

Abstract
Signaling by immunoreceptors is often initiated by phosphorylation of cytosolic tyrosines, which then recruit effector molecules. In the case of MHC class I-specific inhibitory receptors, phosphorylation of cytosolic tyrosine residues within immunoreceptor tyrosine-based inhibition motifs (ITIM) results in recruitment of a protein tyrosine phosphatase that blocks activation signals. Recent work has shown that signaling by an HLA-C-specific killer cell Ig-like receptor (KIR) is independent of signaling by activation receptors. It is not known how ITIM phosphorylation is initiated and regulated. Here we show that substitution of histidine 36 (His-36) in the first Ig domain of KIR2DL1 with alanine (KIR2DL1-H36A) resulted in constitutive KIR2DL1 self-association and phosphorylation, and recruitment of tyrosine phosphatase SHP-1. Furthermore, substitution of His-36 with a similar bulky amino acid, phenylalanine (KIR2DL1-H36F), maintained the receptor in its unphosphorylated state, suggesting that steric hindrance by the His-36 side chain prevents constitutive KIR2DL1 self-association and ITIM phosphorylation. The equally strong phosphorylation of KIR2DL1 and KIR2DL1-H36A after inhibition of tyrosine phosphatase by pervanadate suggested that KIR2DL1-H36A is selectively protected from dephosphorylation. We propose that KIR phosphorylation is controlled by the accessibility of ITIM to tyrosine phosphatases, and that KIR binding to HLA-C must override the hindrance His-36 puts on KIR2DL1 self-association. Expression of KIR2DL1-H36A on NK cells led to stronger inhibition of lysis of HLA-C+ target cells than expression of wild type KIR2DL1. These results have revealed that ITIM phosphorylation is controlled by self-association of KIR and that His-36 serves as a gatekeeper to prevent unregulated signaling through KIR2DL1.
Keywords: NK cells, KIR2DL1, Inhibitory receptor, Tyrosine phosphorylation
Introduction
Regulation of many cellular processes depends on inhibitory receptors that carry immunoreceptor tyrosine-based inhibition motifs (ITIMs) in their cytoplasmic tail (1). Natural killer (NK) cell cytotoxicity and cytokine secretion is tightly regulated by ITIM-containing inhibitory receptors (2). Engagement of NK cell inhibitory receptors with their MHC class I ligands on target cells leads to ITIM phosphorylation and recruitment of the protein tyrosine phosphatase (PTPase) SHP-1. SHP-1 bound to phosphorylated ITIMs in inhibitory killer cell Ig-like receptors (KIR) is activated and dephosphorylates the guanine nucleotide exchange factor Vav1, thereby inhibiting actin-dependent signals for NK cell activation (2, 3). Inhibition of activation signals by ITIM-bearing receptors is local and transient, and does not globally impair NK cell responses to other stimuli (4, 5). Based on these characteristics, inhibitory receptors have been considered co-inhibition receptors (6). According to earlier models, their phosphorylation is dependent on tyrosine kinases associated with signaling by activation receptors. Kinases of the Src family, such as Lyn and Lck, have been implicated in the phosphorylation of ITIMs (7–9).
Signals from inhibitory receptors in NK cells block activation signals at a very early step, prior to the actin-dependent clustering of activation receptors and the full signaling cascade for activation. For example, inhibition by ITIM-containing receptors blocks inside-out signals from co-activation receptors to β2 integrin (10) and β2 integrin-mediated adhesion (11). Photoactivation of HLA-C-peptide complexes induced microclusters of inhibitory KIR within seconds of ligand binding, and inhibited NK cell attachment to ICAM-1 and the formation of activation receptor clusters (12). These properties of inhibitory KIR are not easily reconciled with models that place the phosphorylation of ITIMs downstream of signaling by activation receptors.
Recent studies have revealed that the binding of inhibitory KIR2DL1 to HLA-C, and of inhibitory receptor CD94-NKG2A to HLA-E, results in tyrosine phosphorylation of the small adaptor molecule Crk (13). As binding of CD94-NKG2A to purified HLA-E is sufficient to trigger Crk phosphorylation, the ITIM-bearing CD94-NKG2A can signal independently of activation receptor signaling (14). Here we studied the regulation of KIR2DL1 phosphorylation and its association with SHP-1. We have identified a gain-of-function, single amino acid mutant of KIR2DL1, which is constitutively phosphorylated. We propose that KIR2DL1, in its basal state, is subjected to a continuous cycle of phosphorylation and dephosphorylation, and that KIR2DL1 self-association facilitates phosphorylation by protecting phosphorylated ITIMs from PTPases, thereby shifting the equilibrium in favor of phosphorylation.
Materials and Methods
Cell lines and reagents
The human NK cell line YTS was transfected with wild type (WT), ITIM tyrosine mutant, wherein both Tyr residues were mutated to Phe (2YF), and His-36 to Ala (H36A) mutant of KIR2DL1, each tagged with Venus at the cytosolic end. The transfectants were selected in 1 μM puromycin. They are referred to as YTS-2DL1-WT-Venus, YTS-2DL1-2YF-Venus, and YTS-2DL1-H36A-Venus in this paper. Expression of KIR2DL1 in these transfectants was comparable to KIR2DL1 in primary NK cells (Supplementary Fig. 1). YTS cells were cultured in RPMI supplemented with glutamine, 10% fetal bovine serum (FBS), and 50 μM 2-mercaptoethanol (R10 medium). YTS cells express HLA-C*01 and HLA-C*08, two group C1 allotypes, which are not ligands for KIR2DL1. The YTS transfectants were cultured in R10 medium supplemented with 1 μM puromycin.
721.221 cell lines (referred to as 221 cells) transfected with HLA-Cw3 and HLA-Cw4 were obtained from J. Gumperz and P. Parham (Stanford University). These 221 transfectants were cultured in R10 medium. TAP deficient 221-HLA-Cw4 cells were generated by transfection of ICP-47-IRES-GFP (15) into 221-HLA-Cw4 cells, and selection in 1 μM puromycin. Cells were sorted for high GFP expression and low HLA-C on the cell surface.
Antibodies
The antibodies used in this study and their sources are as follows: Anti-GFP (11814460001, Roche; A6455, Invitrogen); Anti-phosphotyrosine-biotin (4G10-biotin; Upstate), Anti-SHP-1 (610126, BD Transduction Laboratories; 07-419, Upstate), Anti-HLA-C (F4/326 (IgG2a), a gift from S.Y. Yang (Memorial Sloan-Kettering Cancer Center, New York). The horseradish peroxidase (HRP) conjugated antibodies were from Santa Cruz. Streptavidin-HRP antibody was obtained from GE Healthcare. Allophycocyanin (APC)-conjugated anti-KIR2DL1 antibody used in flow cytometer studies was from Beckman Coulter (A22332).
DNA mutagenesis and transfection
A KIR2DL1 cDNA tagged at the C-terminus with Venus was cloned into a lentiviral vector pCDH-EF1-MCS-T2A-Puro (CD520A-1, System Biosciences) using XbaI and NotI. The resulting plasmid is referred to as p2DL1-WT-Venus. The ITIM tyrosine mutant Y281,311F (2YF) and histidine 36 to alanine (H36A) mutants were generated by site-directed mutagenesis on p2DL1-WT-Venus. Stable YTS transfectants, expressing 2DL1-WT, 2DL1-2YF, and 2DL1-H36A, each tagged with Venus at C-terminus, were generated by transduction of lenti pseudoviral particles packaged with the cDNA into YTS cells, and selection in 1 μM puromycin (System Biosciences). The construct expressing Cerulean-tagged SHP-1 was made by cloning a SHP-1 cDNA, tagged at C-terminus with a glycine-serine linker of 12 residues followed by Cerulean, into pCDH-EF1-MCS-T2A-Puro (CD520A-1, System Biosciences) using XbaI and NotI. The resulting plasmid is referred to as pSHP-1-Cerulean. The cDNA encoding the N-terminal (1-154; VN155) and C-terminal (155-238 with I152L mutation; VC155) fragments of Venus was PCR amplified from the plasmids pBiFC-VN155 and pBiFC-VC155 (Addgene, deposited by Chang-Deng Hu, Purdue University), respectively. The GFP portion of the plasmid 2DL1-TG (16) was replaced with VN155 cDNA or VC155 cDNA. These resulting plasmids are referred to as p2DL1-WT-VN155 and p2DL1-WT-VC155, and express KIR2DL1 tagged at the C-terminus (cytosolic end) with VN155 and VC155, respectively. The H36A and H36F mutants of KIR2DL1 tagged with VN155 or VC155 were generated by site-directed mutagenesis on p2DL1-WT-VN155 or pDL1-WT-VC155. To make the construct expressing VN155-tagged SHP-1, the GFP coding region of pEGFP expression vector (Clontech Laboratories) was replaced with a SHP-1 cDNA, tagged at C-terminus with a glycine-serine linker of 12 residues followed by VN155 using XhoI and NotI sites. The resulting plasmid is referred as pSHP-1-VN155. Transfection for transient protein expression in YTS cells was achieved using nucleofection (VCA-1003, Amaxa Nucleofection Kit V). Typically, 2 million cells and 2–4 μg of plasmid DNA were used for each transfection. The plasmids encoding Venus tagged with K-Ras membrane-targeting sequence (V-K-Ras) have been described (17). In the construct encoding Venus dimer, the two Venus molecules are attached in tandem with a five amino acid linker. Site-directed mutagenesis was performed using the QuickChange site-directed mutagenesis kit from Stratagene. The following primers were used for site-directed mutagenesis. H36A: GTCATGTTTGAAGCCTTCCTTCTGCACAGAGAGGGG and its complement; H36F: GTCATGTTTGAATTCTTCCTTCTGCACAGAGAGGGG and its complement
Bi-molecular fluorescence complementation (BiFC) and flow cytometry
YTS cells were transfected with combinations of VN155 and VC155 constructs using nucleofection (VCA-1003, Amaxa Nucleofection Kit V). After 4 h of incubation at 37 °C, cells were washed once with phosphate buffer saline (PBS) supplemented with 1% FBS. Transfected cells were stained with APC-conjugated anti-KIR2DL1 antibody (A22332, Beckman Coulter) for 30 minutes on ice to probe the receptor surface expression. Fluorescence signals arising from BiFC and anti-KIR2DL1-APC were measured using a flow cytometer (BD FACSCalibur). The data was analyzed on FlowJo software. The BiFC signals from different samples were compared within gates that had the same expression level of the receptor, as determined by the surface staining with the anti-KIR2DL1-APC antibody.
