

Abstract
Context:
Amiodarone (AMIO) is one of the most effective antiarrhythmic drugs available; however, its use is limited by a serious side effect profile, including thyroiditis. The mechanisms underlying AMIO thyroid toxicity have been elusive; thus, identification of novel approaches in order to prevent thyroiditis is essential in patients treated with AMIO.
Objective:
Our aim was to evaluate whether AMIO treatment could induce endoplasmic reticulum (ER) stress in human thyroid cells and the possible implications of this effect in AMIO-induced destructive thyroiditis.
Results:
Here we report that AMIO, but not iodine, significantly induced the expression of ER stress markers including Ig heavy chain-binding protein (BiP), phosphoeukaryotic translation initiation factor 2α (eIF2α), CCAAT/enhancer-binding protein homologous protein (CHOP) and spliced X-box binding protein-1 (XBP-1) in human thyroid ML-1 cells and human primary thyrocytes. In both experimental systems AMIO down-regulated thyroglobulin (Tg) protein but had little effect on Tg mRNA levels, suggesting a mechanism involving Tg protein degradation. Indeed, pretreatment with the specific proteasome inhibitor MG132 reversed AMIO-induced down-regulation of Tg protein levels, confirming a proteasome-dependent degradation of Tg protein. Corroborating our findings, pretreatment of ML-1 cells and human primary thyrocytes with the chemical chaperone 4-phenylbutyric acid completely prevented the effect of AMIO on both ER stress induction and Tg down-regulation.
Conclusions:
We identified ER stress as a novel mechanism contributing to AMIO-induced destructive thyroiditis. Our data establish that AMIO-induced ER stress impairs Tg expression via proteasome activation, providing a valuable therapeutic avenue for the treatment of AMIO-induced destructive thyroiditis.
Amiodarone (AMIO) is a powerful antiarrhythmic drug approved in the United States for the management of various cardiac rhythm disturbances, including potentially lethal ventricular arrhythmias (1). It remains one of the most frequently prescribed antiarrhythmic medications; however, its use has been limited by multiple and serious side effects (2). AMIO is a benzofuran derivative containing two iodine atoms per molecule. The therapeutic drug doses range from 200 to 600 mg daily releasing approximately 7–20 mg of iodine per day, which is about 50- to 100-fold of the recommended daily iodine intake (3). Pharmacokinetic studies in humans indicate that AMIO has a very long elimination half-life, a large volume of distribution, and extensive tissue distribution. Consequently, in cases of AMIO-induced adverse effects stopping therapy usually does not result in resolution of side effects for weeks and sometimes months (4). Due to its benzofuran and biiodinated benzene ring, AMIO is highly lipophilic and is stored in elevated concentration in adipose tissue, myocardium, liver, muscle, and skin as well as in the lungs and thyroid (5). The mechanisms of AMIO toxicity, however, are not fully understood and are generally considered multifactorial (6).
AMIO structure closely resembles that of thyroid hormones, which may explain some of its actions on the thyroid gland (7). In fact, thyroid dysfunction is the most common AMIO complication requiring intervention, occurring in 15%–20% of AMIO-treated patients (8). The molecular mechanisms underlying AMIO-induced thyrotoxicosis (AIT) are still unclear. Because a specific therapy cannot be developed, AIT remains a challenging clinical problem. AIT is commonly classified in two types (9). Type I AIT is a Jod-Basedow phenomenon (iodine induced hyperthyroidism), caused by excessive, uncontrolled biosynthesis of thyroid hormones by autonomously functioning thyroid tissue in response to iodine load (10). Type 2 AIT is a destructive thyroiditis, which is induced directly by AMIO effects on thyroid follicles, triggering an inflammatory reaction via mechanisms that are still unknown (11). The prevalence of type 1 and type 2 AIT varies geographically according to dietary iodine intake. Type 1 AIT is more common in iodine-deficient regions, whereas type 2 AIT is more common in iodine-sufficient regions. However, epidemiological data indicate that even in iodine-deficient populations, type 2 is now the dominant form of AIT (12).
