

Abstract
Immunological responses to pathogens are stringently regulated in the eye to prevent excessive inflammation that damage ocular tissues and compromise vision. Suppressors of cytokine signaling (SOCS) regulate intensity/duration of inflammatory responses. We have used SOCS1-deficient mice and retina-specific SOCS1 transgenic rats to investigate roles of SOCS1 in ocular herpes simplex virus (HSV-1) infection and non-infectious uveitis. We also genetically engineered cell-penetrating SOCS proteins (membrane-translocating sequence (MTS)-SOCS1, MTS-SOCS3) and examined whether they can be used to inhibit inflammatory cytokines. Overexpression of SOCS1 in transgenic rat eyes attenuated ocular HSV-1 infection while SOCS1-deficient mice developed severe non-infectious anterior uveitis, suggesting that SOCS1 may contribute to mechanism of ocular immune privilege by regulating trafficking of inflammatory cells into ocular tissues. Furthermore, MTS-SOCS1 inhibited IFN-γ-induced signal transducers and activators of transcription 1 (STAT1) activation by macrophages while MTS-SOCS3 suppressed expansion of pathogenic Th17 cells that mediate uveitis, indicating that MTS-SOCS proteins maybe used to treat ocular inflammatory diseases of infectious or autoimmune etiology.
Keywords: SOCS1, anterior uveitis, ocular HSV-1 infection, cell-penetrating SOCS, endotoxin-induced uveitis (EIU)
Introduction
The intensity and duration of CNS inflammatory responses must be under stringent regulation to avoid autoimmune pathology in these sensitive immune privileged tissues. In fact, unrestrained neuroinflammatory responses have been implicated in the etiology of CNS autoimmune diseases such as non-infectious uveitis and multiple sclerosis (MS) [1, 2]. Recent studies have shown that inflammatory cells recruited into the uvea and retina during ocular inflammation secrete a variety of cytokines that activate signal transducers and activators of transcription (STAT) pathways [3], and unbridled STAT activation promotes the expansion and persistence of pathogenic T cell subsets implicated in scleritis, autoimmune uveitis, and other ocular inflammatory diseases [4, 5]. Thus, in view of its potential to initiate auto-inflammatory diseases, STAT signaling is under stringent regulation.
The strength and duration of STAT signals are regulated by suppressor of cytokine-signaling (SOCS) proteins, an eight-member family of cytokine-inducible proteins that attenuate or terminate cytokine/growth factor signals, and SOCS1 and SOCS3 are the best characterized members of the family [6, 7]. Inhibitory effects of SOCS1 and SOCS3 derive from direct interactions with cytokine receptors and/or JAKs, targeting them for proteasome-mediated degradation and thereby preventing further recruitment of STATs to the activated cytokine/receptor complex [8]. In a recent study, lymphocytes from patients with scleritis were found to be markedly defective in SOCS1, suggesting that the inability to induce SOCS1 expression may contribute to the persistent inflammation in this potentially blinding disease [9]. In-line with the potential role of SOCS1 in mitigating ocular inflammation, rats and mice with targeted overexpression of SOCS1 in the retina are partially protected from experimental autoimmune uveitis (EAU), and SOCS1 protected their retinal cells from apoptosis [9]. With the establishment of the roles of SOCS proteins in mitigating posterior uveitis, there is interest to examine whether they also contribute to the regulation of host immunity during anterior uveitis and/or ocular inflammatory diseases of infectious etiology. While there is interest in therapeutic use of SOCS mimetics, recent reports indicate that viruses may promote their replication by subverting host anti-viral immune responses through the induction of SOCS1 and SOCS3 [10–12]. Thus, therapeutic use of SOCS mimetics or agonists may pose the risk of promoting virus replication and persistence or exacerbating of viral infections. However, it is not clear whether increased expression of SOCS1 by ocular cells can promote HSV-1 replication or exacerbate ocular HSV-1 infection.
In this study, we have used a SOCS1-deficient mouse strain to investigate the role of SOCS1 in endotoxin-induced uveitis (EIU), an animal model of human non-infectious anterior uveitis [13, 14]. We have also used transgenic rats with targeted overexpression of SOCS1 in the retina (SOCS1-Tg) to investigate the role of SOCS1 during ocular HSV-1 infection. In addition, we describe the generation of two recombinant cell-penetrating SOCS proteins (membrane-translocating sequence (MTS)-SOCS1 and MTS-SOCS3) that can potentially be used to treat ocular inflammatory diseases of infectious or autoimmune etiology.
Materials and Methods
Animals
Six- to 8-week-old C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). The SOCS1−/−STAT1−/− (SOCS1−/−) or SOCS1+/+STAT1−/− (SOCS1+/+) mice are on C57BL/6 background and have been described previously [15, 16]. The transgenic rat strain on Fischer background expressing full-length mouse SOCS1 complementary DNA (cDNA) under direction of an opsin promoter element has been described previously [9]. Animal care and use were in compliance with NIH guidelines on animals use in research.
Endotoxin-Induced Uveitis (EIU)
EIU was induced in wild-type (WT) or SOCS1−/− mice by subcutaneous (SC) injection of 200 μg/kg of Salmonella typhimurium endotoxin (Difco Laboratories, Detroit, MI) in 50 μl of sterile saline as described previously [17]. Development of EIU was determined by histological analysis of enucleated eyes on day 1 post-injection. In some experiments, histological analysis was also performed on eyes enucleated on day 2 post-injection.