Confocal microscopy
Confocal microscopy was performed on a Zeiss LSM-710 confocal microscope using a 63×/1.4 oil immersion objective. Differential interference contrast images were acquired simultaneously with the fluorescence images of cells. The measurements of fluorescence intensities were performed using ImageJ.
Förster Resonance Energy Transfer (FRET)
YTS cells stably expressing 2DL1-WT-Venus, 2DL1-2YF-Venus, and 2DL1-H36A-Venus were transfected with pSHP-1-Cerulean. After 4 h at 37 °C, cells were washed and re-suspended in RPMI supplemented with 10% FBS (R10) medium. The cells were applied to poly-L-lysine coated chambers, fixed with 4% paraformaldehyde, and mounted with Prolong gold (Life Technologies). FRET measurements were performed using a confocal microscope (Zeiss LSM-710). Cerulean molecules (FRET donor) were excited using a 440 nm laser. The fluorescence intensity of Cerulean, at the cell surface, before and after photobleaching (using a laser of 514 nm) of Venus molecules was monitored. The samples were recorded at two time points before and two time points after the photobleaching of Venus. % FRET signal was calculated as % change in the fluorescence intensity of Cerulean after photobleaching of Venus. The values of fluorescence intensities were determined using ImageJ. A small amount of spontaneous photobleaching was observed during acquisition. Therefore, a few cells were seen with negative FRET signal.
Steady-state anisotropy
4 × 106 cells in phenol red-free RPMI solution supplemented with 2% FBS (R2 medium) were plated on to 35 mm glass bottom dish coated with Poly-L Lysine. The dish was washed twice with the R2 medium imaged. Images were acquired on DCS-120 Confocal Scanning FLIM Systems (Becker & Hickl GmbH). The system was equipped with a Ti-Sapphire pulsed laser (Chameleon, Coherent Inc.), Observer.Z1 (Zeiss GmbH) microscope with 40X/1.2 W objective, and MH-100-40 hybrid detectors (Hamamatsu Inc.). The images were acquired for 2 minutes in Time Correlated Single Photon Counting (TCSPC) mode on two detectors, detecting parallel and perpendicular emissions. The parallel and perpendicular images were recorded simultaneously, and were processed later for anisotropy and intensity images, using Igor Pro software, version-6.34A (WaveMetrics).
The following equations were used to calculate Intensity (Itotal) and Anisotropy (r)
| (1) |
| (2) |
In equations (1) and (2), I|| is the count from parallel detector, I⊥ is the count from perpendicular detector, and G is the instrumentation factor (calculated to be 0.99). The intensity and anisotropy images were then color-coded to create a representative image. The mean and SEM were calculated from the non-zero pixel values of the image.
Immuno-precipitation and western blotting
20 × 106 cells were lysed in 1 mL of lysis buffer (0.5% Triton X-100, 150 mM NaCl, 50 mM Tris (pH 7.4), protease inhibitor cocktail (11836170001, Roche), 2 mM EGTA, 1 mM NaVO3) for 30 minutes on ice. Lysate was centrifuged and supernatants were incubated with antibody-coated protein G sepharose beads for 2.5 h. Beads were washed thrice in lysis buffer, and re-suspended in 1× NuPage LDS sample buffer (Invitrogen). After addition of 10 mM DTT, the samples were heated at 70°C for 10 min. The samples were run on SDS-PAGE, and transferred to PVDF membrane (Millipore) for 1.5 h at 40 V. Membrane was blocked in PBS containing 5% BSA and 0.5% Tween 20 for 1 h, and then incubated with primary antibody for 1 h. After three washes (each wash of 5 min) with PBS containing 350 mM NaCl and 1% Tween 20, the membrane was incubated with secondary antibody for 1 h. Then, the membrane was washed three times (each wash of 5 min) with PBS containing 1% Tween 20. The membrane was developed with Super Signal West Dura (Pierce). The intensities of bands were calculated using ImageJ.
Expression and purification of KIR2DL1 (1-224)
For bacterial expression of KIR2DL1, a cDNA encoding the extracellular portion (1-224) of KIR2DL1 was cloned into pET30a (+) using the restriction sites NdeI and XhoI. The resulting plasmid is referred to as p2DL1 (1-224). The H36A mutant variant was generated by site-directed mutagenesis on p2DL1 (1-224). The plasmids were expressed in the E. coli strain BL21-Gold (DE3) (Agilent Technologies). The protein was purified as described (18). The concentration of protein was determined by measuring absorbance at 280 nm, using ε280 nm = 52921 M−1 cm−1 (18).
Fluorescence spectroscopy
The intrinsic fluorescence spectra were collected on Fluoromax-3 spectrofluorometer (Jobin Yvon). The protein was dissolved in 10 mM Tris (pH 7.4) with 50 mM NaCl at a concentration of 1.5 μM. The intrinsic tryptophan residues in the protein were excited at 295 nm, and emission at different wavelengths was monitored. The excitation and emission bandwidths were set at 1 and 10 nm, respectively.
Circular dichroism
The far-UV CD spectra were collected on a Jasco J-815 spectropolarimeter. The protein was dissolved in 10 mM Tris (pH 7.4) with 50 mM NaCl at a concentration of 5 μM. The spectra were acquired using a 1 mm pathlength cuvette. Each spectrum was averaged over 10 scans.
Cytotoxicity assay
Propidium iodide (PI)-based cytotoxicity assays were performed. The TAP deficient 221-Cw4 target cells were incubated with increasing amounts of HLA-Cw4-specific peptide QYDDAVYKL (Biosource) (19) for 16 h at 26 °C. The surface expression of HLA-C at different peptide concentrations was determined using an anti-HLA-C antibody, F4/326. YTS-2DL1-WT-Venus and YTS-2DL1-H36A-Venus cells were labeled with Vybrant® DiD Cell-Labeling Solution (V-22887, Invitrogen), and mixed with TAP deficient 221-HLA-Cw4 target cells, loaded with different amounts of the peptide, at an effector to target ratio of 6:1 in a V-bottom 96 well plate. The plate was centrifuged at 48× g for 3 min, and incubated for 5 h at 37°C. The cells were analyzed on flow cytometer (BD FACSCalibur). The percentage of target cell killing was determined as the percentage of target cells showing PI fluorescence minus % spontaneous killing of target cells in the absence of NK cells.
Statistical analysis
Statistical analysis was performed by nonparametric t test using Prism (GraphPad software).
Results
His-36 of KIR2DL1 is a negative regulator of SHP-1 recruitment
To test the role of histidine residues in the D1 domain of KIR2DL1 in the inhibitory function of the receptor, we employed bi-molecular fluorescence complementation (BiFC) to measure KIR2DL1 association with the phosphatase SHP-1 using wild type and single His to Ala mutants of the receptor. The BiFC method is based on complementation of two non-fluorescent fragments of Venus, which refold into a fluorescent Venus when the proteins fused with these fragments are in close proximity (20, 21). Once refolded, the reconstituted Venus is stable. Therefore, the BiFC signal is cumulative and is well suited for detection of transient interactions. However, this method has inherent noise arising from random encounters, particularly at higher expression levels.