In the current study, we sought to examine whether AMIO causes endoplasmic reticulum (ER) stress in thyroid cells and whether ER stress participates in the molecular mechanisms underlying type 2 AIT.
Materials and Methods
Cell cultures and reagents
Human thyroid ML-1 cells (13) were cultured in DMEM + GlutaMAX-1 medium (Thermo Scientific) supplemented with 10% fetal bovine serum (Sigma) and maintained at 37°C in a humidified atmosphere with 5% CO2. Human primary thyrocytes were isolated as described previously (14) and cultured with medium 199/Earle's balanced salt solution medium (HyClone) supplemented with 10% fetal bovine serum (Sigma) and 1% antibiotic antimycotic solution (HyClone). All experiments were performed after obtaining approval by the Icahn School of Medicine Institutional Review Board. Briefly, tissue specimens were obtained from the unaffected contralateral lobes of thyroid papillary carcinomas or from internodular tissue of nodular goiters undergoing thyroidectomy. Thyroid tissues were minced and digested with type II collagenase (Worthington Biochemical Corp) for 3 hours at 37°C. Cells were pelleted by centrifugation at 1000 rpm for 5 minutes, washed twice in complete medium 199/Earle's balanced salt solution medium, seeded in a 75-cm2 flask (Corning) and cultured in a 5% CO2 atmosphere at 37°C. For different experiments, cells were seeded on 10-cm dishes, 6-cm dishes, or 12-mm-diameter glass coverslips in 24-well plates. Twenty-four hours later, the cells were vehicle treated or treated with the indicated drugs at the indicated experimental settings. Thapsigargin (THAP), AMIO, 4-phenylbutyric acid (PBA) and MG132 were purchased from Sigma, iodine (NaI) was purchased from Fisher Scientific, and tauroursodeoxycholic acid (TUDCA) was purchased from Calbiochem. The AMIO concentrations used in all experiments are identical to the therapeutic levels of AMIO in the blood (5–10 μM).
Real-time RT-PCR
Real-time RT-PCR was performed as previously described (15). In brief, total RNA from ML-1 cells or human primary thyrocytes was isolated using TRIzol reagent (Thermo Scientific) in combination with the RNeasy minikit (QIAGEN) followed by deoxyribonuclease treatment. Five hundred nanograms of total RNA were retrotranscribed using the Superscript III kit (Thermo Scientific) following the manufacturer's instructions. The cDNAs obtained after retrotranscription were used as templates for quantitative PCR, run on an AbiPRISM 7300 Fast real-time cycler using the power SYBR Green real-time PCR master mix kit and quantified by built-in SYBR Green Analysis (all from Applied Biosystems). The relative amount of specific mRNA was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primer sequences (Integrated DNA Technologies) used in this study for gene analysis were as follows (forward, reverse): 5′-AGGCAGGAGAATCACTCGAA-3′ and 5′-GGGCTGGAGTACAGTGGTGT-3′ for Ig heavy chain-binding protein (BiP); 5′-TCTGATTGACCGAATGGTGA-3′ and 5′-TCTGGGAAAGGTGGGTAGTG for CCAAT/enhancer-binding protein homologous protein (CHOP); 5′-CGAATGAGTGAGCTGGAACA-3′ and 5′-CCAAGCGCTGTCTTAACTCC for X-box binding protein-1 (XBP-1); 5′-TGCTGAGTCCGCAGCAGGTG-3′ and 5′-GAGATGTTCTGGAGGGGTGA-3′ for spliced XBP-1; 5′-AACCCCATTGTGTTCTCAGC-3′ and 5′-ATGATGGCACCTCCTTGAAC-3′ for thyroglobulin (Tg); and 5′-ACAGTCAGCCGCATCTTCT-3′ and 5′-ACGACCAAATCCGTTGACTC-3′ for GAPDH.
