

Abstract
Accumulating evidence points to altered GABAergic parvalbumin-expressing interneurons and impaired myelin/axonal integrity in schizophrenia. Both findings could be due to abnormal neurodevelopmental trajectories, affecting local neuronal networks and long-range synchrony and leading to cognitive deficits. In this review, we present data from animal models demonstrating that redox dysregulation, neuroinflammation and/or NMDAR hypofunction (as observed in patients) impairs the normal development of both parvalbumin interneurons and oligodendrocytes. These observations suggest that a dysregulation of the redox, neuroimmune, and glutamatergic systems due to genetic and early-life environmental risk factors could contribute to the anomalies of parvalbumin interneurons and white matter in schizophrenia, ultimately impacting cognition, social competence, and affective behavior via abnormal function of micro- and macrocircuits. Moreover, we propose that the redox, neuroimmune, and glutamatergic systems form a “central hub” where an imbalance within any of these “hub” systems leads to similar anomalies of parvalbumin interneurons and oligodendrocytes due to the tight and reciprocal interactions that exist among these systems. A combination of vulnerabilities for a dysregulation within more than one of these systems may be particularly deleterious. For these reasons, molecules, such as N-acetylcysteine, that possess antioxidant and anti-inflammatory properties and can also regulate glutamatergic transmission are promising tools for prevention in ultra-high risk patients or for early intervention therapy during the first stages of the disease.
Keywords: parvalbumin interneurons, oligodendrocytes, myelination, N-acetylcysteine, development, oxidative stress
Introduction
Schizophrenia is considered a disorder with an important neurodevelopmental component. Various genetic and environmental risk factors can affect brain developmental processes including maturation of interneurons and oligodendrocytes, which could eventually contribute to the emergence of the symptoms during adolescence and early adulthood (Insel, 2010). Our current understanding of the neurobiological processes involved in schizophrenia remains, however, limited. Many hypotheses have been proposed, but a consensus among the research community is lacking. Prominent hypotheses include disturbance of glutamatergic neurotransmission in the form of hypofunction of NMDA receptors (NMDARs) (Coyle et al., 2012; Kantrowitz and Javitt, 2012; Krystal et al., 1994; Steiner et al., 2013), neuroinflammation (Meyer, 2013; Potvin et al., 2008; Saetre et al., 2007; van Berckel et al., 2008), and redox dysregulation (Clay et al., 2011; Do et al., 2009a, b; Gysin et al., 2011; Martins-de-Souza et al., 2011; Yao and Keshavan, 2011). We propose that dysregulation of redox homeostasis, neuroimmune, and glutamatergic systems induced by different etiological factors constitute, via their reciprocal interactions, one “central hub” as a common final pathway contributing to this disorder (Fig. 1). Here, we review the effect of dysregulation of each of these systems and their interactions on excitatory/inhibitory balance of local neuronal circuits (microcircuits), as well as the connections between distant brain areas (macrocircuits). In particular, we propose that dysfunction in these systems has deleterious effects on normal development of cortical and hippocampal parvalbumin-expressing interneurons (PVI), which are essential for fast local neuronal synchronization, and on oligodendrocytes, which form myelin sheets around axons providing fast signal conduction between brain regions. Anomalies of PVI and oligodendrocytes are indeed widely recognized in schizophrenia and considered to contribute to abnormal brain connectivity leading to cognitive, affective, and social deficits.
Fig. 1.

Proposed “hub” formed of the redox, neuroimmune, and glutamatergic systems whose dysregulation during development could disrupt maturation of parvalbumin interneurons (PVI) and oligodendrocytes, two cell types affected in schizophrenia and critical for short- and long-range neuronal network synchronization. This could impact structural and functional connectivity circuits affecting diverse aspects of cognitive, affective and social functioning (Buckholtz and Meyer-Lindenberg, 2012). Genetic risk factors combined with environmental insults can affect the homeostasis of one or several of the “hub” systems which in turn could impact the others through reciprocal interactions (reciprocal arrows). Genetic vulnerability to redox dysregulation in schizophrenia is supported by polymorphisms and copy number variations in genes related to the GSH metabolism (Gravina et al., 2011; Gysin et al., 2007; Mehta et al., 2013; Rodriguez-Santiago et al., 2010; Tosic et al., 2006). In addition, impaired function of proteins coded by other plausible risk genes, including DISC1, PROD, G72, NRG, DTNBP1, indirectly leads to oxidative stress often via mitochondrial dysfunction (Clay et al., 2011; Gokhale et al., 2012; Goldshmit et al., 2001; Johnson et al., 2013; Krishnan et al., 2008; Park et al., 2010). Genes related to the immune system have also been identified as potent risk genes for schizophrenia, in particular the major histocompatibility complex (MHC) genes, one of the most replicated genetic risk factors for schizophrenia disorder (Smyth and Lawrie, 2013; Stefansson et al., 2009). Finally, genetic vulnerability for NMDAR hypofunction seems to be more associated with potent risk genes encoding proteins that indirectly influence the function of this receptor; this includes d-amino acid oxidase, G72, dysbindin, and neuregulin (see Coyle et al., 2012), mGluR5 and proteins belonging to the postsynaptic NMDAR complex (Fromer et al., 2014; Kirov et al., 2012; Purcell et al., 2014; Timms et al., 2013). Developmental insults that are known to increase the risk for schizophrenia cause redox dysregulation/oxidative stress (Do et al., 2009b; Walter et al., 2002) and/or neuroinflammation (Brenhouse and Andersen, 2011; Garate et al., 2013; Kaur et al., 2013; Schiavone et al., 2009). Note that the dopaminergic or serotoninergic (5-HT) systems (and others = X Y) modulated by risk-factor genes and environment could also impact micro- and macrocircuits either directly or indirectly via interactions with the above “hub”. Dotted arrows depict impact of genetic risk factors. E/I balance: excitatory/inhibitory balance; PNN: perineuronal net surrounding PVI.
1. A “hub” formed by the redox, glutamatergic, and neuroimmune systems
A dysregulation of the redox, glutamatergic, and neuroimmune systems has all been reported in schizophrenia. Genetic and/or environmental risk factors can contribute to disturbances within each of these tightly interdependent systems (see Fig. 1 and its legend for more details). In particular, redox pathways represent a central node via their numerous reciprocal interactions with the glutamatergic and immune systems. Oxidative stress is defined as an imbalance between antioxidants and pro-oxidants (reactive oxygen species (ROS) and reactive nitrogen species (RNS)), resulting in macromolecular damage. In addition, redox signaling plays a key regulating role in many cellular and physiological processes (Jones, 2008). A redox dysregulation can affect cell proliferation/differentiation, energy metabolism, and neurotransmission via an alteration of redox-sensitive protein function, redox-dependent gene expression, and epigenetic mechanisms (Cyr and Domann, 2011; Ray et al., 2012; Valko et al., 2007). Several proteins related to glutamatergic neurotransmission contain modulatory redox sites, including glutamine synthase (Mustafa et al., 2007), serine racemase (responsible for synthesis of glycine, a NMDAR co-agonist, (Pinteaux et al., 1996)), and NMDARs (Choi et al., 2001). While redox state modulates NMDAR function, activation of synaptic NMDARs strengthens neuronal antioxidant defense mechanisms (Hardingham and Bading, 2010). Moreover, glutathione (GSH), the main antioxidant and redox regulator, constitutes a neuronal reservoir of glutamate (Koga et al., 2011). These observations indicate that redox and glutamatergic systems are intimately dependent. Likewise, oxidative stress is tightly linked to inflammation. Many inflammatory mediators are activated by oxidative molecules, while activated immune cells such as microglia generate ROS and RNS. The complex interplay between oxidative stress and inflammation is in part governed by the reciprocal interactions between the transcription factors Nrf2 (whose nuclear translocation induces antioxidant phase II gene transcription) and NF-kB (whose translocation to the nucleus promotes transcription of many pro-inflammatory genes) (Buelna-Chontal and Zazueta, 2013). Finally, an imbalance of the immune system may also affect NMDAR function. Human subjects with anti-NMDAR encephalitis develop psychosis (Dalmau et al., 2011) and antibodies against NMDAR have been reported in patients diagnosed with schizophrenia (Steiner et al., 2013). Moreover, inflammatory processes cause increased production of kynurenic acid, an endogenous NMDAR antagonist, via dysregulation of tryptophan/kynurenine metabolism (Muller et al., 2011). Thus, redox, immune, and glutamatergic systems form a triad in which each of its elements can influence the others. Diverse genetic vulnerabilities and environmental risk factors may affect one element of this triad, impacting in turn the other systems. Because of the complex interactions between each element of this triad, it is difficult to untangle the respective contribution of each system in the pathophysiology. To our view, the primary effector may depend on the specific combination of the genetic vulnerability and environmental insults. Therefore, the redox, immune, and glutamatergic systems may be considered together as one “central hub” in which a dysregulation in any of them can lead to a common pathophysiological condition, such as dysconnectivity via impairment of PVI and oligodendrocytes.
2. Parvalbumin interneurons
PVI are GABAergic neurons that form inhibitory synapses onto either the cell body (for parvalbumin-expressing basket cells) or the axon initial segment (for parvalbumin-expressing chandelier cells) of pyramidal neurons (PNs). Basket cells control inputs reaching the soma of PNs, while chandelier cells control PN output. PVI, which are interconnected via gap junctions (Fukuda et al., 2006) and reciprocal GABAergic synapses, constitute a cellular network able to synchronize the excitatory state of large numbers of PNs (Bartos et al., 2007). By way of feedback and feedforward inhibition, fast-spiking interneurons exert precise temporal control on information that can flow through PNs. These interneurons favor summation and transmission of converging inputs arriving synchronously onto a PN. By controlling synchronized excitability state of a network of PNs, PVI also allow the binding of information that reach these different PNs during a defined and narrow time window (Fries et al., 2007). Therefore, PVI strongly influence local neuron-network dynamic. They are critical for high-frequency neuronal synchrony, reflected in gamma band oscillations (30–80 Hz) (Cardin et al., 2009; Fuchs et al., 2007; Gulyas et al., 2010; Massi et al., 2012; Sohal et al., 2009), but can also modulate neuronal activity in the theta band (4–8 Hz), as well as theta-gamma coupling (Korotkova et al., 2010; Wulff et al., 2009). The maturation of PVI and their associated extracellular matrix defines a critical period of cortical network plasticity during postnatal development (Morishita and Hensch, 2008). Moreover, plasticity within the basket-cell network contributes to memory learning, consolidation and retrieval (Donato et al., 2013) and PVI promote neuronal progeny survival and development in the hippocampus (Song et al., 2013). Furthermore, in prefrontal cortical regions, heavily implicated in schizophrenia pathophysiology, PVI mature during adolescence (Tseng and O’Donnell, 2007; O’Donnell, 2011) and is therefore a neural population with a protracted developmental trajectory that could explain the peri-adolescent onset of schizophrenia symptoms.
2.1. Evidence for abnormal PVI in schizophrenia
Compelling evidence suggests an imbalance between glutamatergic excitation and GABAergic inhibition in schizophrenia (Lisman et al., 2008; O’Donnell, 2011). Anomalies associated with PVI constitute a hallmark of the disease, including reduced density of parvalbumin-immunoreactive cells in the hippocampal formation (Wang et al., 2011; Zhang and Reynolds, 2002) and alterations at the level of basket and chandelier cells in the dorsolateral prefrontal cortex (DLPFC) of post-mortem brains (Lewis et al., 2012). These alterations include reduced expression of parvalbumin and GAD67 (isoform of glutamic acid decarboxylase, the GABA synthesizing enzyme), changes in their pre- and postsynaptic terminals (Lewis et al., 2012), and reduced expression of Kv3.1-containing K+ channels, which play a critical role in their fast-spiking properties (Yanagi et al., 2014). Moreover, the extracellular matrix (perineuronal net: PNN) that surrounds many PVI is reduced in DLPFC (Mauney et al., 2013), entorhinal cortex, and amygdala of schizophrenia patients (Pantazopoulos et al., 2010). Current data suggest an impaired maturation of PVI rather than a deficit due to the chronicity of the illness. The DLPFC of young patients already has low expression levels of parvalbumin and GAD67 (Hoftman et al., 2013), and gene expression pattern in PVI of schizophrenia individuals resembles that of non-mature cells (Gandal et al., 2012). Therefore, dysfunction of the PVI-associated network may lead to abnormal neuronal activity in patients, including oscillatory activity within theta, beta and gamma ranges (McNally et al., 2013; Uhlhaas and Singer, 2010, 2012). Ultimately, interneuron dysfunction could contribute to altered sensory perception (Atallah et al., 2012), deficits in working memory (Korotkova et al., 2010; Roux et al., 2012), attention (Rouhinen et al., 2013), and learning (Carlen et al., 2012).