HSV-1 Infection
The McKrae strain of herpes simplex virus type 1 (HSV-1) was propagated on Vero cell monolayers and purified as previously described [18]. Briefly, confluent monolayers of Vero cells were infected with HSV-1 at multiplicity of infection (moi) of 0.01. After 2-h adsorption, virus was aspirated and the monolayers were washed once with serum-free medium, then, re-fed with fresh serum-free medium. The cells were further incubated at 37 °C. At maximum cytopathic effect, the cells were frozen and thawed three times and centrifuged at 3,000 rpm for 10 min. Supernatant was collected, and the virus was further purified by centrifugation at 14,000 rpm for 90 min in a Sorvall SS 34 rotor. Infectivity of the purified virus was determined by plaque titration with the antibody-overlay method. In brief, Vero cell monolayers on a 96-well plate were infected with log-diluted virus suspension for 2 h; after which, the monolayer was washed once with fresh serum-free minimum essential medium (MEM) and overlaid with serum-free medium supplemented with 2 % human gamma globulins (human IgG Cohn fraction II, III: anti-HSV neutralizing antibody titers by 50 % plaque reduction, 1:640; Sigma). Forty-eight hours post-infection (p.i.), viral plaques were counted and titrated. Virus was further purified by sucrose density-gradient centrifugation (10–60 %w/v) using a Beckman SW28 rotor for 1 h at 11,500 rpm. For anterior chamber inoculation, rats were anesthetized by intramuscular injection of ketamine hydro-chloride and 105 plaque-forming units (pfu) virus in 10-μl MEM was injected into one eye using a 30G needle. Virus titer in HSV-1-infected eyes was measured by the plaque assay. For the virus titration, rat eye globes were enucleated, the lens removed, and weight of the remaining ocular tissue was measured. With a mortar and pestle, the tissues were homogenized to 10 % emulsion and plaque-titrated for infectivity by antibody-overlay method. We detected cytokines chemokine/chemokine secretion by ELISA using Pierce SearchLight technology (Pierce).
FACS Analysis and Intracellular Protein Staining
The freshly isolated peripheral blood mononuclear cells (PBMC) from blood, lymphocytes (LN), and splenocytes were subjected directly to cell surface fluorescence-activated cell sorting (FACS) analysis using the labeled mAbs indicated on the figures. The intracellular cytokine staining assay was performed as previously described cells [19]. Briefly, the cells were stimulated with PMA and ionomycine for 4 h, and Brefeldin A (5 μg/ml) was added during the final 2 h of stimulation. The assay was performed using intracellular cytokine staining kit (BD Bioscience). The FACS analysis was performed in Becton-Dickinson FACSCalibur (BD Pharmingen, San Diego, CA).
Quantitative and Semi-Quantitative RT-PCR Analyses
Total RNA was extracted using the TriZol reagent according to the procedures recommended by the manufacturer (Invitrogen, Gaithersburg, MD). All RNA samples were digested with RNase-free DNase 1 (Life Technologies) for 30 min, purified by phenol/chloroform extractions. RNA integrity was verified by analysis of 18S and 28S ribosomal RNA expression on RNA gels. RNA (10 μg), SuperScript III Reverse Transcriptase (Invitrogen), and oligo(dT)12–16 was used for first-strand synthesis as previously described [20]. First-strand synthesis containing each messenger RNA (mRNA) sample but without reverse transcriptase was performed to control for possible DNA contamination; failure to obtain RT-PCR products with any of the PCR amplimers confirmed the absence of contaminating DNA. All cDNA preparations used were suitable for PCR on the basis of efficient amplification of a β-actin sequence. Real-time PCR was performed on an ABI 7500 (Applied Biosystems), and PCR parameters were as recommended for the TaqMan Universal PCR kit (Applied Biosystems). Primers and probes were purchased from (Applied Biosystems). The sequences of RT-PCR primers were as follows: mCCR6 5′-GGGACTGGAGCTG TTCTTTGG GTT-3′ and 5′-GCAGCAATGCAGGAAAGCCAGGAC CT-3′, mCXCR3 5′AACTCTTCCATTGTGGGCAG-3′ and 5′-AAGGCCCCTGCATAGAAGTT-3′, mMig 5′-TACTTTACCAACAAGCACCC-3′ and 5′-TTGTTGGC TGTGTAGAACAC-3′, mIP-10 10 5′-CTAGCTCAGGCT CGTCAGTTC-3′ and 5′-AACTACCCATTGATACATAC-3′, mLARC 5′-TGCGGTGGCAAGCGT CTG-3′ and 5′-CCCAGCTGTGATCAT TTCCTCCTT-3′, and β-actin 5′-CAAGTCATCACTATTGGCAACGA-3′ and 5′-CCCA AGAAGGAAGGCTGGA-3′.
Ribonuclease Protection Assay (RPA)
RPA was performed with RNA (10 μg), [32P]-UTP-radiolabeled RNA probes, and chemokine/chemokine receptor RPA kit (BD Biosciences, San Diego) as recommended by manufacturer.
Western Blot Analysis
Whole cell lysates were prepared as previously described [21]. Briefly, protein extracts were fractionated on a 4–12 % gradient SDS-PAGE and transferred to a PVDF membrane. Antibodies used were as follows: pSTAT3 (Cell Signaling Technology), SOCS1, and β-actin (Santa Cruz Biotechnology). Pre-immune serum was used in parallel as controls and signals were detected with HRP-conjugated secondary F(ab′)2 Abs (Zymed Labs, San Francisco, CA) using the ECL Plus Kit (Amersham, Arlington Heights, IL).
Histology
Eight-week-old WTor SOCS1-Tg rat eyes were carefully enucleated, fixed in 4 % glutaraldehyde for 30 min and transferred to 10 % buffered formalin for over 24 h. Specimens were dehydrated through graded alcohols and embedded in paraffin. Serial vertical sections through the pupillary-optic nerve plane were cut and stained with hematoxylin and eosin (H&E). Photographs of representative sections were taken on an Olympus photomicroscope.