The wild type (WT) and the indicated His to Ala mutants of KIR2DL1, tagged with the C-terminal fragment of Venus (155-238; VC155), and SHP-1, tagged with the N-terminal fragment of Venus (1-154 with I152L mutation (22); VN155), were co-transfected into YTS cells (Fig. 1A). The association of SHP-1 with KIR2DL1 was monitored by the fluorescence of reconstituted Venus using flow cytometry (Fig. 1B, 1C). With wild type KIR2DL1 (2DL1-WT) as well as H1A, H5A, H40A, H55A and H85A mutants, a basal BiFC signal was observed only at high receptor expression (Fig. 1B). To test whether this BiFC signal at high receptor expression originated from specific association of SHP-1 with phosphorylated ITIMs of KIR2DL1 or from random interactions due to high expression, we tested the ITIM tyrosine mutant variant, in which both Tyr residues were mutated to Phe (2DL1-2YF). The observation that 2DL1-2YF showed the same profile (Fig. 1C) indicated that the BiFC signal at high receptor expression originated from non-specific interactions. To avoid the contribution of this non-specific BiFC signal, we gated and performed analysis on cells with lower expression levels at which the wild type receptor showed no or negligible BiFC signal (Fig. 1B). The values of median fluorescence intensities determined from these gates were compared (Fig. 1C). A BiFC signal for 2DL1-H36A was evident at low receptor expression and was higher in magnitude than that for 2DL1-WT and other His to Ala mutants (Fig. 1B, 1C). This suggested that the H36A mutant of KIR2DL1 has higher constitutive association with SHP-1. Given the absence of KIR2DL1 engagement by HLA-C or by Ab, this was a surprising result.
FIGURE 1.
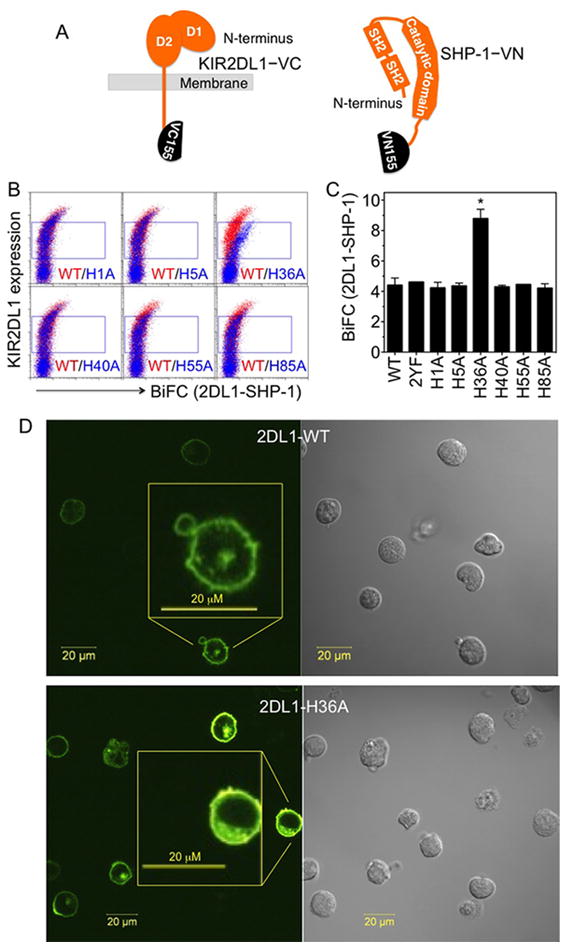
His-36 of KIR2DL1 is a negative regulator of SHP-1 recruitment. (A) Diagram of KIR2DL1 and SHP-1 tagged at C-terminus with the Venus fragments VC155 and VN155, respectively. (B) Constitutive SHP-1 association with KIR2DL1 in transfected YTS cells monitored by BiFC of reconstituted Venus using flow cytometry. BiFC signal of KIR2DL1 wild type (WT, red) and the indicated histidine to alanine mutants (blue) is plotted against surface expression, as detected by anti-2DL1-APC binding. (C) Median fluorescence intensity of BiFC signal in the gates shown in panel B. The error bars represent spread in the values of median BiFC signal determined from two independent experiments. (D) Confocal microscopy images of the BiFC signal in YTS cells transfected with SHP-1 VN and either 2DL1-WT (upper) or 2DL1-H36A (lower). Insets show enlarged views of the indicated cells. Scale bars represent 20 μm.
To visualize the sub-cellular localization of the BiFC signal emanating from association of SHP-1 with KIR2DL1, we performed confocal fluorescence microscopy. The localization of the BiFC signal was similar in the cells expressing the wild type or H36A mutant of KIR2DL1. In both cases, the signal was evenly distributed around the cell periphery and some signal was in a discrete intracellular region, consistent with an association of SHP-1 with KIR2DL1 at the plasma membrane and on intracellular membrane (Fig. 1D).
We validated the role of His-36 of KIR2DL1 in controlling the constitutive recruitment of SHP-1 to the receptor using Förster Resonance Energy Transfer (FRET) measurements. While BiFC is cumulative and produces a signal only after a time delay needed for the folding and maturation of the fluorophore, FRET enables instantaneous detection of protein interactions (23). YTS cells stably expressing 2DL1-WT (YTS-2DL1-WT-Venus), 2DL1-2YF (YTS-2DL1-2YF-Venus), or 2DL1-H36A (YTS-2DL1-H36A-Venus) (Supplemental Fig. 1), each tagged with Venus at the end of the cytosolic tail, were transfected for transient expression of SHP-1 that was tagged at the C-terminus with Cerulean (Fig. 2A). The FRET signal was measured as the increase in donor (Cerulean) fluorescence upon acceptor (Venus) photobleaching (Fig. 2B). In accordance with the BiFC measurements (Fig. 1), the FRET signals arising from association of SHP-1 with 2DL1-WT (1.3% ± 0.6; mean ± SEM) and 2DL1-2YF (0.5% ± 1) were similar, whereas 2DL1-H36A (3.7% ± 0.4) gave a stronger signal (P < 0.002). The FRET signals were not dependent on the strength of the donor (Cerulean) or the acceptor (Venus) fluorescence signals (Fig. 2B). We conclude that the increased association of SHP-1 with 2DL1-H36A was not due to high expression level. The detection of association by FRET confirmed the result obtained by BiFC and showed that 2DL1-H36A SHP-1 association is detectable at a single time point.
FIGURE 2.
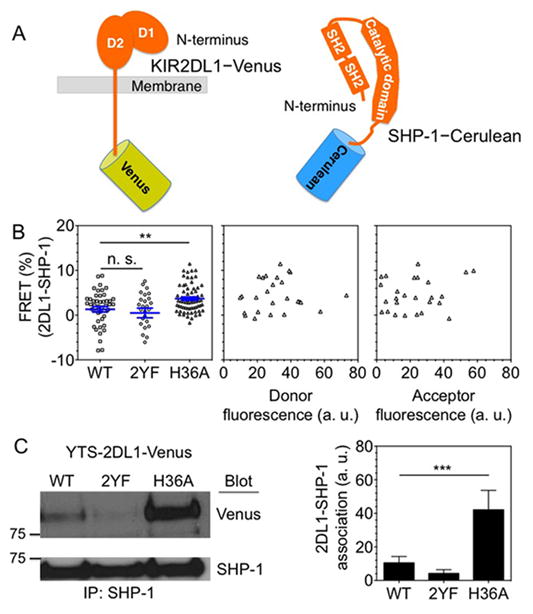
H36A mutation of KIR2DL1 results in constitutive SHP-1 recruitment. (A) Diagram of KIR2DL1 and SHP-1 tagged at their C-terminus with Venus and Cerulean, respectively. (B) SHP-1 proximity to KIR2DL1 monitored by FRET. SHP-1 Cerulean was transiently expressed in YTS cells stably transfected with 2DL1-WT-Venus, 2DL1-2YF-Venus, or 2DL1-H36A-Venus. FRET signal at the plasma membrane was calculated as percentage increase in donor (Cerulean) fluorescence after acceptor (Venus) photobleaching. The negative values of FRET signal are due to unintentional photobleaching of donor fluorescence during acquisition. In the left-most panel, data points from three independent experiments are combined. Horizontal and vertical lines represent the mean and standard error, respectively. The middle and right-most panels show FRET signals of SHP-1 with 2DL1-H36A relative to the expression of FRET donor (SHP-1-Cerulean) and FRET acceptor (2DL1-H36A-Venus). (C) Endogenous SHP-1 was immuno-precipitated from the indicated YTS-2DL1-Venus cells and its association with 2DL1 was probed with an anti-GFP immunoblot. The right panel shows band intensities obtained by scanning immunoblots and calculated using ImageJ. The error bars represent spread in the values of mean obtained from two independent experiments.
Next, we tested the recruitment of SHP-1 to KIR2DL1 by immuno-precipitation and western blotting. Endogenous SHP-1 was immuno-precipitated from YTS-2DL1-WT-Venus, YTS-2DL1-2YF-Venus, or YTS-2DL1-H36A-Venus cells, and its association with the receptor was monitored with an antibody to Venus (Fig. 2C). As expected, 2DL1-2YF was not associated with SHP-1. In contrast, and in accordance with the BiFC and FRET measurements, 2DL1-H36A showed much higher constitutive association with SHP-1 than 2DL1-WT (about 4-fold) (Fig. 2C).