2.2. Mechanisms underlying abnormal PVI
Recent studies have revealed anomalies in hippocampal and/or prefrontal PVI in many preclinical animal models aiming to reproduce genetic vulnerabilities (Carlson et al., 2011; Fazzari et al., 2010; Hikida et al., 2007; Wen et al., 2010) or environmental risk factors (Brown, 2011) such as prenatal maternal stress (Stevens et al., 2013), maternal and perinatal immune challenge (Jenkins et al., 2009; Meyer et al., 2008), hypoxia (Dell’Anna et al., 1996; Komitova et al., 2013), early-life iron deficiency (Callahan et al., 2013), maternal separation (Brenhouse and Andersen, 2011) and social isolation (Harte et al., 2007; Schiavone et al., 2009). These developmental insults cause oxidative stress (Do et al., 2009b; Walter et al., 2002) and/or neuroinflammation (Brenhouse and Andersen, 2011; Garate et al., 2013; Kaur et al., 2013). Furthermore, non-genetic developmental models also result in altered prefrontal PVI (Lodge et al., 2009; Tseng et al., 2008). In rats with a neonatal ventral hippocampal lesion, the normal peri-adolescent maturation of PVI is impaired (Tseng et al., 2008), and in this model PVI show evidence of oxidative stress prior to the onset of behavioral deficits (O’Donnell et al., 2011). Below, we present evidence that PVI are particularly affected during their development by oxidative stress, neuroinflammation, and NMDAR hypofunction.
2.2.1. Vulnerability to redox dysregulation/oxidative stress
To support high-frequency neuronal synchronization, fast-spiking PVI are energy demanding. This requires optimal mitochondrial performance (Kann et al., 2011) with enhanced metabolic activity and oxidative phosphorylation (Harris et al., 2012) leading to elevated mitochondria-generated ROS. Consequently, PVI need well-regulated antioxidant systems to neutralize ROS and maintain proper redox state. Interestingly, the power of β/γ neuronal activity is positively correlated with blood GSH levels in patients (Ballesteros et al., 2013). These cells are vulnerable to redox dysregulation, whether induced by a compromised antioxidant system or ROS overproduction. In an animal model with low GSH content, as reported in the brain of some schizophrenia patients (Do et al., 2000; Gawryluk et al., 2011; Yao et al., 2006), there is a deficit in prefrontal and hippocampal PVI, impairing high-frequency neuronal synchronization (Cabungcal et al., 2013a; Cabungcal et al., 2013b; Steullet et al., 2010). Interestingly, an inhibition of GSH synthesis restricted to PVI is sufficient to affect these interneurons (Cabungcal et al., 2013b) and oxidative stress precedes the PVI deficit (Steullet et al., 2010). PVI can also be affected when antioxidant systems other than GSH are compromised. A reduced number of parvalbumin-immunoreactive cells is observed in mice with a deletion for the selenoprotein P, a glycoprotein with antioxidant properties (Pitts et al., 2012) or for PGC-1α, a transcription factor regulating mitochondria function and ROS metabolism (Lucas et al., 2010). Furthermore, superoxide overproduction by NADPH oxidase (NOX) is also deleterious to PVI (Behrens et al., 2007), and NOX inhibition prevents the PVI impairment induced by social isolation (Schiavone et al., 2009).
Most importantly, prefrontal cortical PVI are more vulnerable to a redox dysregulation during postnatal development than later in life. A pharmacologically induced transient postnatal deficit in GSH causes both immediate and long-term decreased density of parvalbumin-immunoreactive cells in the anterior cingulate cortex (ACC) (Cabungcal et al., 2006; Kulak et al., 2013; Steullet et al., 2011). In mice with a chronic GSH deficit (Gclm KO mice, Kulak et al., 2012), administration of a dopamine re-uptake inhibitor (GBR-12909), which partially mimics dopamine release during psychosocial stress (Lataster et al., 2011) and produces ROS via the catabolism of dopamine (Cadet and Brannock, 1998; Rabinovic and Hastings, 1998), decreases permanently the density of parvalbumin-immunoreactive cells in the ACC when applied during postnatal development, but not adulthood (Cabungcal et al., 2013a). Thus, immature PVI may have a less robust antioxidant defense system than mature cells. Alternatively, molecular mechanisms underlying PVI maturation are highly sensitive to a redox imbalance. Interestingly, the vulnerability of prefrontal immature PVI is associated with the absence of fully mature PNN, which protects these cells against oxidative stress (Cabungcal et al., 2013b). However, excess of oxidative stress also affects PNN (Cabungcal et al., 2013b), which can in turn impact PVI. Indeed, the maturation and phenotypic maintenance of PVI require incorporation of a non-cell autonomous homeobox protein, Otx2, through its affinity with PNN (Beurdeley et al., 2012; Miyata et al., 2012).
The implication of redox dysregulation/oxidative stress for the developmental impairment of PVI has been further substantiated by recent studies on experimental neurodevelopmental models that do not directly manipulate the redox system. First, the widely studied neonatal ventral hippocampal lesion model also displays oxidative stress and PVI defect, both of which are prevented by a juvenile and adolescence treatment with the antioxidant and GSH precursor, N-acetylcysteine (NAC) (O’Donnell et al., 2011; Sullivan and O’Donnell, 2012). Second, a single injection of the DNA-alkylating agent methylazoxymethanol acetate (MAM) during pregnancy, which also causes schizophrenia phenotypes in adult rats, leads to anomalies in PVI and neuronal synchronization (Lodge et al., 2009; Penschuck et al., 2006). MAM-treated rats have also decreased brain GSH levels (Cleland et al., 2013, abstract Neuroscience Meeting). Collectively, these studies demonstrate that a redox dysregulation during a critical developmental period can disrupt normal maturation of PVI.
2.2.2. Vulnerability to NMDAR hypofunction
Numerous studies show that NMDAR blockade in adults disrupts excitatory/inhibitory balance in cortical circuits, affecting PVI (Behrens et al., 2007) and neuronal network activity (Carlen et al., 2012; Homayoun and Moghaddam, 2007; Kocsis et al., 2013; Korotkova et al., 2010; Lazarewicz et al., 2010). However, PVI are especially vulnerable to NMDAR hypofunction during development (Abekawa et al., 2007; Powell et al., 2012; Wang et al., 2008; Wang et al., 2013). Inhibition of NMDARs during early life causes a persistent decrease in number of parvalbumin-immunoreactive cells without cell death (Powell et al., 2012), suggesting that disruption of NMDAR-mediated signaling impairs maturation of these cells. Indeed, the maturation of PVI is activity-dependent (Chattopadhyaya et al., 2004; Patz et al., 2004). Calcium entrance is necessary for the maturation of PNN (Dityatev et al., 2007) and PVI (Jiang and Swann, 2005; Kinney et al., 2006), and activation of NR2A-containing NMDARs contributes to the molecular signaling that leads to the maturation of these cells (Kinney et al., 2006; Zhang and Sun, 2011).
2.2.3. Interactions between NMDAR hypofunction and redox dysregulation
It is intriguing that NMDAR hypofunction and redox dysregulation impair PVI maturation in similar ways. This raises the possibility that both mechanisms interfere with the maturation of PVI cells via related molecular mechanisms. Interestingly, synaptic NMDAR activation boosts GSH, thioredoxin, and peroxiredoxin systems via calcium-mediated signaling involving activation of CREB and inhibition of FOXO (Hardingham and Bading, 2010; Papadia et al., 2008). The work from Nakazawa and colleagues indicates that impaired maturation of PVI induced by NMDAR hypofunction is due to a redox dysregulation. When applied during postnatal development, a deletion of the NR1 subunit of NMDARs in a subpopulation of interneurons (including most PVI) leads to parvalbumin and GAD67 expression deficit along with oxidative stress in PVI (Belforte et al., 2010; Jiang et al., 2013). In these mice, social isolation exacerbates oxidative stress and PVI deficits, both of which are prevented by a NOX inhibitor (Jiang et al., 2013). In NOX-2 knockout mice, PVI are protected from a postnatal ketamine treatment, indicating a crucial role of NOX in the PVI impairment following early-life blockade of NMDAR (Powell et al., 2012). However, the lack of NMDAR-mediated signaling also causes a reduction in PGC-1α levels and expression of several antioxidant enzymes (Jiang et al., 2013), and decreases GSH levels (Stojkovic et al., 2012). These observations suggest that NMDAR hypofunction can weaken antioxidant defenses, contributing to a redox dysregulation and affecting cell maturation. The downregulation of antioxidant systems by NMDAR blockade may be particularly significant in PVI of young individuals, since NMDAR-mediated postsynaptic responses are stronger in immature compared to mature PVI (Rotaru et al., 2011; Wang and Gao, 2009, 2010). However, a redox dysregulation can also downregulate NMDAR function on its own. Functional down-regulation of NMDARs by oxidative conditions can occur via either extracellular redox-sensitive sites located on NR1 and NR2A subunits (Choi et al., 2001; Kohr et al., 1994) or by Ca2+/calmodulin-dependent protein kinase II (Bodhinathan et al., 2010). A transient postnatal GSH deficit results in NMDAR hypofunction (Steullet et al., 2006) and impairs PVI (Cabungcal et al., 2006). Therefore, redox dysregulation and NMDA receptor hypofunction during postnatal development can interact synergistically, creating a vicious circle that is particularly detrimental for PVI.
2.2.4. Vulnerability to neuroinflammation
Because of the tight link between oxidative stress and inflammation, it is not surprising that pro-inflammatory molecules affect PVI. Interleukin-6 mediates ketamine-induced NOX upregulation and subsequently PVI deficits (Behrens et al., 2008). A genome-wide profiling and immunohistological study revealed that reduced PVI density in schizophrenia patients is associated with two modules of genes differentially expressed in patients compared to healthy subjects, among which are many immune/inflammation-related genes (Hwang et al., 2013). Early-life pro-inflammatory conditions, such as maternal and neonatal immune challenges, cause a persistent decrease in number of prefrontal and/or hippocampal parvalbumin-immunoreactive interneurons (Jenkins et al., 2009; Meyer et al., 2008). This deleterious effect could result from a redox dysregulation as maternal immune challenge transiently decreases GSH and vitamin E levels and increase oxidative stress in the hippocampus (Lante et al., 2007). A reduced number of hippocampal and prefrontal parvalbumin-immunoreactive interneurons is also observed in Schnurri-2 KO mice, a genetic model for enhanced neuroinflammation via increased NF-kB-dependent gene expression (Takao et al., 2013). These mice show increased expression of inflammation-related genes and NOX, which affects PVI. The PVI impairment following maternal separation can be prevented by non-steroidal anti-inflammatory drugs (Brenhouse and Andersen, 2011). Nevertheless, the possibility that a ROS scavenger can also protect PVI against the effect of maternal separation cannot be excluded. These data clearly show that neuroinflammation impacts PVI, but the specific role of pro-inflammatory molecules and oxidative stress remains to be established. A combination of vulnerability for pro-inflammatory conditions and redox dysregulation due to environmental and genetic factors may be particularly deleterious for PVI. This combination has been demonstrated in transgenic mice expressing a putative dominant-negative disrupted in schizophrenia 1 (DN-DISC1), which display enhanced prefrontal oxidative stress (Johnson et al., 2013) and shows stronger PVI deficits following a postnatal immune challenge (Ibi et al., 2010).
To conclude, PVI are particularly vulnerable to redox dysregulation/oxidative stress, NMDAR hypofunction, and neuroinflammation during early development. Genetic vulnerabilities and environmental insults that would affect homeostasis of either redox, or glutamatergic, or neuroimmune system could affect the other systems with amplified negative consequences on PVI maturation and subsequently on neuronal network synchronization and information processing.
3. Oligodendrocytes/myelination
3.1. Evidence for impaired oligodendrocytes/myelination in schizophrenia
Oligodendrocytes and myelination are clearly impaired in schizophrenia (Chew et al., 2013; Davis et al., 2003; Takahashi et al., 2011). The observations supporting this claim include decreased expression of oligodendrocyte-related genes (Hakak et al., 2001; Katsel et al., 2005; Tkachev et al., 2003), impairment of oligodendrocyte maturation (Kerns et al., 2010), reduced number and/or density of oligodendrocytes in gray and white matter (Byne et al., 2008; Hof et al., 2003; Uranova et al., 2004), apoptotic oligodendrocytes and ultrastructural alterations in myelinated fibers (Uranova et al., 2001). The anomalies at the level of myelin/axonal integrity increase with illness duration (Uranova et al., 2011). Studies using magnetic resonance techniques such as diffusion tensor imaging (DTI) also suggest abnormal white matter along different fiber tracts, including within and between frontal and temporal areas (Fitzsimmons et al., 2013). Although less consistent than in chronic patients, white matter anomalies are observed in ultra high-risk subjects and first-episode patients (Fitzsimmons et al., 2013; Kyriakopoulos and Frangou, 2009), suggesting a neurodevelopmental component for this impairment. Oligodendrocytic and myelination anomalies in schizophrenia could affect axonal integrity and conduction velocity (Whitford et al., 2011), with the consequence of disrupting temporal control of long-range brain synchronization.