Generation and Characterization of MTS-SOCS1 and MTS-SOCS3 Proteins
Full-length murine SOCS1 or SOCS3 cDNA was ligated to a sequence coding for a MTS composed of 12 amino acids from a hydrophobic signal sequence from fibroblast growth factor 4 as described [22, 23]. The MTS and a His tag (to facilitate purification) were attached as illustrated in figures. The His/SOCS/MTS construct was then ligated to the multiple cloning site of the pET100/DTOPO vector. The recombinant proteins were produced in BL21 Escherichia coli (Invitrogen) and purified on a Ni-NTA column (Qiagen). MTS-SOCS1, MTS-SOCS3, SOCS1, or SOCS3 protein was labeled with fluorescein isothiocyanate (FITC), cultured with RAW macrophage cell line, and FITC-labeled MTS-SOCS proteins were detected by flow cytometry to verify intracellular delivery of the MTS-SOCS proteins.
Statistical Analyses
Student t test is used as indicated in the figure panels. Asterisk indicates p values for their significance while indicates their p values for their very significance.
Results
Innate Immune Cells Are Expanded in Peripheral Blood of SOCS1KO Mice
SOCS1-deficient mice die within 3 weeks after birth from growth retardation, thymic atrophy, fulminant hepatitis, and massive infiltration of mononuclear cells into the lung. However, they can be rescued from neonatal lethality by breeding the socs1−/− strain onto a stat1−/− or ifn-γ−/− background [15, 16]. The socs1−/−/stat1−/− double knockout mouse strain is here forth referred to as SOCS1KO and was used to investigate the role of SOCS1 in regulation of ocular inflammation in the anterior segment of the eye. The stat1−/− mouse strain (referred to as control) is normal by most criteria examined, including their immunological responses, and this strain is routinely used as the appropriate control for the SOCS1KO strain [16, 24]. We verified by Western blot analysis that each SOCS1KO mouse used in this study was indeed deficient in SOCS1 (Fig. 1a). Analysis of peripheral blood mononuclear cells (PBMC) of the SOCS1KO mouse revealed substantial elevation of innate immune cells as indicated by increased frequency of cells expressing monocyte, macrophage and natural killer (NK) cell, and myeloid cell surface markers (Fig. 1b). In contrast to control mice with innate immune cells comprising 2.7 % of the cells in the PBMC, 21.4 % of the cells in the blood of the SOCS1KO mouse were innate immune cells and 43.2 % of the cells were Gr-1+ compared to 10.27 % on control cells (Fig. 1b). DX5 (CD49b) is an integrin alpha subunit involved with cell adherence, and similar to Gr-1, we observed marked increase in expression of this cell-surface protein present on a variety of innate immune cells. The increase in innate immune cell populations was not restricted to the blood as their levels were also elevated in lymph nodes of SOCS1KO mice (Fig. 1c), suggesting that SOCS1 is a negative regulator of activation and expansion of peripheral lymphocytes, monocytes, and granulocytes.
Fig. 1.
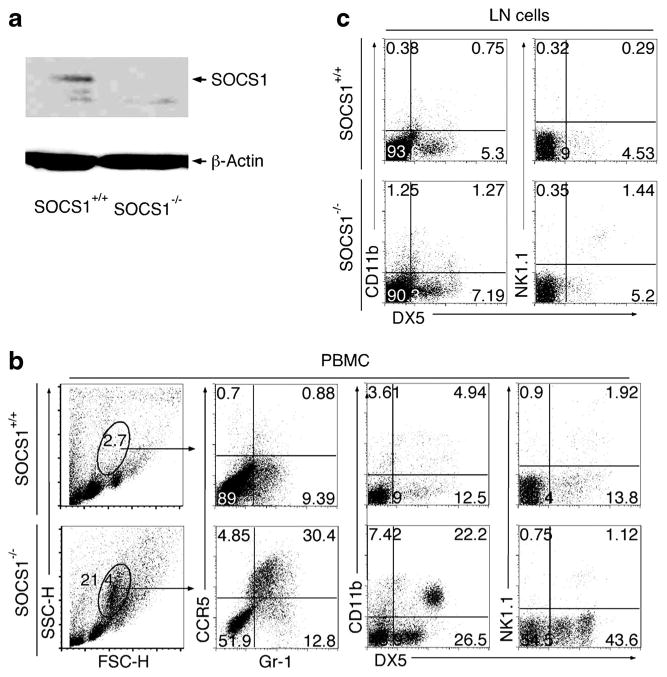
Characterization of innate immune cells of SOCS1-KO mice. a Western blot analysis of protein extracts from PBMC of control or SOCS1KO mice. Freshly isolated PBMC (b) and LN (c) cells from control or SOCS1KO mice were stained with labeled monoclonal Abs specific to cell surface markers of innate immune cells and analyzed by FACS. Numbers in quadrants represent percentage of cells expressing cell surface markers indicated on figure. Data represent at least three independent experiments.
EIU is Exacerbated in SOCS1-Deficient Mice
EIU is a well-characterized model for human acute non-infectious anterior uveitis and is induced by peripheral administration of lipopolysaccharide (LPS) [14]. Similar to the human disease, EIU is characterized by intense acute cellular infiltration (mainly polymorphonuclear neutrophils (PMN) and macrophages), and protein exudation into ocular anterior segment and the inflammatory process in rodents is maximal 24 h post-injection and lasts for 48 h [25-27]. To investigate whether SOCS1 plays a role in regulating inflammatory cells that mediate anterior uveitis, we induced EIU in 8-week-old WT C57BL/6, control (SOCS1+/+), or SOCS1KO mice as described under the section “MATERIALS AND METHODS.” Gross morphological examination of the eyes revealed that the C57BL/6 and control mice responded similarly to LPS as indicated by comparable severities of inflammation in the anterior chamber of the eye (Fig. 2a; left and middle panels). However, in contrast to the relatively mild response of the C57BL/6 and control mice, we observed severe inflammation with obscured anterior chamber in the SOCS1KO mouse (Fig. 2a; right-most panel). Histological examination of the eyes 24 or 48 h post-injection further revealed increased inflammatory cells in the iris, ciliary body, and posterior chamber of the SOCS1KO mice compared to control (Fig. 2b). However, the inflammatory response was most severe at the 24-h time point, and these results are consistent with typical EIU. It is notable that the loss of socs1 also resulted in the recruitment of the inflammatory cells into the posterior vitreous and optic nerve head during EIU (Fig. 2c, d). As indicated, detection of significant numbers of infiltrating inflammatory cells in the anterior segment, posterior vitreous, and optic nerve head region was accompanied by extensive hemorrhage.