Recruitment of SHP-1 to ITIM-bearing receptors is usually dependent on tyrosine phosphorylation of the ITIMs. To test if increased recruitment of SHP-1 by 2DL1-H36A correlated with tyrosine phosphorylation of the receptor, we assessed the phosphorylation of 2DL1-WT, 2DL1-2YF and 2DL1-H36A in these cell lines by immuno-precipitation of the receptor followed by immunoblotting with antibody to pTyr. 2DL1-WT and 2DL1-2YF displayed no or very low phosphorylation, whereas 2DL1-H36A displayed much higher constitutive phosphorylation (Fig. 3A). As ITIM phosphorylation is sufficient for SHP-1 binding (7, 24, 25), this result suggested that SHP-1 association with 2DL1-H36A was a consequence of receptor phosphorylation.
FIGURE 3.
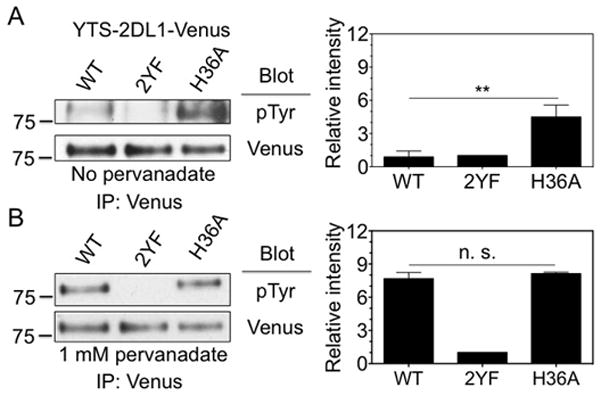
His-36 of KIR2DL1 prevents constitutive ITIM phosphorylation. (A) Constitutive tyrosine phosphorylation of KIR2DL1 monitored by immunoblotting. KIR2DL1 was immuno-precipitated from YTS-2DL1-WT-Venus, YTS-2DL1-2YF-Venus and YTS-2DL1-H36A-Venus cells with an anti-GFP antibody, and tyrosine phosphorylation was detected by immunoblotting with mAb 4G10. The intensity of bands in immunoblots, relative to YTS-2DL1-2YF-Venus cells (set to 1), is displayed on the right. The error bars represent spread in the values of mean determined from two independent experiments. (B) KIR2DL1 tyrosine phosphorylation measured as in panel A after treatment of transfected YTS cells with 1 mM pervanadate for 20 minutes.
The constitutive phosphorylation of 2DL1-H36A could be due to selective recruitment of a tyrosine kinase. Alternatively, as KIR2DL1 may be subjected to a continuous cycle of phosphorylation and dephosphorylation, the self-association of 2DL1-H36A may result in enhanced phosphorylation through the protection of phosphorylated ITIMs from PTPases. We assessed the effect of inhibition of tyrosine phosphatases on receptor phosphorylation. In the case of selective kinase recruitment by 2DL1-H36A, the difference in phosphorylation of 2DL1-WT and 2DL1-H36A may persist after treatment with pervanadate, a PTPase inhibitor. Conversely, protection of 2DL1-H36A from PTPase would be expected to result in equal phosphorylation of 2DL1-WT and 2DL1-H36A after treatment with pervanadate. 2DL1-WT and 2DL1-H36A were phosphorylated to the same extent after pervanadate treatment (Fig. 3B). This suggested that the constitutive phosphorylation of 2DL1-H36A was due to selective protection from PTPase activity rather than selective recruitment of tyrosine kinase.
H36A mutation does not perturb the overall structure of KIR2DL1
It was conceivable that the H36A mutation in KIR2DL1 caused a conformational change of the receptor, which could protect it from ITIM dephosphorylation, and enhance constitutive SHP-1 recruitment. To test this possibility, we compared the spectroscopic characteristics using intrinsic tryptophan fluorescence and circular dichroism spectroscopy, of the purified extra-cellular portions (amino acid 1-224) of 2DL1-WT and 2DL1-H36A, which had been expressed in E. coli, refolded and purified (Fig. 4A).
FIGURE 4.
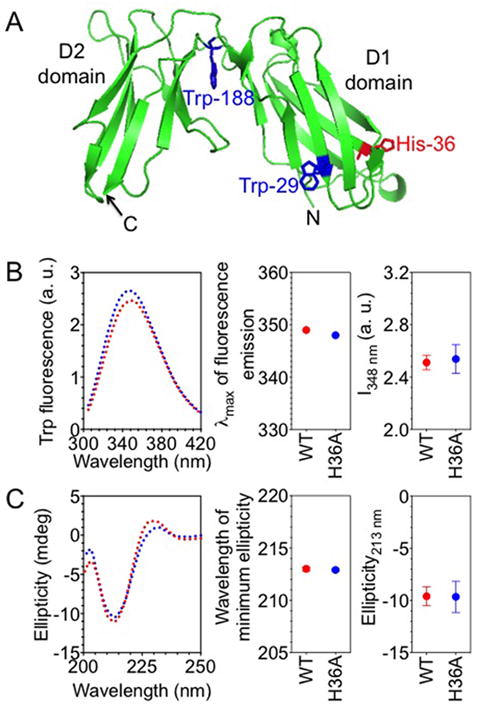
H36A mutation does not perturb the overall structure of KIR2DL1. (A) Ribbon diagram structure of KIR2DL1. Tryptophan residues 29, 188, and His-36 are shown as sticks. Tryptophan residue 207 is not visible in KIR2DL1 structure, probably due to a disordered stem (201–224) structure. The diagram was generated from the PDB file 1NKR using pymol. (B) Intrinsic tryptophan fluorescence of purified, soluble KIR2DL1. The left panel shows the spectra of the wild type (2DL1-WT) (red) and the H36A mutant (2DL1-H36A) (blue) of KIR2DL1 (1-224). Maximum wavelengths of emission and fluorescence intensities at 348 nm (I348 nm) are shown in the middle and the right-most panels, respectively. The error bars represent spread in the values of mean determined from two independent experiments. (C) Far-UV circular dichroism of purified, soluble KIR2DL1. The left panel shows the far-UV circular dichroism spectra of 2DL1-WT (red) and 2DL1-H36A (blue). The positions of minimum ellipticity and the ellipticity values at 213 nm are plotted in the middle and the right-most panels, respectively. The error bars represent spread in the values of mean determined from two independent experiments.
Intrinsic tryptophan fluorescence spectroscopy probes the environment of tryptophan residues in a protein. The emission maximum as well as the quantum yields of intrinsic tryptophan fluorescence spectra is sensitive to the solvent exposure of the tryptophan residues (26, 27). A tryptophan residue buried in the hydrophobic core of a protein shows emission maximum at ~ 330 nm, upon excitation at 295 nm. A solvent-exposed tryptophan residue has the emission maximum at 355–360 nm, and has a lower quantum yield of fluorescence. KIR2DL1 (1-224) has three tryptophan residues, at positions 29 in the D1 domain, 188 in the D2 domain, and 207 in the stem region (Fig. 4A). A difference in the conformation of 2DL1-WT and 2DL1-H36A may lead to a change in the environment of tryptophan residue(s), and thus difference in the tryptophan fluorescence spectrum. 2DL1-WT (1-224) showed emission maximum at ~ 348 nm, indicative of tryptophan residue(s) residing in an environment that is exposed partially to the solvent (Fig. 4B). The wavelength of emission maximum as well as the quantum yield of fluorescence was the same in 2DL1-H36A (1-224), showing that the mutation did not affect the overall environment of the tryptophan residues in the receptor (Fig. 4B).
Far-UV circular dichroism measures secondary structure (α-helix and βstrands) in proteins and peptides (28). The far-UV circular dichroism spectrum of 2DL1-WT (1-224) had a peak at 213 nm, indicative of a protein rich in β-strands (Fig. 4C), consistent with the known X-ray structure of the receptor (Fig. 4A). The spectrum of 2DL1-H36A (1-224) was similar (Fig. 4C), indicated that the mutation did not have a major effect on the secondary structure of the receptor.
His-36 is pointing out in the KIR2DL1 crystal structure and is positioned away from the HLA-C binding site (29). His-36 is not required for binding to HLA-C (30). As His-36 resides in a solvent accessible region of the protein, it is more likely to control inter-molecular interactions than the receptor structure.
