A. Detection of Chikungunya virus in Aedes aegypti during 2011 outbreak in Al Hodayda, Yemen. Acta Trop. 2012;123:62–66. doi: 10.1016/j.actatropica.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Horwood PF, Reimer LJ, Dagina R, Susapu M, Bande G, Katusele M, Koimbu G, Jimmy S, Ropa B, Siba PM, Pavlin BI. Outbreak of chikungunya virus infection, Vanimo, Papua New Guinea. Emerg Infect Dis. 2013;19:1535–1538. doi: 10.3201/eid1909.130130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dupont-Rouzeyrol M, Caro V, Guillaumot L, Vazeille M, D'Ortenzio E, Thiberge JM, Baroux N, Gourinat AC, Grandadam M, Failloux AB. Chikungunya virus and the mosquito vector Aedes aegypti in New Caledonia (South Pacific Region) Vector Borne Zoonotic Dis. 2012;12:1036–1041. doi: 10.1089/vbz.2011.0937. [DOI] [PubMed] [Google Scholar]
- 12.Leparc-Goffart I, Nougairede A, Cassadou S, Prat C, de Lamballerie X. Chikungunya in the Americas. Lancet. 2014;383:514. doi: 10.1016/S0140-6736(14)60185-9. [DOI] [PubMed] [Google Scholar]
- 13.Van Bortel W, Dorleans F, Rosine J, Blateau A, Rousset D, Matheus S, Leparc-Goffart I, Flusin O, Prat C, Cesaire R, Najioullah F, Ardillon V, Balleydier E, Carvalho L, Lemaitre A, Noel H, Servas V, Six C, Zurbaran M, Leon L, Guinard A, van den Kerkhof J, Henry M, Fanoy E, Braks M, Reimerink J, Swaan C, Georges R, Brooks L, Freedman J, Sudre B, Zeller H. Chikungunya outbreak in the Caribbean region, December 2013 to March 2014, and the significance for Europe. Euro Surveill. 2014;19 doi: 10.2807/1560-7917.es2014.19.13.20759. [DOI] [PubMed] [Google Scholar]
- 14.Borgherini G, Poubeau P, Jossaume A, Gouix A, Cotte L, Michault A, Arvin-Berod C, Paganin F. Persistent arthralgia associated with chikungunya virus: a study of 88 adult patients on reunion island. Clin Infect Dis. 2008;47:469–475. doi: 10.1086/590003. [DOI] [PubMed] [Google Scholar]
- 15.Lakshmi V, Neeraja M, Subbalaxmi MV, Parida MM, Dash PK, Santhosh SR, Rao PV. Clinical features and molecular diagnosis of Chikungunya fever from South India. Clin Infect Dis. 2008;46:1436–1442. doi: 10.1086/529444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suhrbier A, La Linn M. Clinical and pathologic aspects of arthritis due to Ross River virus and other alphaviruses. Curr Opin Rheumatol. 2004;16:374–379. doi: 10.1097/01.bor.0000130537.76808.26. [DOI] [PubMed] [Google Scholar]
- 17.Pialoux G, Gauzere BA, Jaureguiberry S, Strobel M. Chikungunya, an epidemic arbovirosis. Lancet Infect Dis. 2007;7:319–327. doi: 10.1016/S1473-3099(07)70107-X. [DOI] [PubMed] [Google Scholar]
- 18.Economopoulou A, Dominguez M, Helynck B, Sissoko D, Wichmann O, Quenel P, Germonneau P, Quatresous I. Atypical Chikungunya virus infections: clinical manifestations, mortality and risk factors for severe disease during the 2005–2006 outbreak on Reunion. Epidemiol Infect. 2009;137:534–541. doi: 10.1017/S0950268808001167. [DOI] [PubMed] [Google Scholar]
- 19.Staples JE, Breiman RF, Powers AM. Chikungunya fever: an epidemiological review of a re-emerging infectious disease. Clin Infect Dis. 2009;49:942–948. doi: 10.1086/605496. [DOI] [PubMed] [Google Scholar]
- 20.Rudd PA, Wilson J, Gardner J, Larcher T, Babarit C, Le TT, Anraku I, Kumagai Y, Loo YM, Gale M, Jr., Akira S, Khromykh AA, Suhrbier A. Interferon response factors 3 and 7 protect against Chikungunya virus hemorrhagic fever and shock. J Virol. 2012;86:9888–9898. doi: 10.1128/JVI.00956-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schilte C, Buckwalter MR, Laird ME, Diamond MS, Schwartz O, Albert ML. Cutting edge: independent roles for IRF-3 and IRF-7 in hematopoietic and nonhematopoietic cells during host response to Chikungunya infection. J Immunol. 2012;188:2967–2971. doi: 10.4049/jimmunol.1103185. [DOI] [PubMed] [Google Scholar]