Fig. 2.
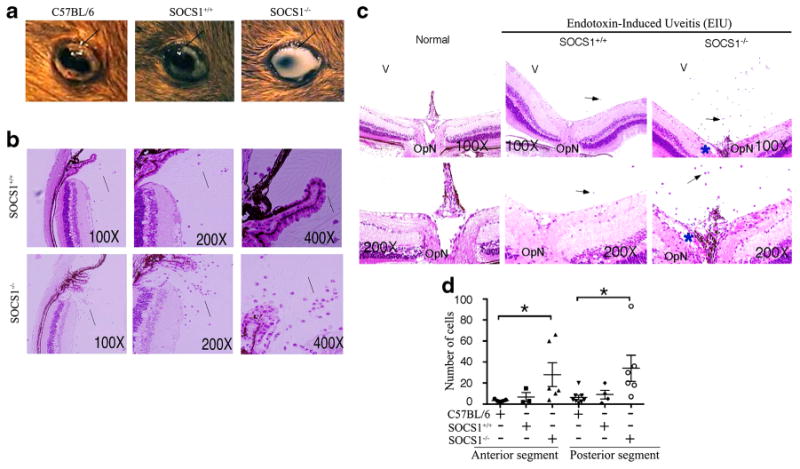
Loss of SOCS1 exacerbates EIU. EIU was induced as described in the “MATERIALS AND METHODS” section. a Morphology of eyes at 48 h after induction of EIU. H&E sections of anterior portion (b) and posterior segment (c) of eyes enucleated 24 h after induction of EIU. Arrows indicate inflammatory cells in vitreous (b, c). d Number of inflammatory cells detected in the anterior or posterior segment of EIU eyes. *P<0.02).
Severe EIU in SOCS1 KO Mice Correlates with Increases in Recruitment of Macrophages and Ocular Expression of Chemokines/Chemokine Receptors
We next examined whether increased recruitment of inflammatory cells into the eye derived from aberrant induction of the expression of chemotactic molecules that promote extravasation of inflammatory cells into tissues. We isolated and purified PBMC from control (SOCS1+/+) or SOCS1KO mice 24 h after induction of EIU and analyzed the freshly isolated cells by FACS. We found that a substantial proportion of the cells detected were macrophages and PMN on the basis of their cell surface markers, and their levels were much higher in the SOCS1KO mice (Fig. 3a). In order to understand mechanisms that might underlie the increased recruitment of inflammatory monocytes/granulocytes into the eyes of the SOCS1KO mice, we examined the expression of chemokines and chemokine receptors in mouse cornea and retina. We isolated RNA from the retina of the control or SOCS1KO mice, converted them to cDNA, and performed RT-PCR or RT-qPCR analysis. We show here that the expression of chemokines [CXCL9 (Mig), CXCL10 (IP-10), CCL20 (LARC/MIP-3α)] and chemokine receptors [CCR6, CXCR3] that mediates the recruitment of inflammatory cells to sites of inflammation is markedly upregulated in the retinas of SOCS1-deficient mice compared to those of C57BL/6 or control mice with EIU (Fig. 3b). To determine whether SOCS1 regulates chemokine and chemokine receptor expression, we analyzed activated AE.7 Th1 cell line and the D10.G4.1 Th2 cells that express chemokine characteristic of these two T cell subsets and examined the effect of SOCS1 overexpression. We overexpressed SOCS1 in the D10.G4.1 cells, and D10.G4.1 cells expressing the empty vector served as controls. The RPA analysis showed that overexpression of SOCS1 induces a substantial reduction of IP-10, eotaxin, and RANTES in the Th2 cells (Fig. 3c), suggesting that some chemokines and chemokine receptors are indeed under feedback regulation by SOCS1. Interestingly, the expression of the pro-inflammatory cytokine, IL-17, and the CCL2 chemokine was also increased in the SOCS1-deficient mouse retina while the level of IL-10 mRNA transcript was significantly lower compared to the level in control mice (Fig. 3d). Taken together, these results suggest that SOCS1 may confer protection from anterior uveitis by inhibiting the expression of molecules that promote trafficking of inflammatory cells into the immune privileged environment of the anterior uvea and retina.
Fig. 3.
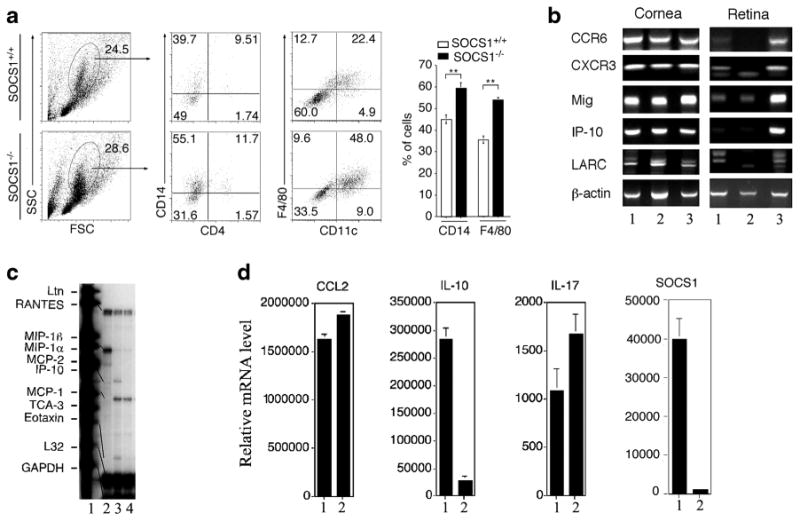
Severe EIU in SOCS1KO eyes correlated with alterations in the expression of chemokines, chemokine receptors, and inflammatory proteins in the retina. a Analysis of freshly isolated PBMC from control or SOCS1KO mice 48 h after induction of EIU by FACS. Numbers in quadrants represent percentage of cells expressing cell surface markers indicated on figure. b RT-PCR analysis of RNA isolated from the cornea or retina of control or SOCS1KO mice 48 h after induction of EIU—WT C57BL/6 mice (lane 1), control mice with EIU (lane 2), and SOCS1KO mice with EIU (lane 3). c RPA analysis of the expression of chemokine or chemokine receptors— probe (lane 1), RNA from AE7 Th1 cells (lane 2), mouse D10.G4.1 Th2 cells stably transfected with pBabe vector alone (lane 3), and pBabe-SOCS1 (lane 4). d RT-qPCR analysis of RNA isolated from the retina of control or SOCS1KO mice 24 h after induction of EIU—control mice with EIU (lane 1) and SOCS1KO with EIU (lane 2). Data represent three independent experiments (five mice/group).