Phe mimics His at position 36 in KIR2DL1
The side-chain of a histidine residue has several physico-chemical properties that could affect protein interactions, such as zinc-binding ability and the ability to create steric hindrance through its bulky imidazole ring. KIR2DL1 is known to bind zinc (31), and zinc contributes to inhibitory signaling through the receptor (30). To test whether constitutive SHP-1 recruitment to 2DL1-H36A results from an altered zinc binding, we assessed constitutive association of SHP-1 with 2DL1-WT and 2DL1-H36A by co-immunoprecipitation and BiFC, in the presence of 200 μM supplemental ZnCl2 or 1 mM EDTA. The constitutive association of SHP-1 with 2DL1-H36A, and its low basal association with 2DL1-WT, did not change upon treatment with either 200 μM ZnCl2 or 1 mM EDTA (Supplemental Fig. 2). Therefore the prevention of constitutive SHP-1 recruitment, and constitutive ITIM phosphorylation by His-36 is not dependent on zinc.
To test if His-36 may prevent KIR2DL1 phosphorylation by steric hindrance we replaced it with another bulky amino acid, phenylalanine. We monitored the constitutive recruitment of SHP-1 to 2DL1-WT, 2DL1-H36A or 2DL1-H36F with BiFC assays using flow cytometry (Fig. 5A, 5B). As seen earlier, 2DL1-H36A showed greater association with SHP-1 than 2DL1-WT. In contrast, constitutive SHP-1 association with 2DL1-H36F was similar to that with 2DL1-WT (Fig. 5A, 5B). We monitored the constitutive association of SHP-1 with KIR2DL1 at the plasma membrane with confocal fluorescence microscopy. In accordance with BiFC, SHP-1 association was higher with 2DL1-H36A than 2DL1-WT, and that with 2DL1-H36F was similar to 2DL1-WT (Fig. 5C).
FIGURE 5.
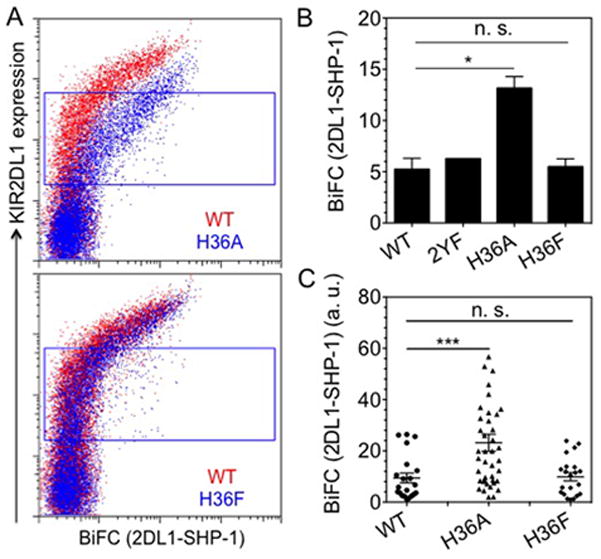
A large side chain at amino acid 36 of KIR2DL1 prevents constitutive SHP-1 recruitment. (A) Constitutive SHP-1 association with KIR2DL1 in transfected YTS cells monitored by BiFC of reconstituted Venus using flow cytometry. BiFC signal of 2DL1-WT (red) and 2DL1-H36A (top) or 2DL1-H36F (bottom) (blue) is plotted against surface expression, as detected by anti-2DL1-APC binding. (B) Median fluorescence intensity of BiFC signal in the gates shown in panel A. The error bars represent spread in the values of median BiFC signal determined from two independent experiments. (C) Constitutive SHP-1 association with KIR2DL1 in transfected YTS cells monitored by BiFC of reconstituted Venus using confocal microscopy. BiFC signal of 2DL1-WT, 2DL1-H36A, and 2DL1-H36F, determined at the plasma membrane, is plotted. The horizontal and vertical lines represent mean and standard error, respectively.
A residue at position 36 with similar bulk as His (Phe) did prevent SHP-1 recruitment to the receptor, suggesting that steric hindrance by the imidazole ring of His-36 of KIR2DL1 may prevent interactions that result in constitutive SHP-1 recruitment to the receptor.
His-36 prevents KIR2DL1 self-association
His-36 or Phe-36 may prevent interaction of KIR2DL1 with itself (self-association) or with something else. We first tested the role of His-36 in regulating KIR2DL1 self-association. Constitutive self-association of 2DL1-WT, 2DL1-H36A or 2DL1-H36F was assessed by a BiFC assay with YTS cells co-expressing KIR2DL1 tagged, at cytosolic tail, with either VN155 or VC155 (Fig. 6A). Self-association of 2DL1-WT was evident only at higher levels of receptor expression (Fig. 6B), probably due to random encounters in the 2-dimensional surface of the plasma membrane. However, the self-association propensity of 2DL1-H36A was clearly higher than 2DL1-WT, and occurred at much lower expression levels (Fig. 6B, 6C). The propensity of self-association of 2DL1-H36F was similar to that of 2DL1-WT. As observed with the KIR2DL1 SHP-1 association assay (Fig. 5), H36F had the same property as 2DL1-WT and prevented KIR2DL1 self-association (Fig. 6B, 6C).
FIGURE 6.
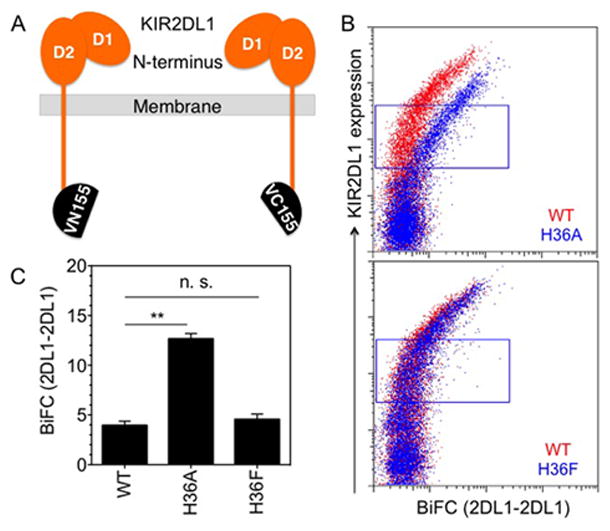
A bulky side chain at amino acid 36 of KIR2DL1 prevents constitutive receptor self-association. (A) Diagram of KIR2DL1 tagged at the C-terminus with Venus fragments VN155 and VC155. (B) Constitutive KIR2DL1 self-association in transfected YTS cells monitored by BiFC. BiFC signal of 2DL1-WT (red) and 2DL1-H36A (top) or 2DL1-H36F (bottom) (blue) is plotted against surface expression, as detected by anti-2DL1-APC binding. (C) Median fluorescence intensity of BiFC signal in the gates shown in panel B. The error bars represent spread in the values of median BiFC signal determined from two independent experiments.
To validate the constitutive self-association of 2DL1-H36A, steady-state anisotropy imaging was performed using YTS-2DL1-WT-Venus and YTS-2DL1-H36A-Venus cells (Fig. 7). Fluorescence anisotropy measures how well the orientation (polarization) of emitted light correlates with the orientation of the linearly polarized light used for excitation (Fig. 7A). When a high molecular weight fluorophore, such as Venus, is excited, the orientation of emitted light is typically highly correlated with the orientation of the electric field of the light used for excitation, resulting in a high anisotropy value (32). This arises primarily because large fluorophores, such as GFP and its derivatives, have slow rotational motion relative to their fluorescence lifetime. However, if two such fluorophores are in close proximity, energy absorbed by one fluorophore can be transferred to the second fluorophore by a phenomenon called homo-FRET. Homo FRET, per se, does not alter the emission intensity or spectrum. However, as the emission arises in part from a molecule other than the one that was photo-selected, which potentially has a different orientation, the orientation of emitted light tends to be less correlated with the excitation light source. Thus, when two fluorophores are in close proximity homo-FRET may occur, and typically this can be observed as a drop in anisotropy.
FIGURE 7.

Self-association of 2DL1-WT and 2DL1-H36A monitored by anisotropy measurements. (A) Diagram depicting how receptor self-association generates homo-FRET and a reduced fluorescence anisotropy. In the absence of homo-FRET, the emission of Venus is detected mostly in the parallel plane (||) of polarization. If homo-FRET occurs between two Venus molecules in close proximity, the emission will become more evenly distributed between both parallel and perpendicular (⊥) planes of polarization, and anisotropy will drop. (B) Anisotropy (top) and fluorescence intensity (bottom) images of YTS-2DL1-WT-Venus and YTS-2DL1-H36A-Venus cells. White arrows show YTS-2DL1-WT-Venus and YTS-2DL1-H36A-Venus cells with matched fluorescence intensity (expression level), but distinct anisotropy values. (C) Quantitation of anisotropy and fluorescence intensity of YTS-2DL1-WT-Venus (red) and YTS-2DL1-H36A-Venus (blue) cells shown in panel B. Individual pixel anisotropy values are plotted parametrically as a function of fluorescence intensity. Quantitation was performed using the software IGOR Pro (WaveMetrics). Right panel shows the same data potted separately for YTS-2DL1-WT-Venus (red) and YTS-2DL1-H36A-Venus (blue) cells. (D) Median pixel anisotropy values (n = 350) are plotted as a function of fluorescence intensity. The left panel shows YTS-2DL1-WT-Venus (red) and YTS-2DL1-H36A-Venus (blue) cells. In the right panel, anisotropy of membrane tethered Venus monomer (V-K-Ras) and Venus dimer (VV-K-Ras) is shown. The median pixel anisotropy values were determined from three independent images.