- 22.Gardner CL, Burke CW, Higgs ST, Klimstra WB, Ryman KD. Interferon-alpha/beta deficiency greatly exacerbates arthritogenic disease in mice infected with wild-type chikungunya virus but not with the cell culture-adapted live-attenuated 181/25 vaccine candidate. Virology. 2012;425:103–112. doi: 10.1016/j.virol.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, Guivel-Benhassine F, Kraxner A, Tschopp J, Higgs S, Michault A, Arenzana-Seisdedos F, Colonna M, Peduto L, Schwartz O, Lecuit M, Albert ML. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J Exp Med. 2010;207:429–442. doi: 10.1084/jem.20090851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lidbury BA, Rulli NE, Musso CM, Cossetto SB, Zaid A, Suhrbier A, Rothenfluh HS, Rolph MS, Mahalingam S. Identification and characterization of a ross river virus variant that grows persistently in macrophages, shows altered disease kinetics in a mouse model, and exhibits resistance to type I interferon. J Virol. 2011;85:5651–5663. doi: 10.1128/JVI.01189-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Couderc T, Chretien F, Schilte C, Disson O, Brigitte M, Guivel-Benhassine F, Touret Y, Barau G, Cayet N, Schuffenecker I, Despres P, Arenzana-Seisdedos F, Michault A, Albert ML, Lecuit M. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008;4:e29. doi: 10.1371/journal.ppat.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seay AR, Kern ER, Murray RS. Interferon treatment of experimental Ross River virus polymyositis. Neurology. 1987;37:1189–1193. doi: 10.1212/wnl.37.7.1189. [DOI] [PubMed] [Google Scholar]
- 27.Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol. 2000;74:3366–3378. doi: 10.1128/jvi.74.7.3366-3378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryman KD, Meier KC, Gardner CL, Adegboyega PA, Klimstra WB. Non-pathogenic Sindbis virus causes hemorrhagic fever in the absence of alpha/beta and gamma interferons. Virology. 2007;368:273–285. doi: 10.1016/j.virol.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 29.Kam YW, Simarmata D, Chow A, Her Z, Teng TS, Ong EK, Renia L, Leo YS, Ng LF. Early appearance of neutralizing immunoglobulin G3 antibodies is associated with chikungunya virus clearance and long-term clinical protection. J Infect Dis. 2012;205:1147–1154. doi: 10.1093/infdis/jis033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lum FM, Teo TH, Lee WW, Kam YW, Renia L, Ng LF. An essential role of antibodies in the control of Chikungunya virus infection. J Immunol. 2013;190:6295–6302. doi: 10.4049/jimmunol.1300304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawman DW, Stoermer KA, Montgomery SA, Pal P, Oko L, Diamond MS, Morrison TE. Chronic joint disease caused by persistent Chikungunya virus infection is controlled by the adaptive immune response. J Virol. 2013;87:13878–13888. doi: 10.1128/JVI.02666-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teo TH, Lum FM, Claser C, Lulla V, Lulla A, Merits A, Renia L, Ng LF. A pathogenic role for CD4+ T cells during Chikungunya virus infection in mice. J Immunol. 2013;190:259–269. doi: 10.4049/jimmunol.1202177. [DOI] [PubMed] [Google Scholar]
- 33.Couderc T, Khandoudi N, Grandadam M, Visse C, Gangneux N, Bagot S, Prost JF, Lecuit M. Prophylaxis and therapy for Chikungunya virus infection. J Infect Dis. 2009;200:516–523. doi: 10.1086/600381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fric J, Bertin-Maghit S, Wang CI, Nardin A, Warter L. Use of human monoclonal antibodies to treat Chikungunya virus infection. J Infect Dis. 2013;207:319–322. doi: 10.1093/infdis/jis674. [DOI] [PubMed] [Google Scholar]