3.2. Mechanisms underlying impaired oligodendrocytes/myelination
Genes related to oligodendrocytes and myelination have been associated with schizophrenia (Takahashi et al., 2011), suggesting that white matter anomalies in this disorder could have a direct genetic origin. However, other mechanisms could also impact white matter integrity. Perinatal insults and psychosocial stress during childhood and adolescence are correlated with structural changes in white matter (Chew et al., 2013; Eluvathingal et al., 2006; Huang et al., 2012). In rodents, early-life insults, most of which cause PVI impairment, also affect oligodendrocytes and myelination. These insults include maternal and early postnatal immune challenge (Fan et al., 2005; Paintlia et al., 2008), perinatal hypoxia (Oorschot et al., 2013), hypoxia-ischemic insults (Robinson et al., 2005), and social isolation (Liu et al., 2012; Makinodan et al., 2012). These observations suggest that some of the biological causes for the developmental PVI and white matter anomalies could be similar.
3.3.1. Vulnerability to redox dysregulation/oxidative stress
In vitro studies show that oligodendrocytes are susceptible to oxidative stress due to their high metabolic activity and iron content combined with low antioxidant levels (Back et al., 1998; Baud et al., 2004; Fragoso et al., 2004). Furthermore, the intracellular redox state controls the proliferation and differentiation of oligodendrocytes (Li et al., 2007; Smith et al., 2000; Do et al., 2012), and low GSH levels affect oligodendrocyte maturation (French et al., 2009). Peripubertal Gclm KO mice (which have low brain GSH content) present a deficit in myelin-associated proteins and mature oligodendrocytes in the ACC (Monin et al., 2013, abstract Neuroscience Meeting, 255.03). This deficit, which recovers in adulthood, is accompanied by an increase in prefrontal N-acetylaspartate (das Neves Duarte et al., 2012), suggesting an impaired myelin lipid synthesis during prefrontal cortical maturation (Kulak et al., 2013). Interestingly during this period, genes associated with myelin and lipid synthesis, antioxidant response systems, mitochondria function and glycolysis are highly expressed (Harris et al., 2009). Therefore, a proper redox state controlled by GSH may be critical for adequate myelination in prefrontal gray matter during this period of high metabolic activity. In addition, a decrease in fractional anisotropy is observed along a few fiber tracts in Gclm KO mice, also suggesting white matter anomalies (Corcoba Garcia et al., 2013, abstract Neuroscience Meeting, 729.24). Interestingly, we found a positive correlation between GSH content in the ACC and fractional anisotropy along the cingulum bundle in young adult human subjects (Monin et al., 2013, abstract Neuroscience Meeting, 255.03). Taken together, these findings indicate that a redox dysregulation can cause oligodendrocytic developmental anomalies and/or delay in gray and white matter which may eventually contribute to abnormal myelin sheath and axonal integrity.
3.3.2. Vulnerability to NMDAR dysfunction
Postnatal inhibition of NMDARs causes deficit not only in PVI, but also in myelination (Zhang et al., 2012). It is, however, unclear whether the myelination impairment results from the redox dysregulation also observed with this manipulation (Stojkovic et al., 2012) or from the loss of NMDAR signaling in oligodendrocytes. NMDARs are indeed expressed in immature and mature oligodendrocytes. Activation of these receptors in oligodendrocytes modulates their metabolism, promotes their differentiation from immature into mature stage, and favors myelination around axons (Cao and Yao, 2013; Li et al., 2013). Interestingly, NMDAR activation promotes the differentiation of cultured oligodendrocytes via NOX-mediated ROS (Cavaliere et al., 2012). A combination of redox dysregulation and NMDAR hypofunction could be therefore deleterious for oligodendrocyte differentiation and myelination.
3.3.3. Vulnerability to neuroinflammation
Oligodendrocytes, like PVI, are also vulnerable to early-life neuroinflammation (Chew et al., 2013). Neonatal administration of inflammatory cytokines such as Il-1β reduces the number of developing oligodendrocytes (Cai et al., 2004; Fan et al., 2009). Maternal and early postnatal immune challenges impair myelination (Fan et al., 2005; Makinodan et al., 2008; Paintlia et al., 2008) and long-range synchronization (Dickerson et al., 2010). As microglial cells and cytokines participate in normal brain development (Bilbo and Schwarz, 2012; Kettenmann et al., 2013), a dysregulation of cytokine-mediated pathways could disrupt normal developmental processes. However, current data indicate that redox dysregulation contributes to myelination impairment due to early-life inflammation. Alpha-phenyl-n-tert-butyl-nitrone (PBN), a free radical scavenger, protects oligodendrocytes and myelination against neonatal immune challenge (Fan et al., 2008). Likewise, NAC prevents this deficit by attenuating the dysfunction of peroxisomes, organelles important for ROS detoxification and myelin-lipid metabolism (Paintlia et al., 2008). Moreover, the decreased expression of myelin-related genes induced by cytokines (Il-1β and TNF-α) in human primary oligodendrocytes is blocked by NAC (Jana and Pahan, 2005), suggesting that these inflammatory molecules act through redox dysregulation. Finally, the tetracycline antibiotic, minocycline, prevents hypomyelination induced by immune challenge, and its protective effect is associated with decreased microglial activation, cytokine levels, and oxidative stress (Fan et al., 2005). In a genetic model of enhanced pro-inflammation (Schnurri-2 KO mice), the expression of myelin-associated proteins is reduced, which could be due to a NOX upregulation (Takao et al., 2013). These data show that the impact of early-life neuroinflammation on oligodendrocytes and myelination is in part due to the generation of ROS/RNS. Thus as for PVI, a vulnerability to redox dysregulation may further exacerbate the deleterious effect of pro-inflammatory conditions on myelin integrity.
Taken together, it is remarkable that PVI and oligodendrocytes are affected by similar early-life insults and by the disruption of redox, neuroimmune and glutamatergic systems. One reason for this shared vulnerability could be related to the high metabolic requirement of these two cell types. The data compiled in the present review strongly suggest that genetic and environmental risk factors, which lead to dysregulation of the “hub” comprised of redox, neuroimmune and glutamatergic systems, would invariably affect PVI and white matter, and consequently impair long- and short-range neuronal network connectivity. Given the pivotal role of PVI, PNN, and myelin in regulating brain plasticity dynamics and active epochs (McGee et al., 2005; Miyata et al., 2012; Morishita and Hensch, 2008), the dysregulation of this “hub” could yield the slow emergence of clinical symptoms by altering the timing of key windows of critical period of brain plasticity (Morishita et al., 2010, abstract Neuroscience Meeting, 62.30). Because of the complex interplay between redox, immune, and glutamatergic systems, we propose that combinations of genetic and environmental risk factors could generate a vicious circle of dysregulation within all these systems, ultimately giving rise to abnormal functional and structural connectivity deficits, as observed in schizophrenia. We have however to acknowledge that this hypothesis is based mostly on observations and experiments in rodent models and postulates that the mechanisms are similar in humans. Moreover, we have to emphasize that other systems such as the dopamine system known to be implicated in schizophrenia and their reciprocal interactions with the proposed “hub” (see: Avshalumov et al., 2007; Baker et al., 2002; Kulak et al., 2013; Lodge and Grace, 2011; Meiser et al., 2013; Meyer and Feldon, 2009; Moller et al., 2013; Steullet et al., 2008) certainly participate to the pathology. Increasing evidence points to altered stress-reactivity as a vulnerability marker for psychosis (Myin-Germeys et al., 2003; Lataster et al., 2013). The biological mechanism underlying psychotic reactivity to stress could be related to hyper-reactivity of dopamine neurons to environmental stimuli and stress (Lataster et al., 2011; Mizrahi et al., 2012; Myin-Germeys et al., 2005). As dopamine catabolism is known to generate ROS, its excess would induce an oxidative stress. Metabolomic approach in cells derived from patients and controls highlights the possibility of using metabolic signatures of reactivity to oxidative stress as biomarkers for early psychosis (Fournier et al., 2014).
4. N-acetylcysteine, a potential therapeutic or prevention drug
Novel strategies aiming to regulate redox, immune, and glutamatergic systems could therefore be potentially useful to prevent or attenuate developmental anomalies yielding schizophrenia pathophysiology. To date, clinical trials using molecules targeting either the glutamatergic system (mGluR5, mGluR2/3, glycine site of NMDARs) (Javitt, 2012; Kantrowitz and Javitt, 2012; Poels et al., 2014; Vinson and Conn, 2012), the immune (Leza et al., 2014; Muller et al., 2013), or antioxidant systems (Leza et al., 2014; Reddy and Reddy, 2011) have produced mixed results, some encouraging and others inconclusive. Several promising clinical phase III trials aiming to act on the glutamatergic system proved inconclusive, but other relatively small scale clinical studies targeting the glycine site of NMDAR using D-serine indicate positive effects (Kantrowitz and Javitt, 2012). The efficacy of such molecules may however depend on the disease stages but also on patient subgroups. Future trials should focuse on unmedicated patients in the early phase of the disease and target not only clinical but also biological measures such as mismatch negativity as readouts.
Several antioxidants have been tested in relatively small clinical trials as add-on to antipsychotics; this includes vitamin C, vitamin E, and N-acetylcysteine (NAC) (see for reviews: Leza et al., 2014; Reddy and Reddy, 2011). Vitamins C and E are non-enzymatic antioxidants that scavenge free radicals in the cytosol and at the level of cell membranes, respectively. GSH is required for the recycling of oxidized vitamin C into its active form, while vitamin C is itself needed to reactivate oxidized vitamin E. Therefore, the efficacy of these vitamins may greatly depend on the integrity of the GSH system and the intrinsic redox status. This might explain the limited or the lack of efficacy of these compounds. However, these vitamins have also been given with some success together with 3-omega fatty acids which are key components of membrane phospholipids and have anti-inflammatory properties among others (see reviews: Leza et al., 2014; Muller et al., 2013; Sinn et al., 2010). Interestingly, Bentsen et al. (2013) found that vitamins and 3-omega fatty acids, when given separately, can be deleterious in a subgroup of patients. In the present review, we will however focus on NAC because it has antioxidant and anti-inflammatory properties, and can regulate glutamatergic neurotransmission. NAC is already used as antioxidant and GSH precursor to treat GSH deficiency in a wide range of infections, genetic defects, and metabolic disorder (Atkuri et al., 2007; Zafarullah et al., 2003). Therefore, it represents a safe and potential compound for the prevention or treatment of schizophrenia and other psychiatric disorders (Berk et al., 2013). NAC is deacetylated to form cysteine, the rate-limiting precursor of GSH, and therefore yields upregulation of GSH synthesis when cells face an excess of ROS production. NAC also participates to the control of the intracellular redox state by supplying cysteine into the cystine/cysteine redox couple (Mandal et al., 2010). In addition, NAC has anti-inflammatory effects, likely via its antioxidant properties. Finally, NAC upregulates the activity of the astrocytic cystine/glutamate antiporter, leading to cellular entry of cystine (which can be reduced to cysteine and incorporated into GSH) and extracellular release of glutamate (Bridges et al., 2012). This antiporter plays an important role in the regulation of extrasynaptic glutamate levels, which in turn regulate synaptic glutamate release via presynaptic mGluR2/3 (Baker et al., 2002). Thus, NAC could be useful to reduce synaptic glutamate release and to indirectly enhance NMDAR function via its antioxidant and redox regulator properties. Studies on several preclinical models and a few clinical trials on schizophrenia patients have recently provided a proof of concept that NAC could be a useful therapeutic tool.
4.1. NAC in preclinical models
In mice with a weakened GSH synthesis (Gclm KO mice), NAC prevents PVI and PNN deficits induced by an oxidative insult during postnatal development (Cabungcal et al., 2013a), although it does not increase GSH levels because of the Gclm deletion (das Neves Duarte et al., 2012). Furthermore, in young Gclm KO mice, NAC normalizes most of the neurochemical profiles, including the glutamime/glutamate ratio known to be altered in a similar way in first-episode schizophrenia patients (das Neves Duarte et al., 2012). Likewise, NAC reduces oxidative stress, protects prefrontal PVI, and prevents deficits in mismatch negativity and pre-pulse inhibition in the neonatal ventral hippocampal lesion rat model (O’Donnell et al., 2011; Sullivan and O’Donnell, 2012). NAC also prevents myelin impairment following a maternal immune challenge (Paintlia et al., 2008), re-establishes normal function of the cystine/glutamate antiporter and GSH levels in MAM-injected rats (Cleland et al. 2013, abstract Neuroscience Meeting, 428.10), normalizes extracellular glutamate levels and attenuates behavioral anomalies in phencyclidine-treated rats (Lutgen et al., 2013), reduces oxidative stress and rescues abnormal behavioral phenotype in G72/G30 transgenic mice (Otte et al., 2011), and reverses the social isolation-induced changes in corticostriatal monoamine levels (Moller et al., 2013). Thus, NAC has beneficial effects on very diverse animal models relevant to schizophrenia.