To control for the homo-FRET signals resulting from high expression rather than receptor self-association, anisotropy and fluorescence intensity (receptor expression) were acquired simultaneously. We performed simultaneous two-photon anisotropy and fluorescence intensity imaging using YTS-2DL1-WT-Venus or 2DL1-H36A-Venus cells (Fig. 7B; Supplemental Fig. 3). Images were color coded to highlight changes in anisotropy and intensity values. The anisotropy values of 2DL1-H36A were overall lower than those of 2DL1-WT (Fig. 7B; top panels). Further, Cells expressing 2DL1-WT or 2DL1-H36A with matched fluorescence intensity (Fig. 7B; white arrows) showed lower anisotropy values for the H36A mutant. A more quantitative comparison was obtained by plotting the individual pixel anisotropy values as a function of fluorescence intensity (Fig. 7C, 7D). As expected from Poissonian counting statistics (33), variance in the anisotropy values was higher at low fluorescence intensity. In this low intensity region, the anisotropy values were independent of receptor expression level, and thus they indicated the true aggregation state of the receptor. Importantly, at these low intensity values, the anisotropy for 2DL1-H36A were lower than for 2DL1-WT. At higher fluorescence intensity values, the anisotropy values of both samples dropped, presumably due to overcrowding of the fluorophores. However, throughout the intensity range, 2DL1-H36A (0.261 ± 0.004; mean ± SEM) had consistently lower anisotropy values than 2DL1-WT (0.284 ± 0.004; mean ± SEM), indicating higher homo-FRET, and thus self-association, of 2DL1-H36A.
We reasoned that if 2DL1-WT-Venus is a monomer, it should have an anisotropy value similar to that of a membrane-tethered monomeric Venus tagged at the N-terminus with a membrane targeting sequence from K-Ras. We performed the measurements on YTS cells expressing monomeric Venus or dimeric Venus (two Venus molecules joined by a linker of 5 amino acids) tagged with the membrane targeting sequence of K-Ras. They are referred as V-K-Ras and VV-K-Ras. The anisotropy values of 2DL1-WT were similar to those of V-K-Ras (Fig. 7D), suggesting that 2DL1-WT exist as monomer at the cell surface. 2DL1-H36A had lower anisotropy values than V-K-Ras, indicating that H36A mutation facilitates KIR2DL1 self-association, and conversely, His-36 prevents KIR2DL1 self-association. As expected for a covalent dimer, the anisotropy of the membrane-targeted dimeric Venus (VV-K-Ras) was lower (i.e. higher homo-FRET) than that of 2DL1-H36A. The proportion of 2DL1-H36A receptors that are self-associated at any one time is not known. In addition, it is possible that the homo-FRET efficiency is greater between the covalently joined Venus molecules in VV-K-Ras, than between Venus molecules at the C-terminal end of self-associated 2DL1-H36A. Finally, FRET and anisotropy depend also on the relative orientation of Venus pairs.
Overall, our results from BiFC and anisotropy measurements indicated that 2DL1-WT exists as monomer at the cell surface, and that the H36A mutation facilitates KIR2DL1 self-association.
Inhibition of lysis of HLA-C+ target cells is stronger with YTS-2DL1-H36A cells than with YTS-2DL1-WT cells
An important question is how the mutation H36A, which resulted in constitutive KIR2DL1 self-association, phosphorylation, and recruitment of SHP-1, would affect the inhibitory function of KIR2DL1 during interaction of NK cells with sensitive target cells that express HLA-C ligands. We first tested killing of 221 target cells expressing HLA-Cw3, an HLA-C allotype that does not bind KIR2DL1, or HLA-Cw4, a ligand for KIR2DL1, by YTS-2DL1-WT-Venus and YTS-2DL1-H36A-Venus cells (Supplemental Fig. 4). YTS-2DL1-WT-Venus and YTS-2DL1-H36A-Venus had a similar ability to kill 221-HLA-Cw3 target cells, indicating that the constitutive self-association and phosphorylation of 2DL1-H36A did not affect NK cell cytotoxicity. Inhibition of killing of 221 cells expressing HLA-Cw4 was efficient and similar for wild type and the H36A mutant of KIR2DL1 (Supplemental Fig. 4). Therefore, the inhibitory function of KIR2DL1 was not affected by the mutation at His-36.
To expose the role of His-36, if any, in regulating inhibitory KIR2DL1 function, we developed a quantitative assay for the strength of inhibition. We generated a 221-HLA-Cw4 cell line stably expressing ICP47, an inhibitor of TAP that is expressed by Herpes simplex virus (34). HLA-Cw4 at the surface of TAP-deficient cells can be stabilized by the addition of exogenous HLA-Cw4 specific peptide, providing an opportunity to titrate the amount of peptide-loaded HLA-Cw4 available to KIR2DL1 for inhibition of NK cell activation (Fig. 8A). 2DL1-WT and 2DL1-H36A YTS cells had a similar ability to kill 221-Cw4-ICP47 cells in the absence of peptide, indicating that, as seen with normal (TAP sufficient) 221-HLA-Cw3 cells, the H36A mutation did not affect the cytotoxicity activity of the NK cell (Fig. 8B). Expression of 2DL1-WT or 2DL1-H36A inhibited the cytotoxicity in a peptide concentration dependent manner. However, 2DL1-H36A inhibited NK cell activation at much lower peptide concentrations (Fig. 8B, 8C). This showed that 2DL1-H36A inhibits NK cell cytotoxicity more efficiently than the wild type receptor. In other words, His-36 of KIR2DL1 impedes inhibitory signaling through KIR2DL1 upon HLA-C binding. Consistent with the observation with TAP sufficient 221 target cells with higher HLA-C expression, where there is comparable inhibition of killing by 2DL1-WT and 2DL1-H36A (Supplemental Fig. 4), at higher levels of peptide-loaded HLA-Cw4 (5–10 μM of the peptide), the ability to inhibit killing by NK cells was similar for 2DL1-WT and 2DL1-H36A (Fig. 8B).
FIGURE 8.
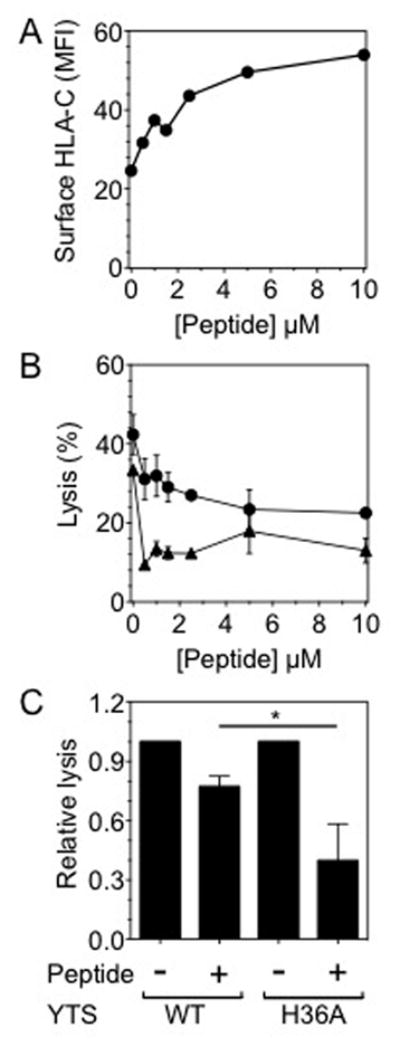
Inhibition of lysis of HLA-C+ target cells is stronger with YTS-2DL1-H36A cells than with YTS-2DL1-WT cells. (A) Surface expression of HLA-C on TAP deficient 221-HLA-Cw4 target cells that were loaded with an exogenous HLA-Cw4-specific peptide was determined with anti-HLA-C antibody F4/326. (B) Lysis of TAP deficient 221-Cw4 cells by YTS-2DL1-WT-Venus (circle) and YTS-2DL1-H36A-Venus (triangle) cells after loading with peptides at different concentrations. The NK to target ratio was 6. Error bars represent spread in the values of mean determined from duplicate samples. (C) Relative lysis of TAP deficient 221-HLA-Cw4 cells by YTS-2DL1-WT and YTS-2DL1-H36A, after loading with 0.5 μM peptide. To compare different experiments, lysis of 221-HLA-Cw4 cells in the absence of exogenous peptide was set to 1. Bars represent the values of mean and error bars represent standard deviations obtained from three independent experiments.