- 35.Pal P, Dowd KA, Brien JD, Edeling MA, Gorlatov S, Johnson S, Lee I, Akahata W, Nabel GJ, Richter MK, Smit JM, Fremont DH, Pierson TC, Heise MT, Diamond MS. Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus. PLoS Pathog. 2013;9:e1003312. doi: 10.1371/journal.ppat.1003312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu H, Das SC, Fuchs JF, Suresh M, Weaver SC, Stinchcomb DT, Partidos CD, Osorio JE. Deciphering the protective role of adaptive immunity to CHIKV/IRES a novel candidate vaccine against Chikungunya in the A129 mouse model. Vaccine. 2013;31:3353–3360. doi: 10.1016/j.vaccine.2013.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plante K, Wang E, Partidos CD, Weger J, Gorchakov R, Tsetsarkin K, Borland EM, Powers AM, Seymour R, Stinchcomb DT, Osorio JE, Frolov I, Weaver SC. Novel chikungunya vaccine candidate with an IRES-based attenuation and host range alteration mechanism. PLoS Pathog. 2011;7:e1002142. doi: 10.1371/journal.ppat.1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holzer GW, Coulibaly S, Aichinger G, Savidis-Dacho H, Mayrhofer J, Brunner S, Schmid K, Kistner O, Aaskov JG, Falkner FG, Ehrlich H, Barrett PN, Kreil TR. Evaluation of an inactivated Ross River virus vaccine in active and passive mouse immunization models and establishment of a correlate of protection. Vaccine. 2011;29:4132–4141. doi: 10.1016/j.vaccine.2011.03.089. [DOI] [PubMed] [Google Scholar]
- 39.Akahata W, Yang ZY, Andersen H, Sun S, Holdaway HA, Kong WP, Lewis MG, Higgs S, Rossmann MG, Rao S, Nabel GJ. A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat Med. 2010;16:334–338. doi: 10.1038/nm.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Partidos CD, Weger J, Brewoo J, Seymour R, Borland EM, Ledermann JP, Powers AM, Weaver SC, Stinchcomb DT, Osorio JE. Probing the attenuation and protective efficacy of a candidate chikungunya virus vaccine in mice with compromised interferon (IFN) signaling. Vaccine. 2011;29:3067–3073. doi: 10.1016/j.vaccine.2011.01.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levine B, Hardwick JM, Trapp BD, Crawford TO, Bollinger RC, Griffin DE. Antibody-mediated clearance of alphavirus infection from neurons. Science. 1991;254:856–860. doi: 10.1126/science.1658936. [DOI] [PubMed] [Google Scholar]
- 42.Levine B, Griffin DE. Persistence of viral RNA in mouse brains after recovery from acute alphavirus encephalitis. J Virol. 1992;66:6429–6435. doi: 10.1128/jvi.66.11.6429-6435.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wauquier N, Becquart P, Nkoghe D, Padilla C, Ndjoyi-Mbiguino A, Leroy EM. The acute phase of Chikungunya virus infection in humans is associated with strong innate immunity and T CD8 cell activation. J Infect Dis. 2011;204:115–123. doi: 10.1093/infdis/jiq006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoarau JJ, Jaffar Bandjee MC, Krejbich Trotot P, Das T, Li-Pat-Yuen G, Dassa B, Denizot M, Guichard E, Ribera A, Henni T, Tallet F, Moiton MP, Gauzere BA, Bruniquet S, Jaffar Bandjee Z, Morbidelli P, Martigny G, Jolivet M, Gay F, Grandadam M, Tolou H, Vieillard V, Debre P, Autran B, Gasque P. Persistent chronic inflammation and infection by Chikungunya arthritogenic alphavirus in spite of a robust host immune response. J Immunol. 2010;184:5914–5927. doi: 10.4049/jimmunol.0900255. [DOI] [PubMed] [Google Scholar]