4.2. NAC in clinical trials
Although beneficial effects of a compound in rodent models do not necessarily translate into an efficient therapeutic drug in humans, the few published clinical studies using NAC show some promises. In a first randomized double-blind placebo-controlled trial, an add-on treatment of NAC in chronic patients diminished negative symptoms and improved global functioning (Berk et al., 2008). Two additional studies also demonstrated that chronic patients improved with add-on NAC, particularly in their negative symptoms (Bulut et al., 2009; Farokhnia et al., 2013). In addition, NAC normalized neuronal activity and connectivity and improved mismatch-negativity (Lavoie et al., 2008), an auditory-related, NMDA-dependent evoked potential typically impaired in schizophrenia (Umbricht et al., 2000). NAC also increased phase synchronization of neuronal activity over the left parieto-temporal, the right temporal, and the bilateral prefrontal regions (Carmeli et al., 2012). However, the beneficial effect of NAC has to be taken with caution since the current data is based on only a few studies showing relatively moderate clinical improvement in chronic schizophrenia patients, probably due to the low bioavailability and membrane permeability of NAC which enters the brain at a very modest rate (Farr et al., 2003). The development of other molecules with better bioavailability and blood-brain barrier permeability are therefore needed. As vitamins C and E or 3-omega fatty acids are detrimental for a subgroup of patients (Bentsen et al., 2013), it would be also advisable using biomarkers to identify patients that would most benefit from an antioxidant treatment. Moreover, NAC or other molecules that target redox, immune, and glutamatergic systems may be more beneficial for young subjects at risk than for chronic patients because the defects in PVI and oligodendrocytes/myelination may precede illness onset. Finally, it would be also worth investigating compounds such as sulforaphane (Shirai et al., 2012) that up-regulate Nrf2-dependent phase II detoxification enzymes and antioxidant proteins which include the cystine/glutamate antiporter and enzymes of the GSH system (Lavoie et al., 2009).
5. Concluding remarks
Brain development is dependent upon sequences of events: proliferation, differentiation, migration, formation, and maturation of neuronal circuitry. The pace of development varies among brain structures, with the prefrontal cortex being the last to mature. Brain maturation mechanisms are under genetic control and influenced by environmental insults, suggesting that different brain regions could be vulnerable to a dysregulation of the “hub” during specific developmental periods. For instance, under a GSH deficit, the most susceptible periods for oxidative stress differ in the ACC, the ventral and dorsal hippocampus (Cabungcal et al., 2013a; Steullet et al., 2010). Moreover, inflammation induced at different prenatal periods can lead to distinct adult phenotypes (Meyer et al., 2008). Therefore, the timing of environmental insults during development combined with specific genetic vulnerability could differentially affect circuit connectivity and cognition, social competence, and affective behavior, leading to heterogeneous clinical phenotypes (Fig. 2).
Fig. 2.
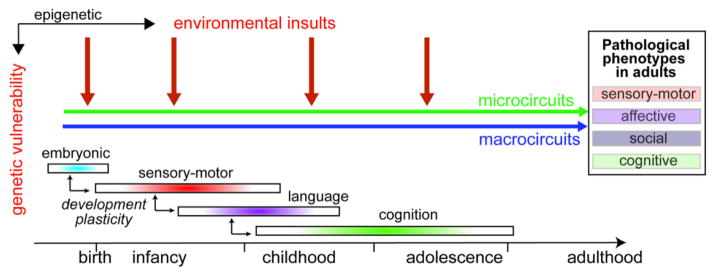
The timing of environmental insults levied upon an individual (at risk) during his development, may determine which brain region microcircuits and which macrocircuits connecting distant brain areas are structurally and functionally affected. The period(s) of vulnerability of a micro- or macrocircuit may vary according to the genetic risk factors and the nature of the environmental stress and may be influenced by the developmental trajectory of other brain areas. In addition, microcircuits might be particularly susceptible prior to their final maturation during the period of enhanced plasticity. Therefore, the timing of environmental insults during development combined with specific genetic vulnerability could differentially affect circuit connectivity associated with sensory-motor function, social competence, affective behavior, and cognition leading to heterogeneous clinical phenotypes. An early insult could lead to more severe and wide-spectrum clinical phenotypes than a later insult.
The body of knowledge reviewed above suggests that it would be worth to intervene early during brain development on all three elements of the proposed “hub”. The genetic vulnerability factors, although important as potential biomarkers for high-risk individuals, may not lend themselves to therapeutic interventions. In contrast, we propose that targeting neuroinflammation, oxidative stress, and NMDAR hypofunction at critical developmental periods and early in the disease may reduce neuropathological anomalies and alleviate the risk of emergence of clinical manifestations.
Acknowledgments
We thank our financial supports: Swiss National Science Foundation (# 31-116689 to KQD and # 310030_135736/1 to KQD and PS), National Center of Competence in Research (NCCR) “SYNAPSY - The Synaptic Bases of Mental Diseases” from the Swiss National Science Foundation (n° 51AU40_125759 to KQD), Avina Foundation, Damm-Etienne Foundation, “Loterie Romande”, Alamaya Foundation. Dr. O’Donnell was supported by the University of Maryland School of Medicine and by the National Institute of Mental Health (R01 MH057683) and a NARSAD Distinguished Investigator Award
Abreviations
- NAC
N-acetylcysteine
- PN
pyramidal neuron
- PNN
perineuronal net
- PVI
parvalbumin interneurons
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
Footnotes
Contributors: PS: wrote the drafts of the manuscript; JHC, AM, DD, POD, MC, and KQD contributed to the writing of the manuscript and approved its final version
Conflict of interest: P. O. is employee and stockholder of Pfizer, Inc. All other authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abekawa T, Ito K, Nakagawa S, Koyama T. Prenatal exposure to an NMDA receptor antagonist, MK-801 reduces density of parvalbumin-immunoreactive GABAergic neurons in the medial prefrontal cortex and enhances phencyclidine-induced hyperlocomotion but not behavioral sensitization to methamphetamine in postpubertal rats. Psychopharmacology. 2007;192 (3):303–316. doi: 10.1007/s00213-007-0729-8. [DOI] [PubMed] [Google Scholar]
- Atallah BV, Bruns W, Carandini M, Scanziani M. Parvalbumin-expressing interneurons linearly transform cortical responses to visual stimuli. Neuron. 2012;73 (1):159–170. doi: 10.1016/j.neuron.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-Acetylcysteine--a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol. 2007;7 (4):355–359. doi: 10.1016/j.coph.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avshalumov MV, Bao L, Patel JC, Rice ME. H2O2 signaling in the nigrostriatal dopamine pathway via ATP-sensitive potassium channels: issues and answers. Antioxid Redox Sign. 2007;9 (2):219–231. doi: 10.1089/ars.2007.9.219. [DOI] [PubMed] [Google Scholar]
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18 (16):6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22 (20):9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros A, Summerfelt A, Du X, Jiang P, Chiappelli J, Tagamets M, O’Donnell P, Kochunov P, Hong LE. Electrophysiological intermediate biomarkers for oxidative stress in schizophrenia. Clin Neurophysiol. 2013;124 (11):2209–2215. doi: 10.1016/j.clinph.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nature reviews Neuroscience. 2007;8 (1):45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Baud O, Greene AE, Li J, Wang H, Volpe JJ, Rosenberg PA. Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J Neurosci. 2004;24 (7):1531–1540. doi: 10.1523/JNEUROSCI.3989-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318 (5856):1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dugan LL. Interleukin-6 mediates the increase in NADPH-oxidase in the ketamine model of schizophrenia. J Neurosci. 2008;28 (51):13957–13966. doi: 10.1523/JNEUROSCI.4457-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nature Neurosci. 2010;13 (1):76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentsen H, Osnes K, Refsum H, Solberg DK, Bohmer T. A randomized placebo-controlled trial of an omega-3 fatty acid and vitamins E+C in schizophrenia. Translational psychiatry. 2013;3:e335. doi: 10.1038/tp.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk M, Copolov D, Dean O, Lu K, Jeavons S, Schapkaitz I, nderson-Hunt M, Judd F, Katz F, Katz P, Ording-Jespersen S, Little J, Conus P, Cuenod M, Do KQ, Bush AI. N-acetyl cysteine as a glutathione precursor for schizophrenia--a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64 (5):361–368. doi: 10.1016/j.biopsych.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Berk M, Malhi GS, Gray LJ, Dean OM. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol Sci. 2013;34 (3):167–177. doi: 10.1016/j.tips.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Beurdeley M, Spatazza J, Lee HH, Sugiyama S, Bernard C, Di Nardo AA, Hensch TK, Prochiantz A. Otx2 binding to perineuronal nets persistently regulates plasticity in the mature visual cortex. J Neurosci. 2012;32 (27):9429–9437. doi: 10.1523/JNEUROSCI.0394-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM. The immune system and developmental programming of brain and behavior. Front Neuroendocrin. 2012;33 (3):267–286. doi: 10.1016/j.yfrne.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodhinathan K, Kumar A, Foster TC. Intracellular redox state alters NMDA receptor response during aging through Ca2+/calmodulin-dependent protein kinase II. J Neurosci. 2010;30 (5):1914–1924. doi: 10.1523/JNEUROSCI.5485-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenhouse HC, Andersen SL. Nonsteroidal anti-inflammatory treatment prevents delayed effects of early life stress in rats. Biol Psychiatry. 2011;70 (5):434–440. doi: 10.1016/j.biopsych.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges R, Lutgen V, Lobner D, Baker DA. Thinking outside the cleft to understand synaptic activity: contribution of the cystine-glutamate antiporter (System xc-) to normal and pathological glutamatergic signaling. Pharmacol Rev. 2012;64 (3):780–802. doi: 10.1124/pr.110.003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS. The environment and susceptibility to schizophrenia. Prog Neurobiol. 2011;93 (1):23–58. doi: 10.1016/j.pneurobio.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckholtz JW, Meyer-Lindenberg A. Psychopathology and the human connectome: toward a transdiagnostic model of risk for mental illness. Neuron. 2012;74 (6):990–1004. doi: 10.1016/j.neuron.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Buelna-Chontal M, Zazueta C. Redox activation of Nrf2 & NF-kappaB: a double end sword? Cell Signal. 2013;25 (12):2548–2557. doi: 10.1016/j.cellsig.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Bulut M, Savas HA, Altindag A, Virit O, Dalkilic A. Beneficial effects of N-acetylcysteine in treatment resistant schizophrenia. World J Biol Psychia. 2009;10 (4 Pt 2):626–628. doi: 10.1080/15622970903144004. [DOI] [PubMed] [Google Scholar]
- Byne W, Tatusov A, Yiannoulos G, Vong GS, Marcus S. Effects of mental illness and aging in two thalamic nuclei. Schizophr Res. 2008;106 (2–3):172–181. doi: 10.1016/j.schres.2008.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabungcal JH, Nicolas D, Kraftsik R, Cuenod M, Do KQ, Hornung JP. Glutathione deficit during development induces anomalies in the rat anterior cingulate GABAergic neurons: Relevance to schizophrenia. Neurobiol Dis. 2006;22 (3):624–637. doi: 10.1016/j.nbd.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Cabungcal JH, Steullet P, Kraftsik R, Cuenod M, Do KQ. Early-life insults impair parvalbumin interneurons via oxidative stress: reversal by N-acetylcysteine. Biol Psychiatry. 2013a;73 (6):574–582. doi: 10.1016/j.biopsych.2012.09.020. [DOI] [PubMed] [Google Scholar]
- Cabungcal JH, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK, Do KQ. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci U S A. 2013b;110 (22):9130–9135. doi: 10.1073/pnas.1300454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet JL, Brannock C. Free radicals and the pathobiology of brain dopamine systems. Neurochem Int. 1998;32 (2):117–131. doi: 10.1016/s0197-0186(97)00031-4. [DOI] [PubMed] [Google Scholar]