Discussion
The very proximal block of NK activation signals by inhibitory receptors (10–12) is not easily reconciled with models that place the phosphorylation of ITIMs downstream of signaling by activation receptors. The question we addressed here is how phosphorylation of ITIMs in KIR is regulated. As ITIMs in inhibitory receptors are readily phosphorylated in cells treated with pervanadate (a general inhibitor of cysteine-dependent PTPase), ITIMs must be subjected to a constitutive cycle of phosphorylation and dephosphorylation, with an equilibrium that favors dephosphorylation. If so, how could this cycle be regulated to control ITIM phosphorylation? The PTPase SHP-1 is the key effector in the inhibitory function of ITIM-bearing KIR (2). SHP-1 is maintained in a self-inhibited conformation in the cytosol, and SHP-1 recruitment by phosphorylated ITIMs results in its activation and ability to dephosphorylate components of activation pathways. Binding of phosphorylated ITIMs to SH2 domains of SHP-1 protects them from tyrosine phosphatases. Therefore, SHP-1 may not be the primary PTPase responsible for the maintenance of ITIMs in a dephosphorylated state.
In this study, we have identified a gain-of-function mutant of KIR2DL1, which is phosphorylated in its basal, ligand-free state. In a screen of histidine residues in the D1 domain of KIR2DL1 for their role in receptor function, we noticed that a His to Ala mutation at position 36 (H36A) resulted in constitutive association of KIR2DL1 with SHP-1, constitutive ITIM phosphorylation, and KIR2DL1 self-association. These unusual properties of 2DL1-H36A are independent of HLA-C ligands, given that KIR2DL1 exhibits exclusive specificity for group 2 HLA-C allotypes, and that YTS cells express two group 1 HLA-C allotypes. The constitutive phosphorylation of 2DL1-H36A implies a change in the balance between phosphorylation and dephosphorylation of this mutant KIR. Our results suggest that KIR2DL1 phosphorylation is controlled by the accessibility of ITIM to tyrosine phosphatases, and that this access is denied by 2DL1-H36A self-association.
The constitutive tyrosine phosphorylation of 2DL1-H36A measured in biochemical experiments, in which immunoprecipitated receptor was immunoblotted for phosphotyrosine, was much greater than that of KIR2DL1. Constitutive tyrosine phosphorylation of KIR2DL1 was very low, similar to the negative control of KIR2DL1 lacking tyrosines in both ITIMs. However, in cells treated with pervanadate, tyrosine phosphorylation of KIR2DL1 and of 2DL1-H36A was the same, indicating that 2DL1-H36A has acquired some level of protection from the constitutive dephosphorylation the wild-type receptor is subjected to in the absence of pervanadate. Constitutive association of SHP-1 with 2DL1-H36A was evident in similar biochemical experiments. This association was also detected using both BiFC and FRET between appropriately tagged KIR2DL1 and SHP-1 molecules. The constitutive association of 2DL1-H36A with SHP-1 is not due to the specific loss of histidine at position 36, because the similarly bulky amino acid phenylalanine at position 36 behaved like wild-type receptor. The folding of a purified, soluble H36A mutant of KIR2DL1 was tested by intrinsic tryptophan fluorescence and far-UV circular dichroism. By both methods, 2DL1-H36A was indistinguishable from purified, soluble wild type KIR2DL1. Those results, and the fact that 2DL1-H36A is a gain-of-function mutant, do not support the possibility that 2DL1-H36A is a misfolded receptor.
The constitutive tyrosine phosphorylation of 2DL1-H36A could be due to the loss of a His-36-dependent binding to a PTPase or to a protein complex that includes a PTPase. The maintenance of wild-type KIR2DL1 in a dephosphorylated state due to this His-36-dependent association would have to be overcome when KIR2DL1 binds to HLA-C. An alternative is that His-36 may prevent an interaction with a protein or a protein complex that favors tyrosine phosphorylation of KIR2DL1. In support of this interpretation, substitution of His-36 by a Phe, which has an equally bulky side chain, resulted in a receptor that was not constitutively associated with SHP-1, just as wild-type KIR2DL1. Using BiFC assays with YTS cells we found that 2DL1-H36A was associated with itself, whereas wild-type 2DL1 and 2DL1-H36F did not undergo self-association. For those experiments, cells were transfected with an equal amount of two plasmids, each encoding KIR2DL1 tagged with a different half of unfolded Venus. The refolding of Venus due to KIR2DL1 self-association was monitored at low KIR2DL1 expression levels, in order to avoid the crowding effect at high receptor densities.
To confirm the propensity of 2DL1-H36A to self-associate, we performed anisotropy measurements on KIR2DL1 tagged at the C-terminus with an intact Venus molecule. Anisotropy measures the amount of fluorophore emission that has a polarization (or orientation) similar to the polarized light used for excitation. Close proximity (<10 nm) of two Venus molecules results in homo-FRET, which can lead to depolarization of fluorescence emission, and can be detected by a reduction in anisotropy. This technique is different from standard FRET experiments, which measure changes in the wavelength or in the lifetime of emission, and eliminates complications associated with the use of two distinct fluorophores in standard FRET measurements. Emission that has become unpolarized, due to homo-FRET from one Venus molecule to another Venus molecule with a different orientation, reduces anisotropy. 2DL1-H36A had reduced mean anisotropy values over a wide range of fluorescence intensity, which was below that of wild-type 2DL1 at any fluorescence intensity, clearly demonstrating that 2DL1-H36A has the propensity to self-associate.
Our results clearly show that His-36 prevents self-association of the wild type 2DL1 receptor. Mutation of His-36 to an alanine resulted in constitutive receptor self-association and tyrosine phosphorylation. We propose that receptor self-association is a mechanism that protects ITIMs from dephosphorylation by PTPases. In this model, wild-type KIR2DL1 is continuously exposed to tyrosine kinases and PTPases, with an equilibrium that favors dephosphorylation, and thus disfavors SHP-1 recruitment. The negative effect of His-36 on receptor self-association must be overcome upon binding to HLA-C on target cells. Whether KIR2DL1 bound to HLA-C forms complexes similar to the constitutive self-association of 2DL1-H36A is not known. However, the striking ability of mutant 2DL1-H36A to inhibit YTS cells at a much lower HLA-C-peptide density on target cells than the HLA-C-peptide density required to inhibit through wild-type KIR2DL1 clearly demonstrates that the constitutive 2DL1-H36A receptor complexes have strong inhibitory potential. The steric hindrance imposed by His-36 on receptor self-association may serve as a gatekeeper to prevent unregulated inhibition of NK cell activation. Activation of signaling receptors by the relief of structural constraints upon ligand binding has precedents, notably in growth factor receptors (35).
In the kinetic partitioning model proposed to explain TCR phosphorylation, regulation of phosphorylation occurs at the level of the distribution of the trans-membrane phosphatase CD45 and its exclusion from the center of the immunological synapse, and specific recruitment of a tyrosine kinase to the immunoreceptor is not required (36). Our model of KIR phosphorylation is similar in that ligand-induced receptor phosphorylation is achieved by protection of phosphorylated tyrosines from phosphatase. However, in our model for KIR phosphorylation, the receptor itself switches into a state that protects its cytosolic tyrosine residues from local tyrosine phosphatases.
His-36 is positioned away from the HLA-C binding region of the receptor, and is not involved in KIR binding to HLA-C (29, 30). This conserved histidine is present in the D1 domain of 11 out of 12 D1-containing KIR family members. Its role in inhibitory signaling by other ITIM-bearing KIRs or in signaling by activating KIRs remains to be tested. A precise understanding of how the hindrance of KIR2DL1 self-association by His-36 is overcome, or bypassed, upon binding to HLA-C ligands on target cells, will await future investigation. Solving the structure of a 2DL1-H36A self-associated complex could shed light on this question.
The observation made here that 2DL1-H36A has a greater ability to inhibit NK cell activation than the wild type receptor is remarkable, considering that ITIM-bearing KIRs are potent inhibitors of NK cell cytotoxicity. It shows that KIRs have evolved to achieve an optimal degree of inhibition rather than the maximum possible. Proper tuning of KIR inhibitory signals may also be relevant to their role in maintaining NK cells in a state of high responsiveness (37, 38).
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program at the National Institutes of Health, National Institute of Allergy and Infectious Diseases and National Institute on Alcohol Abuse and Alcoholism. M. J. W. S is supported by an NIH-Wellcome trust studentship.
We thank David Narum and Karine Reiter for assistance with circular dichroism, Joseph Brzostowski for advice on FRET measurements, Rajat Varma for advice on nucleofection, Emmanuel Wiertz for the plasmid pLZRS-ICP47-IRES-eGFP, and Mary Carrington for HLA typing of YTS cells.