- 45.Messaoudi I, Vomaske J, Totonchy T, Kreklywich CN, Haberthur K, Springgay L, Brien JD, Diamond MS, Defilippis VR, Streblow DN. Chikungunya virus infection results in higher and persistent viral replication in aged rhesus macaques due to defects in anti-viral immunity. PLoS Negl Trop Dis. 2013;7:e2343. doi: 10.1371/journal.pntd.0002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoarau JJ, Gay F, Pelle O, Samri A, Jaffar-Bandjee MC, Gasque P, Autran B. Identical Strength of the T Cell Responses against E2, nsP1 and Capsid CHIKV Proteins in Recovered and Chronic Patients after the Epidemics of 2005–2006 in La Reunion Island. PLoS One. 2013;8:e84695. doi: 10.1371/journal.pone.0084695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozden S, Huerre M, Riviere JP, Coffey LL, Afonso PV, Mouly V, de Monredon J, Roger JC, El Amrani M, Yvin JL, Jaffar MC, Frenkiel MP, Sourisseau M, Schwartz O, Butler-Browne G, Despres P, Gessain A, Ceccaldi PE. Human muscle satellite cells as targets of Chikungunya virus infection. PLoS One. 2007;2:e527. doi: 10.1371/journal.pone.0000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soden M, Vasudevan H, Roberts B, Coelen R, Hamlin G, Vasudevan S, La Brooy J. Detection of viral ribonucleic acid and histologic analysis of inflamed synovium in Ross River virus infection. Arthritis Rheum. 2000;43:365–369. doi: 10.1002/1529-0131(200002)43:2<365::AID-ANR16>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 49.Morrison TE, Whitmore AC, Shabman RS, Lidbury BA, Mahalingam S, Heise MT. Characterization of Ross River virus tropism and virus-induced inflammation in a mouse model of viral arthritis and myositis. J Virol. 2006;80:737–749. doi: 10.1128/JVI.80.2.737-749.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morrison TE, Fraser RJ, Smith PN, Mahalingam S, Heise MT. Complement contributes to inflammatory tissue destruction in a mouse model of Ross River virus-induced disease. J Virol. 2007;81:5132–5143. doi: 10.1128/JVI.02799-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morrison TE, Oko L, Montgomery SA, Whitmore AC, Lotstein AR, Gunn BM, Elmore SA, Heise MT. A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am J Pathol. 2011;178:32–40. doi: 10.1016/j.ajpath.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P, Schroder WA, Higgs S, Suhrbier A. Chikungunya virus arthritis in adult wild-type mice. J Virol. 2010;84:8021–8032. doi: 10.1128/JVI.02603-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rowell JF, Griffin DE. Contribution of T cells to mortality in neurovirulent Sindbis virus encephalomyelitis. J Neuroimmunol. 2002;127:106–114. doi: 10.1016/s0165-5728(02)00108-x. [DOI] [PubMed] [Google Scholar]
- 54.Kimura T, Griffin DE. The role of CD8(+) T cells and major histocompatibility complex class I expression in the central nervous system of mice infected with neurovirulent Sindbis virus. J Virol. 2000;74:6117–6125. doi: 10.1128/jvi.74.13.6117-6125.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Binder GK, Griffin DE. Interferon-gamma-mediated site-specific clearance of alphavirus from CNS neurons. Science. 2001;293:303–306. doi: 10.1126/science.1059742. [DOI] [PubMed] [Google Scholar]
- 56.Linn ML, Mateo L, Gardner J, Suhrbier A. Alphavirus-specific cytotoxic T lymphocytes recognize a cross-reactive epitope from the capsid protein and can eliminate virus from persistently infected macrophages. J Virol. 1998;72:5146–5153. doi: 10.1128/jvi.72.6.5146-5153.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Doherty RI, Whitehead RH, Gorman BM, O'Gower AK. The isolation of a third group A arbovirus in Australia, with preliminary observations on its relationships to epidemic polyarthritis. Aust J Sci. 1963;26:183–184. [Google Scholar]
- 58.Kuhn RJ, Niesters HG, Hong Z, Strauss JH. Infectious RNA transcripts from Ross River virus cDNA clones and the construction and characterization of defined chimeras with Sindbis virus. Virology. 1991;182:430–441. doi: 10.1016/0042-6822(91)90584-x. [DOI] [PubMed] [Google Scholar]