Western blot assay
Protein samples from ML-1 cells and human primary thyrocytes were lysed in the following assay buffer (50 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1% Nonidet P-40; 0.5% sodium deoxycholate; 0.1% sodium dodecyl sulfate; 1 mM phenylmethylsulfonyl fluoride; 10 mM NaF; and 0.5 M Na3OV4). Lysates were centrifuged at 13 000 rpm for 45 minutes. Supernatants were collected and equal amount of proteins (50 μg per sample) were subjected to electrophoresis on 12% (for BiP antibody) and 6% (for Tg antibody) sodium dodecyl sulfate-polyacrylamide gel and transferred to Immobilon-P membranes (Millipore) for 2 hours at 100 V on ice. Membranes were incubated in Odyssey blocking buffer (LI-COR) for 60 minutes at room temperature and then reacted overnight at 4°C with the following primary antibodies: rabbit monoclonal anti-BiP (Cell Signaling; 1:1000), rabbit monoclonal anti-Tg (Abcam; 1:10 000), mouse monoclonal anti-β-actin (Cell Signaling; 1:1000), rabbit monoclonal antiphosphoeukaryotic translation initiation factor 2α (eIF2α) (Cell Signaling; 1:1000), mouse monoclonal anti-CHOP (Cell Signaling; 1:1000). All immunoblots were developed with the LI-COR Odyssey system, using infrared-labeled antirabbit and antimouse IgG (LI-COR; 1:5000) secondary antibodies.
Immunofluorescence
The 1.5 × 105 ML-1 cells or human primary thyrocytes were seeded on poly-D-lysine-coated, 12-mm glass coverslips in 24-well plates and allowed to attach for 24 hours at 37°C. Cells were then vehicle treated or treated with the indicated drugs at the indicated experimental settings and fixed in 4% paraformaldehyde (Polysciences, Inc) in PBS for 15 minutes at room temperature. After being permeabilized by 0.5% Triton X-100 (Bio-Rad Laboratories) in PBS for 10 minutes, cells were blocked for 1 hour with 1% normal goat serum (Cell Signaling) in PBS (both steps were done at room temperature). Cells were subsequently immunostained overnight at 4°C with the primary antibodies diluted in blocking buffer. Indirect immunofluorescence was performed using rabbit monoclonal anti phospho-eIF2α (Cell Signaling; 1:100), mouse monoclonal anti-CHOP (Cell Signaling; 1:500), rabbit monoclonal anti-Tg (Abcam; 1:10 000). Secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 568 (1:500; Thermo Scientific) were used for 1 hour at room temperature. After final washes with PBS, the coverslips were mounted on microscope slides with ProLong Gold antifade reagent containing 4′,6′-diamino-2-phenylindole (DAPI) to counterstain the nuclei (Cell Signaling). The samples were examined with a Leica DM6000 microscope.
Statistics
All experiments were performed at least in triplicate. Data are presented as means ± SEM. Statistical differences were determined using a two-tailed Student's t test. Statistical analysis was performed using GraphPad Prism (version 5.01; GraphPad Software Inc). A value of P < .05 was considered statistically significant.