Abbreviations
- KIR
killer cell Ig-like receptor
- HLA
human leukocyte antigen
- BiFC
bi-molecular fluorescence complementation
- FRET
Förster resonance energy transfer
- IP
immuno-precipitation
- n. s
not significant
- DiD
1,19-dioctadecyl-3,3,39,39-tetramethylindodicar-bocyanine
- PI
propidium iodide
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev. 2008;224:70–84. doi: 10.1111/j.1600-065X.2008.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol Cell Biol. 2003;23:6291–6299. doi: 10.1128/MCB.23.17.6291-6299.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blery M, Delon J, Trautmann A, Cambiaggi A, Olcese L, Biassoni R, Moretta L, Chavrier P, Moretta A, Daeron M, Vivier E. Reconstituted killer cell inhibitory receptors for major histocompatibility complex class I molecules control mast cell activation induced via immunoreceptor tyrosine-based activation motifs. J Biol Chem. 1997;272:8989–8996. doi: 10.1074/jbc.272.14.8989. [DOI] [PubMed] [Google Scholar]
- 5.Eriksson M, Leitz G, Fallman E, Axner O, Ryan JC, Nakamura MC, Sentman CL. Inhibitory receptors alter natural killer cell interactions with target cells yet allow simultaneous killing of susceptible targets. J Exp Med. 1999;190:1005–1012. doi: 10.1084/jem.190.7.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson CC, Sinclair NR. FcR-mediated inhibition of cell activation and other forms of coinhibition. Crit Rev Immunol. 1998;18:525–544. doi: 10.1615/critrevimmunol.v18.i6.30. [DOI] [PubMed] [Google Scholar]
- 7.Burshtyn DN, Scharenberg AM, Wagtmann N, Rajagopalan S, Berrada K, Yi T, Kinet JP, Long EO. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity. 1996;4:77–85. doi: 10.1016/s1074-7613(00)80300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Binstadt BA, Brumbaugh KM, Dick CJ, Scharenberg AM, Williams BL, Colonna M, Lanier LL, Kinet JP, Abraham RT, Leibson PJ. Sequential involvement of Lck and SHP-1 with MHC-recognizing receptors on NK cells inhibits FcR-initiated tyrosine kinase activation. Immunity. 1996;5:629–638. doi: 10.1016/s1074-7613(00)80276-9. [DOI] [PubMed] [Google Scholar]
- 9.Chan VW, Lowell CA, DeFranco AL. Defective negative regulation of antigen receptor signaling in Lyn-deficient B lymphocytes. Curr Biol. 1998;8:545–553. doi: 10.1016/s0960-9822(98)70223-4. [DOI] [PubMed] [Google Scholar]
- 10.Bryceson YT, Ljunggren HG, Long EO. Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood. 2009;114:2657–2666. doi: 10.1182/blood-2009-01-201632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burshtyn DN, Shin J, Stebbins C, Long EO. Adhesion to target cells is disrupted by the killer cell inhibitory receptor. Curr Biol. 2000;10:777–780. doi: 10.1016/s0960-9822(00)00568-6. [DOI] [PubMed] [Google Scholar]
- 12.Abeyweera TP, Merino E, Huse M. Inhibitory signaling blocks activating receptor clustering and induces cytoskeletal retraction in natural killer cells. J Cell Biol. 2011;192:675–690. doi: 10.1083/jcb.201009135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterson ME, Long EO. Inhibitory receptor signaling via tyrosine phosphorylation of the adaptor Crk. Immunity. 2008;29:578–588. doi: 10.1016/j.immuni.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D, Peterson ME, Long EO. The adaptor protein Crk controls activation and inhibition of natural killer cells. Immunity. 2012;36:600–611. doi: 10.1016/j.immuni.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oosten LE, Koppers-Lalic D, Blokland E, Mulder A, Ressing ME, Mutis T, van Halteren AG, Wiertz EJ, Goulmy E. TAP-inhibiting proteins US6, ICP47 and UL49.5 differentially affect minor and major histocompatibility antigen-specific recognition by cytotoxic T lymphocytes. Int Immunol. 2007;19:1115–1122. doi: 10.1093/intimm/dxm082. [DOI] [PubMed] [Google Scholar]
- 16.Borszcz PD, Peterson M, Standeven L, Kirwan S, Sandusky M, Shaw A, Long EO, Burshtyn DN. KIR enrichment at the effector-target cell interface is more sensitive than signaling to the strength of ligand binding. Eur J Immunol. 2003;33:1084–1093. doi: 10.1002/eji.200323582. [DOI] [PubMed] [Google Scholar]
- 17.Koushik SV, Vogel SS. Energy migration alters the fluorescence lifetime of Cerulean: implications for fluorescence lifetime imaging Forster resonance energy transfer measurements. J Biomed Opt. 2008;13:031204. doi: 10.1117/1.2940367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan QR, Garboczi DN, Winter CC, Wagtmann N, Long EO, Wiley DC. Direct binding of a soluble natural killer cell inhibitory receptor to a soluble human leukocyte antigen-Cw4 class I major histocompatibility complex molecule. Proc Natl Acad Sci USA. 1996;93:7178–7183. doi: 10.1073/pnas.93.14.7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajagopalan S, Long EO. The direct binding of a p58 killer cell inhibitory receptor to human histocompatibility leukocyte antigen (HLA)-Cw4 exhibits peptide selectivity. J Exp Med. 1997;185:1523–1528. doi: 10.1084/jem.185.8.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 21.Shyu YJ, Liu H, Deng X, Hu CD. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques. 2006;40:61–66. doi: 10.2144/000112036. [DOI] [PubMed] [Google Scholar]
- 22.Kodama Y, Hu CD. An improved bimolecular fluorescence complementation assay with a high signal-to-noise ratio. Biotechniques. 2010;49:793–805. doi: 10.2144/000113519. [DOI] [PubMed] [Google Scholar]
- 23.Kerppola TK. Visualization of molecular interactions using bimolecular fluorescence complementation analysis: characteristics of protein fragment complementation. Chem Soc Rev. 2009;38:2876–2886. doi: 10.1039/b909638h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burshtyn DN, Lam AS, Weston M, Gupta N, Warmerdam PA, Long EO. Conserved residues amino-terminal of cytoplasmic tyrosines contribute to the SHP-1-mediated inhibitory function of killer cell Ig-like receptors. J Immunol. 1999;162:897–902. [PubMed] [Google Scholar]
- 25.Olcese L, Lang P, Vely F, Cambiaggi A, Marguet D, Blery M, Hippen KL, Biassoni R, Moretta A, Moretta L, Cambier JC, Vivier E. Human and mouse killer-cell inhibitory receptors recruit PTP1C and PTP1D protein tyrosine phosphatases. J Immunol. 1996;156:4531–4534. [PubMed] [Google Scholar]
- 26.Lakowicz JR. Plasmonics in Biology and Plasmon-Controlled Fluorescence. Plasmonics. 2006;1:5–33. doi: 10.1007/s11468-005-9002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lakowicz JR, Chowdhury MH, Ray K, Zhang J, Fu Y, Badugu R, Sabanayagam CR, Nowaczyk K, Szmacinski H, Aslan K, Geddes CD. Plasmon-controlled fluorescence: A new detection technology. Proc Soc Photo Opt Instrum Eng. 2006;6099:609909. doi: 10.1117/12.673106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan QR, Long EO, Wiley DC. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat Immunol. 2001;2:452–460. doi: 10.1038/87766. [DOI] [PubMed] [Google Scholar]
- 30.Rajagopalan S, Long EO. Zinc bound to the killer cell-inhibitory receptor modulates the negative signal in human NK cells. J Immunol. 1998;161:1299–1305. [PubMed] [Google Scholar]
- 31.Rajagopalan S, Winter CC, Wagtmann N, Long EO. The Ig-related killer cell inhibitory receptor binds zinc and requires zinc for recognition of HLA-C on target cells. J Immunol. 1995;155:4143–4146. [PubMed] [Google Scholar]
- 32.Sarkar P, Koushik SV, Vogel SS, Gryczynski I, Gryczynski Z. Photophysical properties of Cerulean and Venus fluorescent proteins. J Biomed Opt. 2009;14:034047. doi: 10.1117/1.3156842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor JR. An Introduction to Error Analysis. 2. University Science Books; Mill Valley, CA: 1997. The Square-root rule for a counting experiment/the poisson distribution; pp. 48–49.pp. 245–253. [Google Scholar]
- 34.Fruh K, Ahn K, Djaballah H, Sempe P, van Endert PM, Tampe R, Peterson PA, Yang Y. A viral inhibitor of peptide transporters for antigen presentation. Nature. 1995;375:415–418. doi: 10.1038/375415a0. [DOI] [PubMed] [Google Scholar]
- 35.Kavran JM, McCabe JM, Byrne PO, Connacher MK, Wang Z, Ramek A, Sarabipour S, Shan Y, Shaw DE, Hristova K, Cole PA, Leahy DJ. How IGF-1 activates its receptor. Elife. 2014:3. doi: 10.7554/eLife.03772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis SJ, van der Merwe PA. The kinetic-segregation model: TCR triggering and beyond. Nat Immunol. 2006;7:803–809. doi: 10.1038/ni1369. [DOI] [PubMed] [Google Scholar]
- 37.Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, Breso V, Frassati C, Reviron D, Middleton D, Romagne F, Ugolini S, Vivier E. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–342. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 38.Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, French AR, Sunwoo JB, Lemieux S, Hansen TH, Yokoyama WM. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 2005;436:709–713. doi: 10.1038/nature03847. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.