- 59.Dalgarno L, Rice CM, Strauss JH. Ross River virus 26 s RNA: complete nucleotide sequence and deduced sequence of the encoded structural proteins. Virology. 1983;129:170–187. doi: 10.1016/0042-6822(83)90404-x. [DOI] [PubMed] [Google Scholar]
- 60.Homann D, Tishon A, Berger DP, Weigle WO, von Herrath MG, Oldstone MB. Evidence for an underlying CD4 helper and CD8 T-cell defect in B-cell-deficient mice: failure to clear persistent virus infection after adoptive immunotherapy with virus-specific memory cells from muMT/muMT mice. J Virol. 1998;72:9208–9216. doi: 10.1128/jvi.72.11.9208-9216.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marsolais D, Hahm B, Walsh KB, Edelmann KH, McGavern D, Hatta Y, Kawaoka Y, Rosen H, Oldstone MB. A critical role for the sphingosine analog AAL-R in dampening the cytokine response during influenza virus infection. Proc Natl Acad Sci U S A. 2009;106:1560–1565. doi: 10.1073/pnas.0812689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stoermer KA, Burrack A, Oko L, Montgomery SA, Borst LB, Gill RG, Morrison TE. Genetic ablation of arginase 1 in macrophages and neutrophils enhances clearance of an arthritogenic alphavirus. J Immunol. 2012;189:4047–4059. doi: 10.4049/jimmunol.1201240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jupille HJ, Medina-Rivera M, Hawman DW, Oko L, Morrison TE. A tyrosine-to-histidine switch at position 18 of the Ross River virus E2 glycoprotein is a determinant of virus fitness in disparate hosts. J Virol. 2013;87:5970–5984. doi: 10.1128/JVI.03326-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lidbury BA, Simeonovic C, Maxwell GE, Marshall ID, Hapel AJ. Macrophage-induced muscle pathology results in morbidity and mortality for Ross River virus-infected mice. J Infect Dis. 2000;181:27–34. doi: 10.1086/315164. [DOI] [PubMed] [Google Scholar]
- 65.Stoermer Burrack KA, Hawman DW, Jupille HJ, Oko L, Minor M, Shives KD, Gunn BM, Long KM, Morrison TE. Attenuating mutations in nsP1 reveal tissue-specific mechanisms for control of Ross River virus infection. J Virol. 2014;88:3719–3732. doi: 10.1128/JVI.02609-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 67.Marsolais D, Hahm B, Edelmann KH, Walsh KB, Guerrero M, Hatta Y, Kawaoka Y, Roberts E, Oldstone MB, Rosen H. Local not systemic modulation of dendritic cell S1P receptors in lung blunts virus-specific immune responses to influenza. Mol Pharmacol. 2008;74:896–903. doi: 10.1124/mol.108.048769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Slifka MK, Pagarigan R, Mena I, Feuer R, Whitton JL. Using recombinant coxsackievirus B3 to evaluate the induction and protective efficacy of CD8+ T cells during picornavirus infection. J Virol. 2001;75:2377–2387. doi: 10.1128/JVI.75.5.2377-2387.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thomas JM, Klimstra WB, Ryman KD, Heidner HW. Sindbis virus vectors designed to express a foreign protein as a cleavable component of the viral structural polyprotein. J Virol. 2003;77:5598–5606. doi: 10.1128/JVI.77.10.5598-5606.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun C, Gardner CL, Watson AM, Ryman KD, Klimstra WB. Stable, high-level expression of reporter proteins from improved alphavirus expression vectors to track replication and dissemination during encephalitic and arthritogenic disease. J Virol. 2014;88:2035–2046. doi: 10.1128/JVI.02990-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwartz O, Albert ML. Biology and pathogenesis of chikungunya virus. Nat Rev Microbiol. 2010;8:491–500. doi: 10.1038/nrmicro2368. [DOI] [PubMed] [Google Scholar]