Results
AMIO but not iodine induces BiP up-regulation in ML-1 cells
To investigate whether human thyroid ML-1 cells were sensitive to ER stress, we treated them with THAP, a classic ER stress inducer able to inhibit the sarcoplasmic/ER calcium-transporting ATPases. A 24-hour treatment with THAP increased the mRNA of the diagnostic ER stress marker BiP (Figure 1A), a molecular chaperone that resides in the ER (16). The THAP-induced increase of BiP mRNA expression was paralleled by a similar up-regulation of BiP protein levels (Figure 1, B and C). ML-1 cells were then exposed to increasing concentrations of AMIO (5, 10, and 20 μM), and BiP induction was examined by real-time RT-PCR and Western blot analysis. Treatment with AMIO for 24 hours revealed an effect comparable with THAP, increasing mRNA and protein levels of BiP, already at a concentration of 5 μM and with a similar effect at 10 or 20 μM (Figure 1, A–C). In addition, a time-course analysis with 5 μM AMIO showed that BiP mRNA was significantly increased as early as 16 hours after treatment and remained elevated for up to 72 hours (Figure 1D). To determine whether iodine released by AMIO was involved in inducing ER stress, we treated ML-1 cells with increasing concentrations of iodine (10, 100, or 1000 μM). Intriguingly, none of the iodine concentrations caused an up-regulation of BiP, tested both at the mRNA and protein levels (Figure 1, A–C), demonstrating that AMIO did not induce BiP via iodine-mediated mechanisms.
Figure 1.
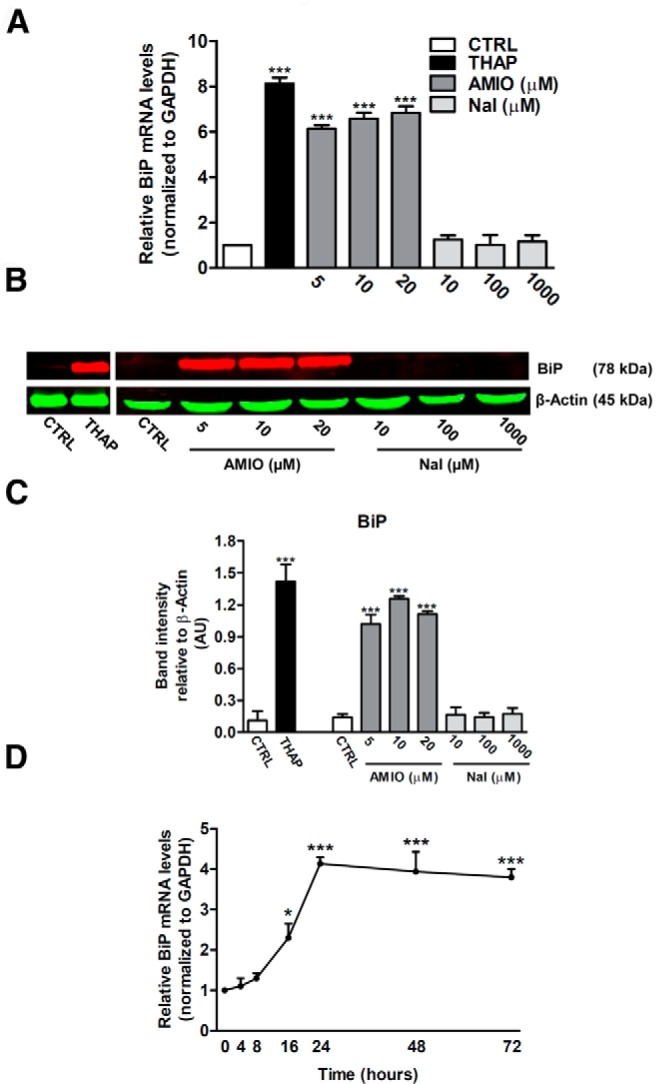
AMIO, but not NaI, triggers BiP induction in ML-1 cells. A and B, ML-1 cells were treated with 0.5 μM THAP or with the indicated concentrations of AMIO or NaI for 24 hours, and the levels of BiP mRNA were determined by real-time RT-PCR analysis of total RNA using GAPDH as internal standard. The mRNA levels in treated cells are relative to those in vehicle-treated cells (CTRL). B, Cells were solubilized and an equal amount of proteins (50 μg per sample) were analyzed by immunoblotting. Filters were probed with antibodies against BiP and β-actin and band intensities were quantified by densitometry using ImageJ software (C) (National Institutes of Health, Bethesda, Maryland). D, Time course of BiP mRNA in ML-1 cells cultured with 5 μM AMIO for the indicated times. Bars represent means ± SEM from four to five independent experiments. *, P < .001; ***, P < .001 compared with CTRL cells.