- Cai Z, Lin S, Pang Y, Rhodes PG. Brain injury induced by intracerebral injection of interleukin-1beta and tumor necrosis factor-alpha in the neonatal rat. Pediatr Res. 2004;56 (3):377–384. doi: 10.1203/01.PDR.0000134249.92944.14. [DOI] [PubMed] [Google Scholar]
- Callahan LS, Thibert KA, Wobken JD, Georgieff MK. Early-life iron deficiency anemia alters the development and long-term expression of parvalbumin and perineuronal nets in the rat hippocampus. Dev Neurosci. 2013;35 (5):427–436. doi: 10.1159/000354178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao N, Yao ZX. Oligodendrocyte N-methyl-D-aspartate receptor signaling: insights into its functions. Mol Neurobiol. 2013;47 (2):845–856. doi: 10.1007/s12035-013-8408-8. [DOI] [PubMed] [Google Scholar]
- Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459 (7247):663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, Ruhlmann C, Jones SR, Deisseroth K, Sheng M, Moore CI, Tsai LH. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17 (5):537–548. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GC, Talbot K, Halene TB, Gandal MJ, Kazi HA, Schlosser L, Phung QH, Gur RE, Arnold SE, Siegel SJ. Dysbindin-1 mutant mice implicate reduced fast-phasic inhibition as a final common disease mechanism in schizophrenia. Proc Natl Acad Sci U S A. 2011;108 (43):E962–970. doi: 10.1073/pnas.1109625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeli C, Knyazeva MG, Cuenod M, Do KQ. Glutathione precursor N-acetyl-cysteine modulates EEG synchronization in schizophrenia patients: a double-blind, randomized, placebo-controlled trial. PloS one. 2012;7 (2):e29341. doi: 10.1371/journal.pone.0029341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaliere F, Urra O, Alberdi E, Matute C. Oligodendrocyte differentiation from adult multipotent stem cells is modulated by glutamate. Cell Death & Disease. 2012;3:e268. doi: 10.1038/cddis.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 2004;24 (43):9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew LJ, Fusar-Poli P, Schmitz T. Oligodendroglial alterations and the role of microglia in white matter injury: relevance to schizophrenia. Dev Neurosci. 2013;35 (2–3):102–129. doi: 10.1159/000346157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Chen HV, Lipton SA. Three pairs of cysteine residues mediate both redox and zn2+ modulation of the nmda receptor. J Neurosci. 2001;21 (2):392–400. doi: 10.1523/JNEUROSCI.21-02-00392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay HB, Sillivan S, Konradi C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosi. 2011;29 (3):311–324. doi: 10.1016/j.ijdevneu.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Basu A, Benneyworth M, Balu D, Konopaske G. Glutamatergic synaptic dysregulation in schizophrenia: therapeutic implications. Handb Exp Pharmacol. 2012;213:267–295. doi: 10.1007/978-3-642-25758-2_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Sign. 2011;15 (2):551–589. doi: 10.1089/ars.2010.3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10 (1):63–74. doi: 10.1016/S1474-4422(10)70253-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- das Neves Duarte JM, Kulak A, Gholam-Razaee MM, Cuenod M, Gruetter R, Do KQ. N-acetylcysteine normalizes neurochemical changes in the glutathione-deficient schizophrenia mouse model during development. Biol Psychiatry. 2012;71 (11):1006–1014. doi: 10.1016/j.biopsych.2011.07.035. [DOI] [PubMed] [Google Scholar]
- Davis KL, Stewart DG, Friedman JI, Buchsbaum M, Harvey PD, Hof PR, Buxbaum J, Haroutunian V. White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch Gen Psychiatry. 2003;60 (5):443–456. doi: 10.1001/archpsyc.60.5.443. [DOI] [PubMed] [Google Scholar]
- Dell’Anna E, Geloso MC, Magarelli M, Molinari M. Development of GABA and calcium binding proteins immunoreactivity in the rat hippocampus following neonatal anoxia. Neurosci Lett. 1996;211 (2):93–96. doi: 10.1016/0304-3940(96)12733-6. [DOI] [PubMed] [Google Scholar]
- Dickerson DD, Wolff AR, Bilkey DK. Abnormal long-range neural synchrony in a maternal immune activation animal model of schizophrenia. J Neurosci. 2010;30 (37):12424–12431. doi: 10.1523/JNEUROSCI.3046-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dityatev A, Bruckner G, Dityateva G, Grosche J, Kleene R, Schachner M. Activity-dependent formation and functions of chondroitin sulfate-rich extracellular matrix of perineuronal nets. Dev Neurobiol. 2007;67 (5):570–588. doi: 10.1002/dneu.20361. [DOI] [PubMed] [Google Scholar]
- Do KQ, Monin A, Klaey M, Butticaz C, Cabungcal JH, Steullet P, Cuenod M. Redox dysregulation affects proliferation, differentiation of oligodendrocyte progenitors and myelination: relevance to dysconnectivity in schizophrenia, Biol. Psychiatry. 2012;71(8):4S. [Google Scholar]
- Do KQ, Bovet P, Cabungcal JH, Conus P, Gysin R, Lavoie S, Steullet P, Cuenod M. Redox dysregulation in schizophrenia: Genetic susceptibility and pathophysiological mechanisms. In: Lajtha A, Javitt DC, Kantrowitz JT, editors. Handbook of neurochemistry and molecular neurobiology. 3. Springer; New York: 2009a. pp. 286–311. Vol. Schizophrenia. [Google Scholar]
- Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol. 2009b;19 (2):220–230. doi: 10.1016/j.conb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Do KQ, Trabesinger AH, Kirsten-Kruger M, Lauer CJ, Dydak U, Hell D, Holsboer F, Boesiger P, Cuenod M. Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci. 2000;12 (10):3721–3728. doi: 10.1046/j.1460-9568.2000.00229.x. [DOI] [PubMed] [Google Scholar]
- Donato F, Rompani SB, Caroni P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature. 2013;504 (7479):272–276. doi: 10.1038/nature12866. [DOI] [PubMed] [Google Scholar]
- Eluvathingal TJ, Chugani HT, Behen ME, Juhasz C, Muzik O, Maqbool M, Chugani DC, Makki M. Abnormal brain connectivity in children after early severe socioemotional deprivation: a diffusion tensor imaging study. Pediatrics. 2006;117 (6):2093–2100. doi: 10.1542/peds.2005-1727. [DOI] [PubMed] [Google Scholar]
- Fan LW, Mitchell HJ, Tien LT, Rhodes PG, Cai Z. Interleukin-1beta-induced brain injury in the neonatal rat can be ameliorated by alpha-phenyl-n-tert-butyl-nitrone. Exp Neurol. 2009;220 (1):143–153. doi: 10.1016/j.expneurol.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan LW, Mitchell HJ, Tien LT, Zheng B, Pang Y, Rhodes PG, Cai Z. alpha-Phenyl-n-tert-butyl-nitrone reduces lipopolysaccharide-induced white matter injury in the neonatal rat brain. Dev Neurobiol. 2008;68 (3):365–378. doi: 10.1002/dneu.20591. [DOI] [PubMed] [Google Scholar]
- Fan LW, Pang Y, Lin S, Rhodes PG, Cai Z. Minocycline attenuates lipopolysaccharide-induced white matter injury in the neonatal rat brain. Neuroscience. 2005;133 (1):159–168. doi: 10.1016/j.neuroscience.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Farokhnia M, Azarkolah A, Adinehfar F, Khodaie-Ardakani MR, Hosseini SM, Yekehtaz H, Tabrizi M, Rezaei F, Salehi B, Sadeghi SM, Moghadam M, Gharibi F, Mirshafiee O, Akhondzadeh S. N-acetylcysteine as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia: a randomized, double-blind, placebo-controlled study. Clin Neuropharmacol. 2013;36 (6):185–192. doi: 10.1097/WNF.0000000000000001. [DOI] [PubMed] [Google Scholar]
- Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E, Butterfield DA, Morley JE. The antioxidants a-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem. 2003;84(5):1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]
- Fazzari P, Paternain AV, Valiente M, Pla R, Lujan R, Lloyd K, Lerma J, Marin O, Rico B. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signaling. Nature. 2010;464 (7293):1376–1380. doi: 10.1038/nature08928. [DOI] [PubMed] [Google Scholar]
- Fitzsimmons J, Kubicki M, Shenton ME. Review of functional and anatomical brain connectivity findings in schizophrenia. Curr Opin Psychiatr. 2013;26 (2):172–187. doi: 10.1097/YCO.0b013e32835d9e6a. [DOI] [PubMed] [Google Scholar]
- Fournier M, Ferrari C, Baumann PS, Polari A, Monin A, Bellier-Teichmann T, Wulff J, Pappan KL, Cuenod M, Conus P, Do KQ. Impaired metabolic reactivity to oxidative stress in early psychosis patients. Schizophr Bull. 2014 doi: 10.1093/schbul/sbu053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragoso G, Martinez-Bermudez AK, Liu HN, Khorchid A, Chemtob S, Mushynski WE, Almazan G. Developmental differences in HO-induced oligodendrocyte cell death: role of glutathione, mitogen-activated protein kinases and caspase 3. J Neurochem. 2004;90 (2):392–404. doi: 10.1111/j.1471-4159.2004.02488.x. [DOI] [PubMed] [Google Scholar]
- French HM, Reid M, Mamontov P, Simmons RA, Grinspan JB. Oxidative stress disrupts oligodendrocyte maturation. J Neurosci Res. 2009;87 (14):3076–3087. doi: 10.1002/jnr.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries P, Nikolic D, Singer W. The gamma cycle. Trends Neurosci. 2007;30 (7):309–316. doi: 10.1016/j.tins.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, Carrera N, Humphreys I, Johnson JS, Roussos P, Barker DD, Banks E, Milanova V, Grant SG, Hannon E, Rose SA, Chambert K, Mahajan M, Scolnick EM, Moran JL, Kirov G, Palotie A, McCarroll SA, Holmans P, Sklar P, Owen MJ, Purcell SM, O’Donovan MC. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506 (7487):179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, Rozov A, Whittington MA, Traub RD, Rawlins JN, Monyer H. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53 (4):591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Kosaka T, Singer W, Galuske RA. Gap junctions among dendrites of cortical GABAergic neurons establish a dense and widespread intercolumnar network. J Neurosci. 2006;26 (13):3434–3443. doi: 10.1523/JNEUROSCI.4076-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Nesbitt AM, McCurdy RM, Alter MD. Measuring the maturity of the fast-spiking interneuron transcriptional program in autism, schizophrenia, and bipolar disorder. PloS one. 2012;7 (8):e41215. doi: 10.1371/journal.pone.0041215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garate I, Garcia-Bueno B, Madrigal JL, Caso JR, Alou L, Gomez-Lus ML, Mico JA, Leza JC. Stress-induced neuroinflammation: role of the Toll-like receptor-4 pathway. Biol Psychiatry. 2013;73 (1):32–43. doi: 10.1016/j.biopsych.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Gawryluk JW, Wang JF, Andreazza AC, Shao L, Young LT. Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int J Neuropsychopharmacol. 2011;14 (1):123–130. doi: 10.1017/S1461145710000805. [DOI] [PubMed] [Google Scholar]
- Gokhale A, Larimore J, Werner E, So L, Moreno-De-Luca A, Lese-Martin C, Lupashin VV, Smith Y, Faundez V. Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex 1. J Neurosci. 2012;32 (11):3697–3711. doi: 10.1523/JNEUROSCI.5640-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldshmit Y, Erlich S, Pinkas-Kramarski R. Neuregulin rescues PC12-ErbB4 cells from cell death induced by H(2)O(2). Regulation of reactive oxygen species levels by phosphatidylinositol 3-kinase. J Biol Chem. 2001;276 (49):46379–46385. doi: 10.1074/jbc.M105637200. [DOI] [PubMed] [Google Scholar]
- Gravina P, Spoletini I, Masini S, Valentini A, Vanni D, Paladini E, Bossu P, Caltagirone C, Federici G, Spalletta G, Bernardini S. Genetic polymorphisms of glutathione S-transferases GSTM1, GSTT1, GSTP1 and GSTA1 as risk factors for schizophrenia. Psychiat Res. 2011;187 (3):454–456. doi: 10.1016/j.psychres.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Gulyas AI, Szabo GG, Ulbert I, Holderith N, Monyer H, Erdelyi F, Szabo G, Freund TF, Hajos N. Parvalbumin-containing fast-spiking basket cells generate the field potential oscillations induced by cholinergic receptor activation in the hippocampus. J Neurosci. 2010;30 (45):15134–15145. doi: 10.1523/JNEUROSCI.4104-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gysin R, Kraftsik R, Boulat O, Bovet P, Conus P, Comte-Krieger E, Polari A, Steullet P, Preisig M, Teichmann T, Cuenod M, Do KQ. Genetic dysregulation of glutathione synthesis predicts alteration of plasma thiol redox status in schizophrenia. Antioxid Redox Signal. 2011;15 (7):2003–2010. doi: 10.1089/ars.2010.3463. [DOI] [PubMed] [Google Scholar]