SOCS1 Overexpression Suppresses HSV-1 Replication and Mitigates Ocular Pathology
Ocular HSV-1 infection is of significant public health interest [18, 28]. Unlike HSV-1 infections at the skin or mucous membrane which may not be progressive and mainly characterized by recurrent, self-limited epithelial lesions, ocular HSV-1 infection can involve the cornea (epithelium, keratocyte, endothelium), anterior uveal tract (iris, ciliary body), and/or the posterior segment (vitreous, retina, choroid, optic nerve) [18, 28]. Interestingly, recent reports indicate that viruses can evade anti-viral immunity by inducing expression of SOCS1 protein by infected host cells [10–12]. Sustained production of high levels of SOCS1 protein in infected cells is thus thought to render infected cells unresponsive to protective cytokine-induced STAT1 signals required in mediating host immunity to viruses and inhibiting viral replication. In this study, we tested the hypothesis that sustained overexpression of SOCS1 in ocular cells would promote HSV-1 replication in ocular resident cells and thus exacerbate ocular HSV-1 infection. We thus used a transgenic rat strain with targeted overexpression of SOCS1 in the retina under the direction of an opsin promoter element [9] to investigate the role of SOCS1 in subverting host immunity to HSV-1 in the eye. Each SOCS1 transgenic rat used in the study was genotyped to verify that it indeed constitutively overexpressed SOCS1 in the retina (Fig. 4a). We infected the SOCS1 transgenic rats by injecting HSV-1 (1 × 105 pfu) into the anterior chamber and monitored the infection over 7 days as described [18, 28]. The intracameral inoculation of HSV-1 produced an intense inflammatory reaction that evolved between 1 and 7 days post-inoculation and was accompanied by pathological changes assessed and scored in a masked fashion by an ophthalmologist and an ocular pathologist. Histological analysis revealed enhanced ocular inflammation at early time points post-infection in the WT compared to the SOCS1-Tg rats (Fig. 4b). Although we observed marked increases in inflammatory cells in the SOCS1-Tg rat eyes by day 7 post-inoculation, the levels were similar to those observed in WT eyes (Fig. 4b), suggesting that overexpression of SOCS1 did not exacerbate ocular inflammation. Consistent with the kinetics of the inflammatory response, ocular pathology was less in the SOCS1-Tg rats at various time points post-infection (Fig. 4c). It is notable that the disease score in the SOCS1-Tg rats steadily increased with time and approached scores observed in WT rat by day 7 (Fig. 4c), suggesting that overexpression of SOCS-1 in retinal cells might have delayed the onset of severe ocular inflammation. Moreover, we detected significant less HSV-1 virus in the eyes of the SOCS1 transgenic rats (Fig. 4d), and the levels of pro-inflammatory molecules were consistently elevated in HSV-1-infected WT rats at each time point compared to their transgenic counterparts (Fig. 4e). Taken together, these results suggest that overexpression of SOCS1 inhibited HSV-1 replication and conferred protection against HSV-1-induced ocular pathology.
Fig. 4.
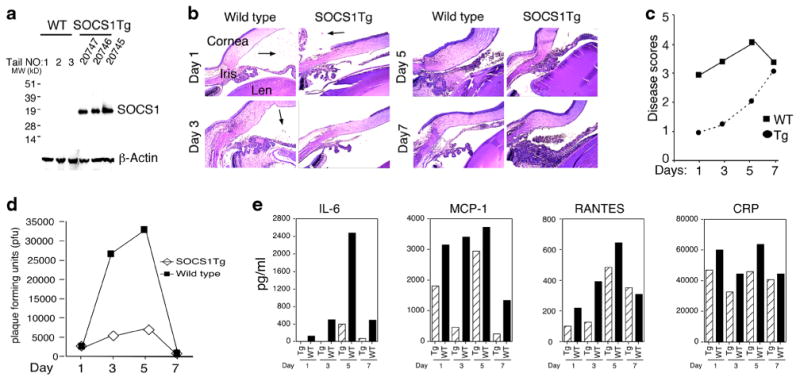
SOCS1-Tg rats had higher viral load and were protected from HSV-1-induced ocular pathology. a Western blot analysis of protein extracts from retina of wild-type or SOCS1 transgenic rats. b Photomicrograph of histological sections from WT or SOCS1 transgenic rat eyes at various days post-infection. c Clinical disease scores. d Quantification of virus titer in the eyes of HSV-1-infected WT or SOCS1 transgenic rats by the plaque assay. e Detection of cytokines in HSV-1-infected WT or SOCS1 TR rat eyes by ELISA. Data represent three independent experiments.