Chemical chaperones prevent AMIO-induced BiP up-regulation in ML-1 cells
To corroborate these findings, we tested the ability of classical ER stress modulators, PBA and TUDCA (17), to prevent the AMIO-induced BiP up-regulation. PBA is a low-molecular-weight nonspecific chemical chaperone known to stabilize protein conformation, improving ER folding capacity, and facilitating the traffic of mutant proteins. Likewise, TUDCA, a taurine-conjugated derivative of bile acid, mitigates ER stress by acting as a chemical chaperone. As shown in Figure 2, A–C, PBA and TUDCA almost completely prevented the effect of AMIO on BiP up-regulation. These data indicate that AMIO up-regulates BiP in thyroid cells by inducing ER stress.
Figure 2.
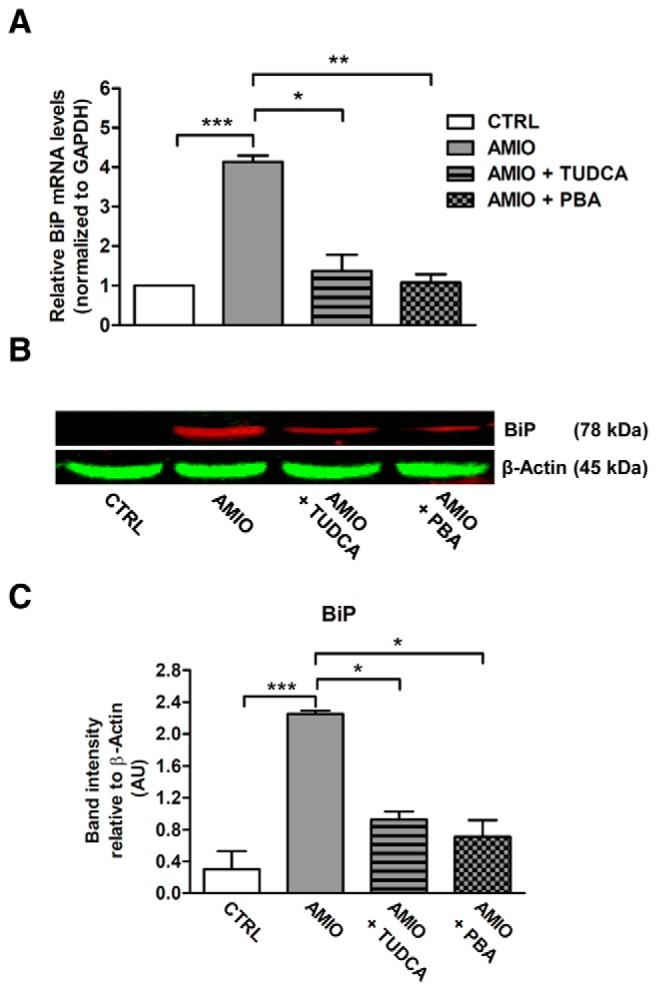
Chemical chaperones prevent AMIO-induced BiP up-regulation in ML-1 cells. A and B, ML-1 cells were pretreated or not for 24 hours with 5 mM TUDCA or with 7.5 mM PBA and then cultured in presence of 5 μM AMIO for 24 hours. A, Real-time RT-PCR analysis of BiP mRNA levels using the total RNAs from ML-1 cells treated as indicated, with GAPDH as internal standard. mRNA levels in treated cells are relative to those in vehicle-treated cells (CTRL). B, Immunoblot using an equal amount of proteins (50 μg per sample) and the indicated antibodies. C, Densitometric quantification of ML-1 cells treated as described; band fluorescence intensity was assessed with ImageJ software. Experiments were repeated three times, and values are mean ± SEM. *, P < .001; ***, P < .001 compared with CTRL cells.