- Gysin R, Kraftsik R, Sandell J, Bovet P, Chappuis C, Conus P, Deppen P, Preisig M, Ruiz V, Steullet P, Tosic M, Werge T, Cuenod M, Do KQ. Impaired glutathione synthesis in schizophrenia: convergent genetic and functional evidence. Proc Natl Acad Sci U S A. 2007;104 (42):16621–16626. doi: 10.1073/pnas.0706778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V, Fienberg AA. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A. 2001;98 (8):4746–4751. doi: 10.1073/pnas.081071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signaling: implications for neurodegenerative disorders. Nature Rev Neurosci. 2010;11 (10):682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75 (5):762–777. doi: 10.1016/j.neuron.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Harris LW, Lockstone HE, Khaitovich P, Weickert CS, Webster MJ, Bahn S. Gene expression in the prefrontal cortex during adolescence: implications for the onset of schizophrenia. BMC Med Genomics. 2009;2:28. doi: 10.1186/1755-8794-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harte MK, Powell SB, Swerdlow NR, Geyer MA, Reynolds GP. Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J Neural Transm. 2007;114 (7):893–898. doi: 10.1007/s00702-007-0627-6. [DOI] [PubMed] [Google Scholar]
- Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, Wu D, Xue R, Andrade M, Tankou S, Mori S, Gallagher M, Ishizuka K, Pletnikov M, Kida S, Sawa A. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci U S A. 2007;104 (36):14501–14506. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof PR, Haroutunian V, Friedrich VL, Jr, Byne W, Buitron C, Perl DP, Davis KL. Loss and altered spatial distribution of oligodendrocytes in the superior frontal gyrus in schizophrenia. Biol Psychiatry. 2003;53 (12):1075–1085. doi: 10.1016/s0006-3223(03)00237-3. [DOI] [PubMed] [Google Scholar]
- Hoftman GD, Volk DW, Bazmi HH, Li S, Sampson AR, Lewis DA. Altered Cortical Expression of GABA-Related Genes in Schizophrenia: Illness Progression vs Developmental Disturbance. Schizophr Bull. 2013 doi: 10.1093/schbul/sbt178. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27 (43):11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Gundapuneedi T, Rao U. White matter disruptions in adolescents exposed to childhood maltreatment and vulnerability to psychopathology. Neuropsychopharmacology. 2012;37 (12):2693–2701. doi: 10.1038/npp.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang Y, Kim J, Shin JY, Kim JI, Seo JS, Webster MJ, Lee D, Kim S. Gene expression profiling by mRNA sequencing reveals increased expression of immune/inflammation-related genes in the hippocampus of individuals with schizophrenia. Transl Psychiatry. 2013;3:e321. doi: 10.1038/tp.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibi D, Nagai T, Koike H, Kitahara Y, Mizoguchi H, Niwa M, Jaaro-Peled H, Nitta A, Yoneda Y, Nabeshima T, Sawa A, Yamada K. Combined effect of neonatal immune activation and mutant DISC1 on phenotypic changes in adulthood. Behav Brain Res. 2010;206 (1):32–37. doi: 10.1016/j.bbr.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR. Rethinking schizophrenia. Nature. 2010;468(7321):187–193. doi: 10.1038/nature09552. [DOI] [PubMed] [Google Scholar]
- Jana M, Pahan K. Redox regulation of cytokine-mediated inhibition of myelin gene expression in human primary oligodendrocytes. Free Radic Biol Med. 2005;39 (6):823–831. doi: 10.1016/j.freeradbiomed.2005.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC. Glycine transport inhibitors in the treatment of schizophrenia. Handb Exp Pharmacol. 2012;(213):367–399. doi: 10.1007/978-3-642-25758-2_12. [DOI] [PubMed] [Google Scholar]
- Jenkins TA, Harte MK, Stenson G, Reynolds GP. Neonatal lipopolysaccharide induces pathological changes in parvalbumin immunoreactivity in the hippocampus of the rat. Behav Brain Res. 2009;205 (2):355–359. doi: 10.1016/j.bbr.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Jiang M, Swann JW. A role for L-type calcium channels in the maturation of parvalbumin-containing hippocampal interneurons. Neuroscience. 2005;135 (3):839–850. doi: 10.1016/j.neuroscience.2005.06.073. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Rompala GR, Zhang S, Cowell RM, Nakazawa K. Social isolation exacerbates schizophrenia-like phenotypes via oxidative stress in cortical interneurons. Biol Psychiatry. 2013;73 (10):1024–1034. doi: 10.1016/j.biopsych.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AW, Jaaro-Peled H, Shahani N, Sedlak TW, Zoubovsky S, Burruss D, Emiliani F, Sawa A, Gallagher M. Cognitive and motivational deficits together with prefrontal oxidative stress in a mouse model for neuropsychiatric illness. Proc Natl Acad Sci U S A. 2013;110 (30):12462–12467. doi: 10.1073/pnas.1307925110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295 (4):C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O, Huchzermeyer C, Kovacs R, Wirtz S, Schuelke M. Gamma oscillations in the hippocampus require high complex I gene expression and strong functional performance of mitochondria. Brain. 2011;134 (Pt 2):345–358. doi: 10.1093/brain/awq333. [DOI] [PubMed] [Google Scholar]
- Kantrowitz J, Javitt DC. Glutamatergic transmission in schizophrenia: from basic research to clinical practice. Curr Opin Psychiatr. 2012;25 (2):96–102. doi: 10.1097/YCO.0b013e32835035b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsel P, Davis KL, Haroutunian V. Variations in myelin and oligodendrocyte-related gene expression across multiple brain regions in schizophrenia: a gene ontology study. Schizophr Res. 2005;79 (2–3):157–173. doi: 10.1016/j.schres.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Kaur C, Rathnasamy G, Ling EA. Roles of activated microglia in hypoxia induced neuroinflammation in the developing brain and the retina. J Neuroimmune Pharm. 2013;8 (1):66–78. doi: 10.1007/s11481-012-9347-2. [DOI] [PubMed] [Google Scholar]
- Kerns D, Vong GS, Barley K, Dracheva S, Katsel P, Casaccia P, Haroutunian V, Byne W. Gene expression abnormalities and oligodendrocyte deficits in the internal capsule in schizophrenia. Schizophr Res. 2010;120 (1–3):150–158. doi: 10.1016/j.schres.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77 (1):10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26 (5):1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, Grozeva D, Fjodorova M, Wollerton R, Rees E, Nikolov I, van de Lagemaat LN, Bayes A, Fernandez E, Olason PI, Bottcher Y, Komiyama NH, Collins MO, Choudhary J, Stefansson K, Stefansson H, Grant SG, Purcell S, Sklar P, O’Donovan MC, Owen MJ. De novo CNV analysis implicates specific abnormalities of postsynaptic signaling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17 (2):142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocsis B, Brown RE, McCarley RW, Hajos M. Impact of ketamine on neuronal network dynamics: translational modeling of schizophrenia-relevant deficits. CNS Neurosci Ther. 2013;19 (6):437–447. doi: 10.1111/cns.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga M, Serritella AV, Messmer MM, Hayashi-Takagi A, Hester LD, Snyder SH, Sawa A, Sedlak TW. Glutathione is a physiologic reservoir of neuronal glutamate. Biochem Biophys Res Commun. 2011;409 (4):596–602. doi: 10.1016/j.bbrc.2011.04.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr G, Eckardt S, Luddens H, Monyer H, Seeburg PH. NMDA receptor channels: subunit-specific potentiation by reducing agents. Neuron. 1994;12 (5):1031–1040. doi: 10.1016/0896-6273(94)90311-5. [DOI] [PubMed] [Google Scholar]
- Komitova M, Xenos D, Salmaso N, Tran KM, Brand T, Schwartz ML, Ment L, Vaccarino FM. Hypoxia-induced developmental delays of inhibitory interneurons are reversed by environmental enrichment in the postnatal mouse forebrain. J Neurosci. 2013;33 (33):13375–13387. doi: 10.1523/JNEUROSCI.5286-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68 (3):557–569. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- Krishnan N, Dickman MB, Becker DF. Proline modulates the intracellular redox environment and protects mammalian cells against oxidative stress. Free Radic Biol Med. 2008;44 (4):671–681. doi: 10.1016/j.freeradbiomed.2007.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51 (3):199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Kulak A, Cuenod M, Do KQ. Behavioral phenotyping of glutathione-deficient mice: relevance to schizophrenia and bipolar disorder. Behav Brain Res. 2012;226 (2):563–570. doi: 10.1016/j.bbr.2011.10.020. [DOI] [PubMed] [Google Scholar]
- Kulak A, Steullet P, Cabungcal JH, Werge T, Ingason A, Cuenod M, Do KQ. Redox dysregulation in the pathophysiology of schizophrenia and bipolar disorder: insights from animal models. Antioxid Redox Sign. 2013;18 (12):1428–1443. doi: 10.1089/ars.2012.4858. [DOI] [PubMed] [Google Scholar]
- Kyriakopoulos M, Frangou S. Recent diffusion tensor imaging findings in early stages of schizophrenia. Cur Opin Psychiatr. 2009;22 (2):168–176. doi: 10.1097/YCO.0b013e328325aa23. [DOI] [PubMed] [Google Scholar]
- Lante F, Meunier J, Guiramand J, Maurice T, Cavalier M, de Jesus Ferreira MC, Aimar R, Cohen-Solal C, Vignes M, Barbanel G. Neurodevelopmental damage after prenatal infection: role of oxidative stress in the fetal brain. Free Radic Biol Med. 2007;42 (8):1231–1245. doi: 10.1016/j.freeradbiomed.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Lataster J, Collip D, Ceccarini J, Haas D, Booij L, van OJ, Pruessner J, Van LK, Myin-Germeys I. Psychosocial stress is associated with in vivo dopamine release in human ventromedial prefrontal cortex: A positron emission tomography study using [(18)F]fallypride. NeuroImage. 2011;58 (4):1081–1089. doi: 10.1016/j.neuroimage.2011.07.030. [DOI] [PubMed] [Google Scholar]
- Lataster T, Valmaggia L, Lardinois M, van Os J, Myin-Germeys I. Increased stress reactivity: a mechanism specifically associated with the positive symptoms of psychotic disorder. Psychol Med. 2013;43 (7):1389–1400. doi: 10.1017/S0033291712002279. [DOI] [PubMed] [Google Scholar]
- Lavoie S, Murray MM, Deppen P, Knyazeva MG, Berk M, Boulat O, Bovet P, Bush AI, Conus P, Copolov D, Fornari E, Meuli R, Solida A, Vianin P, Cuenod M, Buclin T, Do KQ. Glutathione precursor, N-acetyl-cysteine, improves mismatch negativity in schizophrenia patients. Neuropsychopharmacology. 2008;33 (9):2187–2199. doi: 10.1038/sj.npp.1301624. [DOI] [PubMed] [Google Scholar]
- Lavoie S, Chen Y, Dalton TP, Gysin R, Cuenod M, Steullet P, Do KQ. Curcumin, quercetin and tBHQ modulate glutathione levels in astrocytes and neurons: Importance of the glutamate cysteine ligase modifier subunit. J Neurochem. 2009;108 (6):1410–1422. doi: 10.1111/j.1471-4159.2009.05908.x. [DOI] [PubMed] [Google Scholar]
- Lazarewicz MT, Ehrlichman RS, Maxwell CR, Gandal MJ, Finkel LH, Siegel SJ. Ketamine modulates theta and gamma oscillations. J Cognitive Neurosci. 2010;22 (7):1452–1464. doi: 10.1162/jocn.2009.21305. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35 (1):57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leza JC, García Bueno B, Bioque M, Arango C, Parellada M, Do KQ, O’Donnell P, Bernardo M. Oxidative stress and inflammation in schizophrenia. A question of balance. Schizophr Bull. 2014 doi: 10.1016/j.neubiorev.2015.05.014. in press. [DOI] [PubMed] [Google Scholar]
- Li C, Xiao L, Liu X, Yang W, Shen W, Hu C, Yang G, He C. A functional role of NMDA receptor in regulating the differentiation of oligodendrocyte precursor cells and remyelination. Glia. 2013;61 (5):732–749. doi: 10.1002/glia.22469. [DOI] [PubMed] [Google Scholar]
- Li Z, Dong T, Proschel C, Noble M. Chemically diverse toxicants converge on Fyn and c-Cbl to disrupt precursor cell function. PLoS Biol. 2007;5 (2):e35. doi: 10.1371/journal.pbio.0050035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31 (5):234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Dietz K, DeLoyht JM, Pedre X, Kelkar D, Kaur J, Vialou V, Lobo MK, Dietz DM, Nestler EJ, Dupree J, Casaccia P. Impaired adult myelination in the prefrontal cortex of socially isolated mice. Nature Neurosci. 2012;15 (12):1621–1623. doi: 10.1038/nn.3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci. 2009;29 (8):2344–2354. doi: 10.1523/JNEUROSCI.5419-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci. 2011;32 (9):507–513. doi: 10.1016/j.tips.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Markwardt SJ, Gupta S, Meador-Woodruff JH, Lin JD, Overstreet-Wadiche L, Cowell RM. Parvalbumin deficiency and GABAergic dysfunction in mice lacking PGC-1alpha. J Neurosci. 2010;30 (21):7227–7235. doi: 10.1523/JNEUROSCI.0698-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutgen V, Qualmann K, Resch J, Kong L, Choi S, Baker DA. Reduction in phencyclidine induced sensorimotor gating deficits in the rat following increased system xc(−) activity in the medial prefrontal cortex. Psychopharmacology. 2013;226 (3):531–540. doi: 10.1007/s00213-012-2926-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinodan M, Rosen KM, Ito S, Corfas G. A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science. 2012;337 (6100):1357–1360. doi: 10.1126/science.1220845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinodan M, Tatsumi K, Manabe T, Yamauchi T, Makinodan E, Matsuyoshi H, Shimoda S, Noriyama Y, Kishimoto T, Wanaka A. Maternal immune activation in mice delays myelination and axonal development in the hippocampus of the offspring. J Neurosci Res. 2008;86 (10):2190–2200. doi: 10.1002/jnr.21673. [DOI] [PubMed] [Google Scholar]