Genetic Engineering of Cell-Penetrating SOCS1 (MTS-SOCS1) and MTS-SOCS3 Proteins
The critical roles of SOCS1 and SOCS3 in negatively regulating activities of pro-inflammatory cytokines suggest that they can be exploited therapeutically in inflammatory and autoimmune diseases. To regulate cytokine signaling and immune responses by delivering exogenous SOCS proteins into inflammatory cells, we generated cell-penetrating SOCS1 (MTS-SOCS1) and MTS-SOCS3 by fusing the mouse SOCS1 or SOCS3 cDNA sequence to the cDNA sequence coding for a hydrophobic 12 AA sequence of the signal peptide of the Kaposi FGF4 protein (Figs. 5a and 6a). A similar approach has been used to deliver transcription factors into inflammatory cells [22, 23]. The MTS-SOCS1 was purified, and we used Western blot analysis with SOCS-1-specific antibodies to confirm that the recombinant protein was indeed MTS-SOCS1 (Fig. 5b, c). The MTS-SOCS1 or control GST-SOCS1 was then labeled with FITC and incubated with inactivated macrophages. Entry of MTS-SOCS1 into cells was quantified by flow cytometry and suggested that MTS-SOCS-1 was efficiently delivered into macrophage cells (Fig. 5d). To examine whether MTS-SOCS1 functions as a negative feedback regulator, we loaded macrophages with MTS-SOCS1 orGST-SOCS1, stimulated with IFN-γ for 15min, and analyzed by Western blotting. As indicated in Fig. 5e, MTS-SOCS1 efficiently suppressed IFN-γ-induced STAT1 signaling, suggesting that MTS-SOCS1 can be used to inhibit STAT1 signaling pathways of inflammatory cells. We used a similar strategy to produce and characterize MTS-SOCS3 (Fig. 6b, c), and we show here that it can inhibit IL-6-induced activation of STAT3 (Fig. 6d). To examine whether MTS-SOCS3 can potentially be used to inhibit intraocular inflammation, we induced experimental autoimmune uveitis in mice and cultured the pathogenic T cells that mediated uveitis with MTS-SOCS3. We show here that MTS-SOCS3 suppressed the expansion of the pathogenic Th17 cells (Fig. 6e), suggesting that MTS-SOCS3 can potentially be used to suppress uveitis.
Fig. 5.
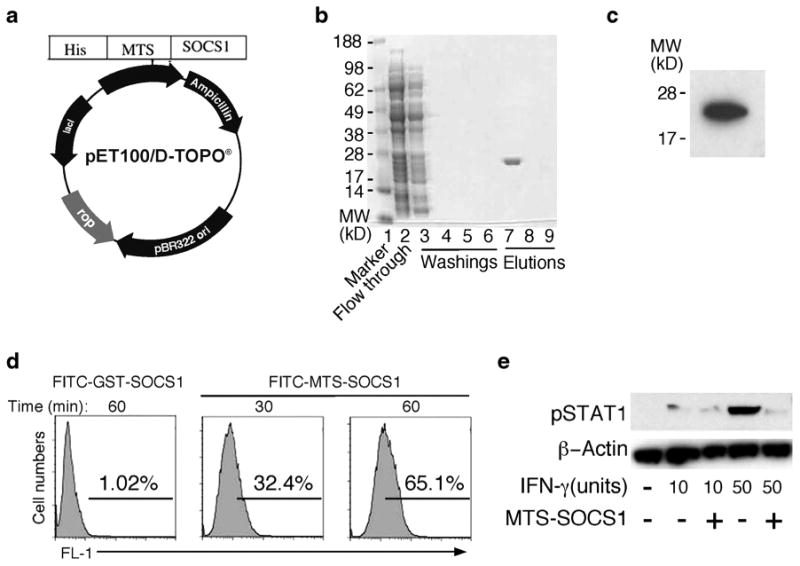
Cell-penetrating recombinant SOCS1 (MTS-SOCS1)—generation and use to suppress STAT1 activation in macrophage. a Schematic illustration of MTS-SOCS1 expression vector. MTS-SOCS1 plasmid contained a membrane-translocating sequences (MTS) and histidine tag for affinity purification (His). MTS (AAVLLPVLLAAP) is a 12 amino acids hydrophobic signal sequence of the fibroblast growth factor 4. b Coomassie blue stained gel at various stages of purification of the MTS-SOCS1. c Western blot analysis of purified MTS-SOCS1. d MTS-SOCS1 or control GST-SOCS1 was labeled with FITC and incubated with inactivated macrophages (“MATERIALS AND METHODS”). Entry of MTS-SOCS1 into cells was quantified by flow cytometry. e Macrophages were loaded with MTS-SOCS1 or GST-SOCS1, stimulated with IFN-γ for 15 min and analyzed by Western blotting. Data represent three independent experiments.
Fig. 6.
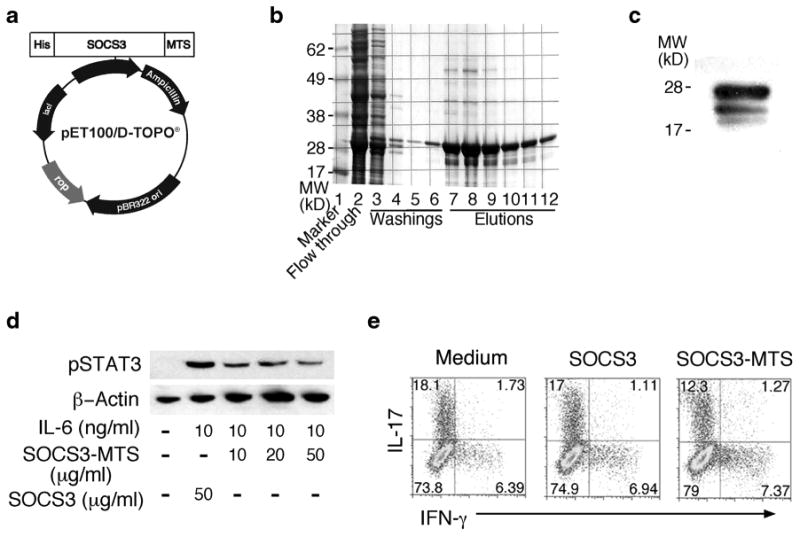
Cell-penetrating SOCS3 (MTS-SOCS3) suppressed the expansion of Th17 cells. a Schematic illustration of MTS-SOCS3 expression vector. b Coomassie blue stained gel at various stages of purification of the MTS-SOCS3.c Western blot analysis of purified MTS-SOCS3. d Macrophages were loaded with MTS-SOCS3 or GST-SOCS3, stimulated with IL-6 for 15 min, and analyzed by Western blotting. e Th17 uveitogenic T cells were cultured in medium containing MTS-SOCS3 or SOCS3 for 4 days, and IL-17 or IFN-γ expression was detected by intracellular cytokine staining and flow cytometry. Data represent three independent experiments.