AMIO induces unfolded protein response activation in ML-1 cells
The accumulation of unfolded proteins in the ER lumen triggers a coordinated adaptive program called the unfolded protein response (UPR), an integrated signal transduction pathway that transmits information about the protein-folding status in the ER to the nucleus and cytosol, to restore ER homeostasis. In mammalian cells the UPR is activated by three transducers, inositol requiring enzyme 1 (IRE1), double-stranded RNA-activated protein kinase-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (18). To examine whether AMIO induced UPR in thyroid cells, we evaluated in ML-1 cells the PERK-dependent phosphorylation of eIF2α, a classical UPR molecular marker (18). Phosphorylation of eIF2α was significantly increased after AMIO treatment when compared with cells treated with vehicle. Moreover, pretreatment with PBA prevented eIF2α phosphorylation upon exposure of ML-1 cells to AMIO (Figure 3, A and B).
Figure 3.
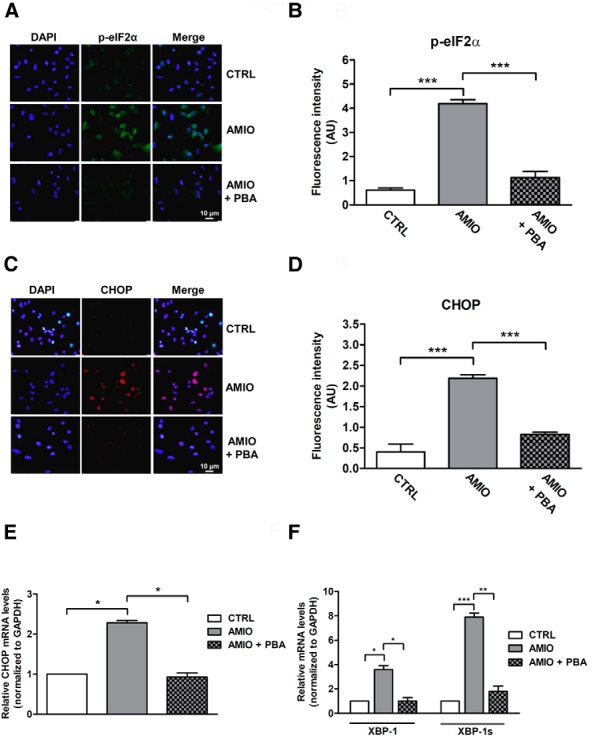
AMIO induces UPR activation in ML-1 cells. A and C, Patterns of expression of phospho-eIF2α and CHOP in ML-1 cells as determined by immunostaining. ML-1 cells were grown on glass coverslips for 24 hours and then pretreated or not with 7.5 mM PBA for 24 hours followed by 24 hours in medium with 5 μM AMIO. Immunofluorescence staining was performed using phospho-eIF2α antibody (green) or CHOP antibody (red). Nuclei were counterstained with DAPI (blue). B and D, Quantification of fluorescence intensity for phospho-eIF2α or CHOP in cultured ML-1 cells treated as described. E and F, The levels of mRNAs for CHOP, XBP-1, and active XBP-1 spliced mRNA were determined by real-time RT-PCR analysis of total RNA from ML-1 cells treated as above. GAPDH was used as an internal standard. Each bar represents the mean ± SEM of four independent experiments, each performed in triplicate. *, P < .05; **, P < .01, ***, P < .001 compared with vehicle-treated cells (CTRL).