- Mandal PK, Seiler A, Perisic T, Kolle P, Banjac Canak A, Forster H, Weiss N, Kremmer E, Lieberman MW, Bannai S, Kuhlencordt P, Sato H, Bornkamm GW, Conrad M. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem. 2010;285 (29):22244–22253. doi: 10.1074/jbc.M110.121327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-de-Souza D, Harris LW, Guest PC, Bahn S. The role of energy metabolism dysfunction and oxidative stress in schizophrenia revealed by proteomics. Antioxid Redox Sign. 2011;15 (7):2067–2079. doi: 10.1089/ars.2010.3459. [DOI] [PubMed] [Google Scholar]
- Massi L, Lagler M, Hartwich K, Borhegyi Z, Somogyi P, Klausberger T. Temporal dynamics of parvalbumin-expressing axo-axonic and basket cells in the rat medial prefrontal cortex in vivo. J Neurosci. 2012;32 (46):16496–16502. doi: 10.1523/JNEUROSCI.3475-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauney SA, Athanas KM, Pantazopoulos H, Shaskan N, Passeri E, Berretta S, Woo TU. Developmental pattern of perineuronal nets in the human prefrontal cortex and their deficit in schizophrenia. Biol Psychiatry. 2013;74 (6):427–435. doi: 10.1016/j.biopsych.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science. 2005;309 (5744):2222–2226. doi: 10.1126/science.1114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally JM, McCarley RW, Brown RE. Impaired GABAergic neurotransmission in schizophrenia underlies impairments in cortical gamma band oscillations. Curr Psychiatry Rep. 2013;15 (3):346. doi: 10.1007/s11920-012-0346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Iwamoto K, Ueda J, Bundo M, Adati N, Kojima T, Kato T. Comprehensive survey of CNVs influencing gene expression in the human brain and its implications for pathophysiology. Neurosci Res. 2013;79:22–33. doi: 10.1016/j.neures.2013.10.009. [DOI] [PubMed] [Google Scholar]
- Meiser J, Weindl D, Hiller K. Complexity of dopamine metabolism. Cell Commun Signal. 2013;11 (1):34. doi: 10.1186/1478-811X-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U. Developmental neuroinflammation and schizophrenia. Prog Neuro-Psychoph. 2013;42:20–34. doi: 10.1016/j.pnpbp.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J. Prenatal exposure to infection: a primary mechanism for abnormal dopaminergic development in schizophrenia. Psychopharmacology. 2009;206 (4):587–602. doi: 10.1007/s00213-009-1504-9. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008;22 (4):469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Mizrahi R, Addington J, Rusjan PM, Suridjan I, Ng A, Boileau I, Pruessner JC, Remington G, Houle S, Wilson AA. Increased stress-induced dopamine release in psychosis. Biol Psychiatry. 2012;71 (6):561–567. doi: 10.1016/j.biopsych.2011.10.009. [DOI] [PubMed] [Google Scholar]
- Miyata S, Komatsu Y, Yoshimura Y, Taya C, Kitagawa H. Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nature Neurosci. 2012;15 (3):414–422. S411–412. doi: 10.1038/nn.3023. [DOI] [PubMed] [Google Scholar]
- Moller M, Du Preez JL, Viljoen FP, Berk M, Harvey BH. N-Acetyl cysteine reverses social isolation rearing induced changes in cortico-striatal monoamines in rats. Metab Brain Dis. 2013;28 (4):687–696. doi: 10.1007/s11011-013-9433-z. [DOI] [PubMed] [Google Scholar]
- Morishita H, Hensch TK. Critical period revisited: impact on vision. Curr Opin Neurobiol. 2008;18 (1):101–107. doi: 10.1016/j.conb.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Muller N, Myint AM, Krause D, Weidinger E, Schwarz MJ. Anti-inflammatory treatment in schizophrenia. Prog Neuro-Psychoph. 2013;42:146–153. doi: 10.1016/j.pnpbp.2012.11.008. [DOI] [PubMed] [Google Scholar]
- Muller N, Myint AM, Schwarz MJ. Kynurenine pathway in schizophrenia: pathophysiological and therapeutic aspects. Curr Pharm Design. 2011;17 (2):130–136. doi: 10.2174/138161211795049552. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Kumar M, Selvakumar B, Ho GP, Ehmsen JT, Barrow RK, Amzel LM, Snyder SH. Nitric oxide S-nitrosylates serine racemase, mediating feedback inhibition of D-serine formation. Proc Natl Acad Sci U S A. 2007;104 (8):2950–2955. doi: 10.1073/pnas.0611620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myin-Germeys I, Marcelis M, Krabbendam L, Delespaul P, van Os J. Subtle fluctuations in psychotic phenomena as functional states of abnormal dopamine reactivity in individuals at risk. Biol Psychiatry. 2005;58 (2):105–110. doi: 10.1016/j.biopsych.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Myin-Germeys I, Peeters F, Havermans R, Nicolson NA, DeVries MW, Delespaul P, Van Os J. Emotional reactivity to daily life stress in psychosis and affective disorder: an experience sampling study. Acta Psychiat Scand. 2003;107 (2):124–131. doi: 10.1034/j.1600-0447.2003.02025.x. [DOI] [PubMed] [Google Scholar]
- O’Donnell P. Adolescent onset of cortical disinhibition in schizophrenia: insights from animal models. Schizophr Bull. 2011;37 (3):484–492. doi: 10.1093/schbul/sbr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell P, Cabungcal JH, Piantadosi PT, Lewis E, Calhoon GG, Do KQ. Oxidative stress during development in prefrontal cortical interneurons in developmental animal models of schizophrenia. Schizophr Bull. 2011;37 (suppl 1):111. [Google Scholar]
- Oorschot DE, Voss L, Covey MV, Goddard L, Huang W, Birchall P, Bilkey DK, Kohe SE. Spectrum of short- and long-term brain pathology and long-term behavioral deficits in male repeated hypoxic rats closely resembling human extreme prematurity. J Neurosci. 2013;33 (29):11863–11877. doi: 10.1523/JNEUROSCI.0342-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otte DM, Sommersberg B, Kudin A, Guerrero C, Albayram O, Filiou MD, Frisch P, Yilmaz O, Drews E, Turck CW, Bilkei-Gorzo A, Kunz WS, Beck H, Zimmer A. N-acetyl Cysteine Treatment Rescues Cognitive Deficits Induced by Mitochondrial Dysfunction in G72/G30 Transgenic Mice. Neuropsychopharmacology. 2011;36 (11):2233–43. doi: 10.1038/npp.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintlia MK, Paintlia AS, Contreras MA, Singh I, Singh AK. Lipopolysaccharide-induced peroxisomal dysfunction exacerbates cerebral white matter injury: attenuation by N-acetyl cysteine. Exp Neurol. 2008;210 (2):560–576. doi: 10.1016/j.expneurol.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantazopoulos H, Woo TU, Lim MP, Lange N, Berretta S. Extracellular matrix-glial abnormalities in the amygdala and entorhinal cortex of subjects diagnosed with schizophrenia. Arch Gen Psychiatry. 2010;67 (2):155–166. doi: 10.1001/archgenpsychiatry.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nature Neurosci. 2008;11 (4):476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YU, Jeong J, Lee H, Mun JY, Kim JH, Lee JS, Nguyen MD, Han SS, Suh PG, Park SK. Disrupted-in-schizophrenia 1 (DISC1) plays essential roles in mitochondria in collaboration with Mitofilin. Proc Natl Acad Sci U S A. 2010;107 (41):17785–17790. doi: 10.1073/pnas.1004361107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patz S, Grabert J, Gorba T, Wirth MJ, Wahle P. Parvalbumin expression in visual cortical interneurons depends on neuronal activity and TrkB ligands during an Early period of postnatal development. Cereb Cortex. 2004;14 (3):342–351. doi: 10.1093/cercor/bhg132. [DOI] [PubMed] [Google Scholar]
- Penschuck S, Flagstad P, Didriksen M, Leist M, Michael-Titus AT. Decrease in parvalbumin-expressing neurons in the hippocampus and increased phencyclidine-induced locomotor activity in the rat methylazoxymethanol (MAM) model of schizophrenia. Eur J Neurosci. 2006;23 (1):279–284. doi: 10.1111/j.1460-9568.2005.04536.x. [DOI] [PubMed] [Google Scholar]
- Pinteaux E, Copin JC, Ledig M, Tholey G. Modulation of oxygen-radical-scavenging enzymes by oxidative stress in primary cultures of rat astroglial cells. Dev Neurosci. 1996;18 (5–6):397–404. doi: 10.1159/000111433. [DOI] [PubMed] [Google Scholar]
- Pitts MW, Raman AV, Hashimoto AC, Todorovic C, Nichols RA, Berry MJ. Deletion of selenoprotein P results in impaired function of parvalbumin interneurons and alterations in fear learning and sensorimotor gating. Neuroscience. 2012;208:58–68. doi: 10.1016/j.neuroscience.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poels EM, Kegeles LS, Kantrowitz JT, Slifstein M, Javitt DC, Lieberman JA, Abi-Dargham A, Girgis RR. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol Psychiatry. 2014;19 (1):20–29. doi: 10.1038/mp.2013.136. [DOI] [PubMed] [Google Scholar]
- Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry. 2008;63 (8):801–808. doi: 10.1016/j.biopsych.2007.09.024. [DOI] [PubMed] [Google Scholar]
- Powell SB, Sejnowski TJ, Behrens MM. Behavioral and neurochemical consequences of cortical oxidative stress on parvalbumin-interneuron maturation in rodent models of schizophrenia. Neuropharmacology. 2012;62 (3):1322–1331. doi: 10.1016/j.neuropharm.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kahler A, Duncan L, Stahl E, Genovese G, Fernandez E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PK, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SG, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll SA, Sklar P. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506 (7487):185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovic AD, Hastings TG. Role of endogenous glutathione in the oxidation of dopamine. J Neurochem. 1998;71 (5):2071–2078. doi: 10.1046/j.1471-4159.1998.71052071.x. [DOI] [PubMed] [Google Scholar]
- Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24 (5):981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy R, Reddy R. Antioxidant therapeutics for schizophrenia. Antioxid Redox Sign. 2011;15 (7):2047–2055. doi: 10.1089/ars.2010.3571. [DOI] [PubMed] [Google Scholar]
- Robinson S, Petelenz K, Li Q, Cohen ML, Dechant A, Tabrizi N, Bucek M, Lust D, Miller RH. Developmental changes induced by graded prenatal systemic hypoxic-ischemic insults in rats. Neurobiol Dis. 2005;18 (3):568–581. doi: 10.1016/j.nbd.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Santiago B, Brunet A, Sobrino B, Serra-Juhe C, Flores R, Armengol L, Vilella E, Gabau E, Guitart M, Guillamat R, Martorell L, Valero J, Gutierrez-Zotes A, Labad A, Carracedo A, Estivill X, Perez-Jurado LA. Association of common copy number variants at the glutathione S-transferase genes and rare novel genomic changes with schizophrenia. Mol Psychiatry. 2010;15 (10):1023–1033. doi: 10.1038/mp.2009.53. [DOI] [PubMed] [Google Scholar]
- Rotaru DC, Yoshino H, Lewis DA, Ermentrout GB, Gonzalez-Burgos G. Glutamate receptor subtypes mediating synaptic activation of prefrontal cortex neurons: relevance for schizophrenia. J Neurosci. 2011;31 (1):142–156. doi: 10.1523/JNEUROSCI.1970-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouhinen S, Panula J, Palva JM, Palva S. Load dependence of beta and gamma oscillations predicts individual capacity of visual attention. J Neurosci. 2013;33 (48):19023–19033. doi: 10.1523/JNEUROSCI.1666-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux F, Wibral M, Mohr HM, Singer W, Uhlhaas PJ. Gamma-band activity in human prefrontal cortex codes for the number of relevant items maintained in working memory. J Neurosci. 2012;32 (36):12411–12420. doi: 10.1523/JNEUROSCI.0421-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E. Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry. 2007;7:46. doi: 10.1186/1471-244X-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavone S, Sorce S, Dubois-Dauphin M, Jaquet V, Colaianna M, Zotti M, Cuomo V, Trabace L, Krause KH. Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol Psychiatry. 2009;66 (4):384–392. doi: 10.1016/j.biopsych.2009.04.033. [DOI] [PubMed] [Google Scholar]