- 73.Burt FJ, Rolph MS, Rulli NE, Mahalingam S, Heise MT. Chikungunya: a re-emerging virus. Lancet. 2012;379:662–671. doi: 10.1016/S0140-6736(11)60281-X. [DOI] [PubMed] [Google Scholar]
- 74.Kam YW, Lee WW, Simarmata D, Harjanto S, Teng TS, Tolou H, Chow A, Lin RT, Leo YS, Renia L, Ng LF. Longitudinal analysis of the human antibody response to Chikungunya virus infection: implications for serodiagnosis and vaccine development. J Virol. 2012;86:13005–13015. doi: 10.1128/JVI.01780-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Warter L, Lee CY, Thiagarajan R, Grandadam M, Lebecque S, Lin RT, Bertin-Maghit S, Ng LF, Abastado JP, Despres P, Wang CI, Nardin A. Chikungunya virus envelope-specific human monoclonal antibodies with broad neutralization potency. J Immunol. 2011;186:3258–3264. doi: 10.4049/jimmunol.1003139. [DOI] [PubMed] [Google Scholar]
- 76.Morrison TE, Simmons JD, Heise MT. Complement receptor 3 promotes severe ross river virus-induced disease. J Virol. 2008;82:11263–11272. doi: 10.1128/JVI.01352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aaskov JG, Dalglish DA, Davies CE, Tucker M, Donaldson MD. Cell-mediated immune response to Ross River virus in mice: evidence for a defective effector cell response. Aust J Exp Biol Med Sci. 1983;61(Pt 5):529–540. doi: 10.1038/icb.1983.50. [DOI] [PubMed] [Google Scholar]
- 78.Mallilankaraman K, Shedlock DJ, Bao H, Kawalekar OU, Fagone P, Ramanathan AA, Ferraro B, Stabenow J, Vijayachari P, Sundaram SG, Muruganandam N, Sarangan G, Srikanth P, Khan AS, Lewis MG, Kim JJ, Sardesai NY, Muthumani K, Weiner DB. A DNA vaccine against chikungunya virus is protective in mice and induces neutralizing antibodies in mice and nonhuman primates. PLoS Negl Trop Dis. 2011;5:e928. doi: 10.1371/journal.pntd.0000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jackson DG. Immunological functions of hyaluronan and its receptors in the lymphatics. Immunol Rev. 2009;230:216–231. doi: 10.1111/j.1600-065X.2009.00803.x. [DOI] [PubMed] [Google Scholar]
- 80.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brooke CB, Deming DJ, Whitmore AC, White LJ, Johnston RE. T cells facilitate recovery from Venezuelan equine encephalitis virus-induced encephalomyelitis in the absence of antibody. J Virol. 2010;84:4556–4568. doi: 10.1128/JVI.02545-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Irani DN, Lin KI, Griffin DE. Regulation of brain-derived T cells during acute central nervous system inflammation. J Immunol. 1997;158:2318–2326. [PubMed] [Google Scholar]
- 83.Paessler S, Yun NE, Judy BM, Dziuba N, Zacks MA, Grund AH, Frolov I, Campbell GA, Weaver SC, Estes DM. Alpha-beta T cells provide protection against lethal encephalitis in the murine model of VEEV infection. Virology. 2007;367:307–323. doi: 10.1016/j.virol.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yun NE, Peng BH, Bertke AS, Borisevich V, Smith JK, Smith JN, Poussard AL, Salazar M, Judy BM, Zacks MA, Estes DM, Paessler S. CD4+ T cells provide protection against acute lethal encephalitis caused by Venezuelan equine encephalitis virus. Vaccine. 2009;27:4064–4073. doi: 10.1016/j.vaccine.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shrestha B, Diamond MS. Role of CD8+ T cells in control of West Nile virus infection. J Virol. 2004;78:8312–8321. doi: 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tomov VT, Osborne LC, Dolfi DV, Sonnenberg GF, Monticelli LA, Mansfield K, Virgin HW, Artis D, Wherry EJ. Persistent enteric murine norovirus infection is associated with functionally suboptimal virus-specific CD8 T cell responses. J Virol. 2013;87:7015–7031. doi: 10.1128/JVI.03389-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.