Discussion
In this study, we have shown that EIU, the animal model of acute anterior uveitis, is exacerbated in SOCS1-deficient mice. This suggests that SOCS1 may confer protection from anterior uveitis. In addition, the innate immune cells of the SOCS1KO mice exhibited pre-activated phenotype indicating that an in vivo function of SOCS1 might be to inhibit or restrain pro-inflammatory activities of myeloid and lymphoid cells. It also suggests that lose of SOCS1 might have contributed to the extensive infiltration of inflammatory cells into the anterior uvea, vitreous, and optic nerve head observed during EIU. Although EIU is generally thought to be limited to the anterior segment of the eye, with minor involvement of the posterior pole, the disease in the SOCS1-deficient mice extended to the posterior segment with significant numbers of infiltrating inflammatory cells and vasculitis in the optic nerve head. Our data also indicate that SOCS1 is a negative feedback regulator of the expression of CXCL9 (MIG), CXCL10 (IP-10), CCR6, and CXCR3 in the retina, and the upregulation of these chemokine and chemokine receptors in the SOCS1-deficient ocular cells might have contributed to increased recruitment of the infiltrating cells. Taken together, these results suggest that SOCS1 may contribute to mechanism of ocular immune privilege by inhibiting trafficking of inflammatory cells into the retina. Therefore, increasing SOCS1 level in retinal cells can be used as a strategy to treat not only posterior but also anterior uveitis.
Induced expression of SOCS1, SOCS3, and vSOCS by viruses has been suggested as a mechanism by which some viruses including varicella-zoster, rhino, influenza, and cytomegalovirus evade host immunological responses [10–12]. In this study, we investigated whether SOCS1 mitigates or exacerbates HSV-1 infection in the eye. We observed significant elevation of HSV-1 titers (pfu) in WT mice infected with HSV-1 compared their counterpart SOCS1 transgenic rat eyes. Thus, in contrast to reports suggesting that viruses may evade host immunological responses through upregulation of SOCS proteins, analysis of the HSV-1-infected SOCS1 transgenic rats indicate that increased expression of SOCS1 in ocular cells does not promote HSV-1 replication during ocular HSV-1 infection. On the other hand, our data suggest that overexpression of SOCS1 attenuated virus-induced inflammation and rendered the ocular cells refractory to STAT1 signaling, thereby protecting the ocular cells from cytotoxic effects of inflammatory proteins elicited by virus infection. The decrease of disease severity observed in the HSV-1-infected SOCS1 transgenic rats compared to their WT counterparts also suggests that enhancing endogenous SOCS1 expression or delivering SOCS1 protein into ocular cells can be exploited to mitigate ocular HSV-1 infection.
Viral infections that cause herpetic stromal keratitis or anterior uveitis present unique challenges to the immune system because a very aggressive response to the pathogen can damage the visual axis while a tepid response may promote chronic persistent inflammation that can spread to the retina and may induce retinal degenerative changes or retinal atrophy. SOCS proteins thus play important roles in this balancing act by regulating the intensity and duration of pro-inflammatory cytokine signals and immune responses. In this study, we have shown that SOCS1 protects ocular cells from inflammation by inhibiting expression of chemotactic molecules that promote trafficking of inflammatory cells into the uvea and retina. We have also successfully engineered the MTS-SOCS1 and MTS-SOCS3 recombinant proteins and shown that delivery of the MTS-SOCS1 into macrophages inhibited IFN-γ-induced STAT1 activation, suggesting that MTS-SOCS1 can be used to mitigate anterior uveitis. In addition, delivery of MTS-SOCS3 into Th17 cells inhibited expansion of this pathogenic T cell subset that mediates posterior uveitis. MTS-SOCS3-mediated suppression of Th17 expansion in vitro has thus laid the foundation for eventually utilizing MTS-SOCS3 in treating posterior uveitis. Taken together, our results suggest that MTS-SOCS proteins and SOCS mimetics may be useful additions to our current armamentarium for treating ocular inflammatory diseases and may be useful in inhibiting HSV-1 infection in the eye.
Footnotes
Cheng-Rong Yu and Kozaburo Hayashi contributed equally to this work.