- Shirai Y, Fujita Y, Hashimoto K. Effects of the antioxidant sulforaphane on hyperlocomotion and prepulse inhibition deficits in mice after phencyclidine administration. Clinical psychopharmacology and neuroscience: the official scientific journal of the Korean College of Neuropsychopharmacology. 2012;10 (2):94–98. doi: 10.9758/cpn.2012.10.2.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Ladi E, Mayer-Proschel M, Noble M. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc Natl Acad Sci U S A. 2000;97 (18):10032–10037. doi: 10.1073/pnas.170209797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth AM, Lawrie SM. The Neuroimmunology of Schizophrenia. Clin Psychopharmacol Neuroscience. 2013;11 (3):107–117. doi: 10.9758/cpn.2013.11.3.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinn A, Milte C, Howe PRC. Oiling the Brain: A Review of Randomized Controlled Trials of Omega-3 Fatty Acids in Psychopathology across the Lifespan. Nutrients. 2010;2:128–170. doi: 10.3390/nu2020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459 (7247):698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Sun J, Moss J, Wen Z, Sun GJ, Hsu D, Zhong C, Davoudi H, Christian KM, Toni N, Ming GL, Song H. Parvalbumin interneurons mediate neuronal circuitry-neurogenesis coupling in the adult hippocampus. Nature Neurosci. 2013;16 (12):1728–1730. doi: 10.1038/nn.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, Werge T, Pietilainen OP, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O, Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J, Lonnqvist J, Paunio T, Borglum AD, Hartmann A, Fink-Jensen A, Nordentoft M, Hougaard D, Norgaard-Pedersen B, Bottcher Y, Olesen J, Breuer R, Moller HJ, Giegling I, Rasmussen HB, Timm S, Mattheisen M, Bitter I, Rethelyi JM, Magnusdottir BB, Sigmundsson T, Olason P, Masson G, Gulcher JR, Haraldsson M, Fossdal R, Thorgeirsson TE, Thorsteinsdottir U, Ruggeri M, Tosato S, Franke B, Strengman E, Kiemeney LA, Genetic R, Melle I, Djurovic S, Abramova L, Kaleda V, Sanjuan J, de Frutos R, Bramon E, Vassos E, Fraser G, Ettinger U, Picchioni M, Walker N, Toulopoulou T, Need AC, Ge D, Yoon JL, Shianna KV, Freimer NB, Cantor RM, Murray R, Kong A, Golimbet V, Carracedo A, Arango C, Costas J, Jonsson EG, Terenius L, Agartz I, Petursson H, Nothen MM, Rietschel M, Matthews PM, Muglia P, Peltonen L, St Clair D, Goldstein DB, Stefansson K, Collier DA Outcome in P. Common variants conferring risk of schizophrenia. Nature. 2009;460 (7256):744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner J, Walter M, Glanz W, Sarnyai Z, Bernstein HG, Vielhaber S, Kastner A, Skalej M, Jordan W, Schiltz K, Klingbeil C, Wandinger KP, Bogerts B, Stoecker W. Increased prevalence of diverse N-methyl-D-aspartate glutamate receptor antibodies in patients with an initial diagnosis of schizophrenia: specific relevance of IgG NR1a antibodies for distinction from N-methyl-D-aspartate glutamate receptor encephalitis. JAMA Psychiatry. 2013;70 (3):271–278. doi: 10.1001/2013.jamapsychiatry.86. [DOI] [PubMed] [Google Scholar]
- Steullet P, Cabungcal JH, Kulak A, Cuenod M, Schenk F, Do KQ. Glutathione deficit in animal models of schizophrenia. In: O’Donnell P, editor. Animal Models Of Schizophrenia and Related Disorders. Humana Press; New York: 2011. pp. 149–188. [Google Scholar]
- Steullet P, Cabungcal JH, Kulak A, Kraftsik R, Chen Y, Dalton TP, Cuenod M, Do KQ. Redox dysregulation affects the ventral but not dorsal hippocampus: impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J Neurosci. 2010;30 (7):2547–2558. doi: 10.1523/JNEUROSCI.3857-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steullet P, Lavoie S, Kraftsik R, Guidi R, Gysin R, Cuenod M, Do KQ. A glutathione deficit alters dopamine modulation of L-type calcium channels via D2 and ryanodine receptors in neurons. Free Radic Biol Med. 2008;44 (6):1042–1054. doi: 10.1016/j.freeradbiomed.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Steullet P, Neijt HC, Cuenod M, Do KQ. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: relevance to schizophrenia. Neuroscience. 2006;137 (3):807–819. doi: 10.1016/j.neuroscience.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Stevens HE, Su T, Yanagawa Y, Vaccarino FM. Prenatal stress delays inhibitory neuron progenitor migration in the developing neocortex. Psychoneuroendocrinology. 2013;38 (4):509–521. doi: 10.1016/j.psyneuen.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojkovic T, Radonjic NV, Velimirovic M, Jevtic G, Popovic V, Doknic M, Petronijevic ND. Risperidone reverses phencyclidine induced decrease in glutathione levels and alterations of antioxidant defense in rat brain. Prog Neuro-Psychoph. 2012;39 (1):192–199. doi: 10.1016/j.pnpbp.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Sullivan EM, O’Donnell P. Inhibitory interneurons, oxidative stress, and schizophrenia. Schizophr Bull. 2012;38 (3):373–376. doi: 10.1093/schbul/sbs052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Sakurai T, Davis KL, Buxbaum JD. Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog Neurobiol. 2011;93 (1):13–24. doi: 10.1016/j.pneurobio.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao K, Kobayashi K, Hagihara H, Ohira K, Shoji H, Hattori S, Koshimizu H, Umemori J, Toyama K, Nakamura HK, Kuroiwa M, Maeda J, Atsuzawa K, Esaki K, Yamaguchi S, Furuya S, Takagi T, Walton NM, Hayashi N, Suzuki H, Higuchi M, Usuda N, Suhara T, Nishi A, Matsumoto M, Ishii S, Miyakawa T. Deficiency of schnurri-2, an MHC enhancer binding protein, induces mild chronic inflammation in the brain and confers molecular, neuronal, and behavioral phenotypes related to schizophrenia. Neuropsychopharmacology. 2013;38 (8):1409–1425. doi: 10.1038/npp.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms AE, Dorschner MO, Wechsler J, Choi KY, Kirkwood R, Girirajan S, Baker C, Eichler EE, Korvatska O, Roche KW, Horwitz MS, Tsuang DW. Support for the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiatry. 2013;70 (6):582–590. doi: 10.1001/jamapsychiatry.2013.1195. [DOI] [PubMed] [Google Scholar]
- Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB, Starkey M, Webster MJ, Yolken RH, Bahn S. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet. 2003;362 (9386):798–805. doi: 10.1016/S0140-6736(03)14289-4. [DOI] [PubMed] [Google Scholar]
- Tosic M, Ott J, Barral S, Bovet P, Deppen P, Gheorghita F, Matthey ML, Parnas J, Preisig M, Saraga M, Solida A, Timm S, Wang AG, Werge T, Cuenod M, Quang DK. Schizophrenia and oxidative stress: glutamate cysteine ligase modifier as a susceptibility gene. Am J Hum Genet. 2006;79 (3):586–592. doi: 10.1086/507566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Lewis BL, Hashimoto T, Sesack SR, Kloc M, Lewis DA, O’Donnell P. A neonatal ventral hippocampal lesion causes functional deficits in adult prefrontal cortical interneurons. J Neurosci. 2008;28 (48):12691–12699. doi: 10.1523/JNEUROSCI.4166-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O’Donnell P. Dopamine modulation of prefrontal cortical interneurons changes during adolescence. Cereb Cortex. 2007;17 (5):1235–1240. doi: 10.1093/cercor/bhl034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nature Rev Neurosci. 2010;11 (2):100–113. doi: 10.1038/nrn2774. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Neuronal dynamics and neuropsychiatric disorders: toward a translational paradigm for dysfunctional large-scale networks. Neuron. 2012;75 (6):963–980. doi: 10.1016/j.neuron.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Umbricht D, Schmid L, Koller R, Vollenweider FX, Hell D, Javitt DC. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry. 2000;57 (12):1139–1147. doi: 10.1001/archpsyc.57.12.1139. [DOI] [PubMed] [Google Scholar]
- Uranova N, Orlovskaya D, Vikhreva O, Zimina I, Kolomeets N, Vostrikov V, Rachmanova V. Electron microscopy of oligodendroglia in severe mental illness. Brain Res Bull. 2001;55 (5):597–610. doi: 10.1016/s0361-9230(01)00528-7. [DOI] [PubMed] [Google Scholar]
- Uranova NA, Vikhreva OV, Rachmanova VI, Orlovskaya DD. Ultrastructural alterations of myelinated fibers and oligodendrocytes in the prefrontal cortex in schizophrenia: a postmortem morphometric study. Schizophr Res Treatment. 2011;2011:325789. doi: 10.1155/2011/325789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium. Schizophr Res. 2004;67 (2–3):269–275. doi: 10.1016/S0920-9964(03)00181-6. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39 (1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, Luurtsema G, Windhorst AD, Cahn W, Lammertsma AA, Kahn RS. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64 (9):820–822. doi: 10.1016/j.biopsych.2008.04.025. [DOI] [PubMed] [Google Scholar]
- Vinson PN, Conn PJ. Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology. 2012;62 (3):1461–1472. doi: 10.1016/j.neuropharm.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter PB, Knutson MD, Paler-Martinez A, Lee S, Xu Y, Viteri FE, Ames BN. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc Natl Acad Sci U S A. 2002;99 (4):2264–2269. doi: 10.1073/pnas.261708798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AY, Lohmann KM, Yang CK, Zimmerman EI, Pantazopoulos H, Herring N, Berretta S, Heckers S, Konradi C. Bipolar disorder type 1 and schizophrenia are accompanied by decreased density of parvalbumin- and somatostatin-positive interneurons in the parahippocampal region. Acta Neuropathol. 2011;122 (5):615–626. doi: 10.1007/s00401-011-0881-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CZ, Yang SF, Xia Y, Johnson KM. Postnatal phencyclidine administration selectively reduces adult cortical parvalbumin-containing interneurons. Neuropsychopharmacology. 2008;33 (10):2442–2455. doi: 10.1038/sj.npp.1301647. [DOI] [PubMed] [Google Scholar]
- Wang HX, Gao WJ. Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology. 2009;34 (8):2028–2040. doi: 10.1038/npp.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HX, Gao WJ. Development of calcium-permeable AMPA receptors and their correlation with NMDA receptors in fast-spiking interneurons of rat prefrontal cortex. J Physiol. 2010;588 (Pt 15):2823–2838. doi: 10.1113/jphysiol.2010.187591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Pinto-Duarte A, Sejnowski TJ, Behrens MM. How Nox2-containing NADPH oxidase affects cortical circuits in the NMDA receptor antagonist model of schizophrenia. Antioxid Redox Sign. 2013;18 (12):1444–1462. doi: 10.1089/ars.2012.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Lu YS, Zhu XH, Li XM, Woo RS, Chen YJ, Yin DM, Lai C, Terry AV, Jr, Vazdarjanova A, Xiong WC, Mei L. Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc Natl Acad Sci U S A. 2010;107 (3):1211–1216. doi: 10.1073/pnas.0910302107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitford TJ, Kubicki M, Ghorashi S, Schneiderman JS, Hawley KJ, McCarley RW, Shenton ME, Spencer KM. Predicting inter-hemispheric transfer time from the diffusion properties of the corpus callosum in healthy individuals and schizophrenia patients: a combined ERP and DTI study. NeuroImage. 2011;54 (3):2318–2329. doi: 10.1016/j.neuroimage.2010.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff P, Ponomarenko AA, Bartos M, Korotkova TM, Fuchs EC, Bahner F, Both M, Tort AB, Kopell NJ, Wisden W, Monyer H. Hippocampal theta rhythm and its coupling with gamma oscillations require fast inhibition onto parvalbumin-positive interneurons. Proc Natl Acad Sci U S A. 2009;106 (9):3561–3566. doi: 10.1073/pnas.0813176106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi M, Joho RH, Southcott SA, Shukla AA, Ghose S, Tamminga CA. Kv3.1-containing K channels are reduced in untreated schizophrenia and normalized with antipsychotic drugs. Mol Psychiatry. 2014;19 (5):573–579. doi: 10.1038/mp.2013.49. [DOI] [PubMed] [Google Scholar]
- Yao JK, Keshavan MS. Antioxidants, redox signaling, and pathophysiology in schizophrenia: an integrative view. Antioxid Redox Sign. 2011;15 (7):2011–2035. doi: 10.1089/ars.2010.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao JK, Leonard S, Reddy R. Altered glutathione redox state in schizophrenia. Dis Markers. 2006;22 (1–2):83–93. doi: 10.1155/2006/248387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60 (1):6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, He J, Zhu S, Zhang H, Wang H, Adilijiang A, Kong L, Wang J, Kong J, Tan Q, Li XM. Myelination deficit in a phencyclidine-induced neurodevelopmental model of schizophrenia. Brain Res. 2012;1469:136–143. doi: 10.1016/j.brainres.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Sun QQ. Development of NMDA NR2 subunits and their roles in critical period maturation of neocortical GABAergic interneurons. Dev Neurobiol. 2011;71 (3):221–245. doi: 10.1002/dneu.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZJ, Reynolds GP. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res. 2002;55 (1–2):1–10. doi: 10.1016/s0920-9964(01)00188-8. [DOI] [PubMed] [Google Scholar]