References
- 1.Caspi RR. A look at autoimmunity and inflammation in the eye. Journal of Clinical Investigation. 2010;120:3073–3083. doi: 10.1172/JCI42440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nature Reviews Immunology. 2009;9:440–447. doi: 10.1038/nri2548. [DOI] [PubMed] [Google Scholar]
- 3.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 4.Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, et al. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nature Medicine. 2007;13:711–718. doi: 10.1038/nm1585. [DOI] [PubMed] [Google Scholar]
- 5.Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. Journal of Experimental Medicine. 2008;205:799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nature Immunology. 2003;4:1169–1176. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- 7.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 8.Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nature Reviews Immunology. 2002;2:410–416. doi: 10.1038/nri818. [DOI] [PubMed] [Google Scholar]
- 9.Yu CR, Mahdi RR, Oh HM, Amadi-Obi A, Levy-Clarke G, Burton J, et al. Suppressor of cytokine signaling-1 (SOCS1) inhibits lymphocyte recruitment into the retina and protects SOCS1 transgenic rats and mice from ocular inflammation. Investigative Ophthalmology & Visual Science. 2011;52:6978–6986. doi: 10.1167/iovs.11-7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramirez-Martinez G, Cruz-Lagunas A, Jimenez-Alvarez L, Espinosa E, Ortiz-Quintero B, Santos-Mendoza T, et al. Seasonal and pandemic influenza H1N1 viruses induce differential expression of SOCS-1 and RIG-I genes and cytokine/chemokine production in macrophages. Cytokine. 2013;62:151–159. doi: 10.1016/j.cyto.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bedke N, Sammut D, Green B, Kehagia V, Dennison P, Jenkins G, et al. Transforming growth factor-beta promotes rhinovirus replication in bronchial epithelial cells by suppressing the innate immune response. PLoS One. 2012;7:e44580. doi: 10.1371/journal.pone.0044580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo CJ, Yang LS, Zhang YF, Wu YY, Weng SP, Yu XQ, et al. A novel viral SOCS from infectious spleen and kidney necrosis virus: interacts with Jak1 and inhibits IFN-alpha induced Stat1/3 activation. PLoS One. 2012;7:e41092. doi: 10.1371/journal.pone.0041092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vos AF, Klaren VN, Kijlstra A. Expression of multiple cytokines and IL-1RA in the uvea and retina during endotoxin-induced uveitis in the rat. Investigative Ophthalmology & Visual Science. 1994;35:3873–3883. [PubMed] [Google Scholar]
- 14.Rosenbaum JT, McDevitt HO, Guss RB, Egbert PR. Endotoxin-induced uveitis in rats as a model for human disease. Nature. 1980;286:611–613. doi: 10.1038/286611a0. [DOI] [PubMed] [Google Scholar]
- 15.Naka T, Tsutsui H, Fujimoto M, Kawazoe Y, Kohzaki H, Morita Y, et al. SOCS-1/SSI-1-deficient NKT cells participate in severe hepatitis through dysregulated cross-talk inhibition of IFN-gamma and IL-4 signaling in vivo. Immunity. 2001;14:535–545. doi: 10.1016/s1074-7613(01)00132-7. [DOI] [PubMed] [Google Scholar]
- 16.Yu CR, Mahdi RM, Liu X, Zhang A, Naka T, Kishimoto T, et al. SOCS1 regulates CCR7 expression and migration of CD4+ T cells into peripheral tissues. Journal of Immunology. 2008;181:1190–1198. doi: 10.4049/jimmunol.181.2.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egwuagu CE, Mahdi RM, Chan CC, Sztein J, Li W, Smith JA, et al. Expression of interferon-gamma in the lens exacerbates anterior uveitis and induces retinal degenerative changes in transgenic Lewis rats. Clinical Immunology. 1999;91:196–205. doi: 10.1006/clim.1999.4701. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi K, Hooper LC, Chin MS, Nagineni CN, Detrick B, Hooks JJ. Herpes simplex virus 1 (HSV-1) DNA and immune complex (HSV-1-human IgG) elicit vigorous interleukin 6 release from infected corneal cells via Toll-like receptors. Journal of General Virology. 2006;87:2161–2169. doi: 10.1099/vir.0.81772-0. [DOI] [PubMed] [Google Scholar]
- 19.Yu CR, Oh HM, Golestaneh N, Amadi-Obi A, Lee YS, Eseonu A, et al. Persistence of IL-2 expressing Th17 cells in healthy humans and experimental autoimmune uveitis. European Journal of Immunology. 2011;41:3495–3505. doi: 10.1002/eji.201141654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee YS, Amadi-Obi A, Yu CR, Egwuagu CE. Retinal cells suppress intraocular inflammation (uveitis) through production of interleukin-27 and interleukin-10. Immunology. 2011;132:492–502. doi: 10.1111/j.1365-2567.2010.03379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egwuagu CE, Yu CR, Zhang M, Mahdi RM, Kim SJ, Gery I. Suppressors of cytokine signaling proteins are differentially expressed in Th1 and Th2 cells: implications for Th cell lineage commitment and maintenance. Journal of Immunology. 2002;168:3181–3187. doi: 10.4049/jimmunol.168.7.3181. [DOI] [PubMed] [Google Scholar]
- 22.Liu D, Zienkiewicz J, DiGiandomenico A, Hawiger J. Suppression of acute lung inflammation by intracellular peptide delivery of a nuclear import inhibitor. Molecular Therapy. 2009;17:796–802. doi: 10.1038/mt.2009.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jo D, Liu D, Yao S, Collins RD, Hawiger J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nature Medicine. 2005;11:892–898. doi: 10.1038/nm1269. [DOI] [PubMed] [Google Scholar]
- 24.Naka T, Matsumoto T, Narazaki M, Fujimoto M, Morita Y, Ohsawa Y, et al. Accelerated apoptosis of lymphocytes by augmented induction of Bax in SSI-1 (STAT-induced STAT inhibitor-1) deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15577–15582. doi: 10.1073/pnas.95.26.15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kozhich AT, Chan CC, Gery I, Whitcup SM. Recurrent intraocular inflammation in endotoxin-induced uveitis. Investigative Ophthalmology & Visual Science. 2000;41:1823–1826. [PubMed] [Google Scholar]
- 26.Shen DF, Buggage RR, Eng HC, Chan CC. Cytokine gene expression in different strains of mice with endotoxin-induced uveitis (EIU) Ocular Immunology and Inflammation. 2000;8:221–225. doi: 10.1076/ocii.8.4.221.6461. [DOI] [PubMed] [Google Scholar]
- 27.Herbort CP, Chan CC, Nussenblatt RB. Endotoxin-induced uveitis in the rat: a hypothesis for preferential involvement of the anterior uvea. Current Eye Research. 1990;9(Suppl):119–124. doi: 10.3109/02713689008999430. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi K, Hooper LC, Hooks JJ. Who (what) pays toll for the development of herpetic stromal keratitis (HSK) Seminars in Ophthalmology. 2008;23:229–234. doi: 10.1080/08820530802111408. [DOI] [PubMed] [Google Scholar]