Next, we measured the expression levels of CHOP, a transcription factor driven by eIF2α phosphorylation that controls genes involved in apoptosis (18). CHOP protein and mRNA levels were also elevated in AMIO-treated cells, and, as shown for phospho-eIF2α, PBA almost completely prevented AMIO-induced CHOP induction (Figure 3, C–E). Finally, we tested the effect of AMIO on the IRE1 pathway of the UPR. IRE1 is a kinase that uses a unique mechanism of nonconventional splicing to transmit the UPR signal. When activated, IRE1 cleaves the mRNA encoding a specific transcription factor called XBP-1, giving rise to an active spliced mRNA that regulates the transcription of genes involved in protein folding (19). RT-PCR analysis showed an increase in both the total and spliced (active) form of XBP-1 mRNA levels after AMIO treatment; this effect was also suppressed by PBA pretreatment (Figure 3F).
Effects of AMIO on human primary thyrocytes
To confirm the results obtained in ML-1 cells, we tested the effects of AMIO on human primary thyrocytes. We first evaluated whether AMIO was able to induce ER stress in this system. As shown in Figure 4A, similar to ML-1 cells, human primary thyrocytes treated with 5 μM AMIO for 24 hours exhibited a significant increase in BiP mRNA levels as measured by RT-PCR and in BiP protein levels as measured by Western blot (Figure 4, B and C). Furthermore, pretreatment with PBA or TUDCA reduced AMIO-induced BiP up-regulation, proving that BiP up-regulation was due to ER stress (Figure 4, A–C). Iodine treatment (10, 100, or 1000 μM) did not cause up-regulation of BiP or UPR activation in primary human thyrocytes, tested both at the mRNA and protein levels (Supplemental Figure 1), confirming the data obtained in the ML-1 cells.
Figure 4.
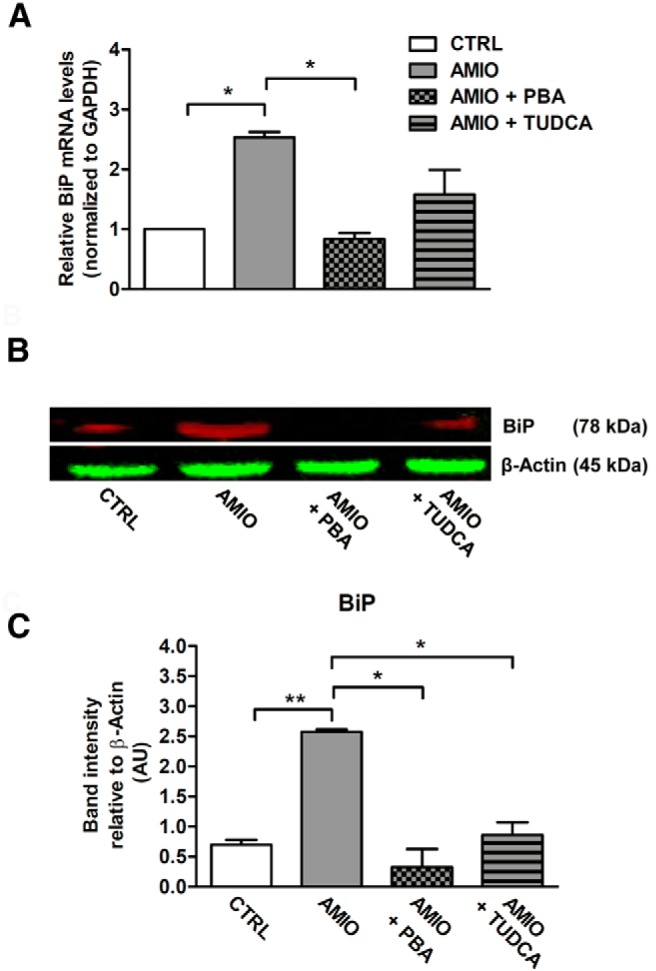
AMIO up-regulates BiP in human primary thyrocytes. A–C, Human primary thyrocytes were pretreated or not for 24 hours with 7.5 mM PBA or 5 mM TUDCA followed by a treatment with 5 μM AMIO for 24 hours. A, BiP mRNA was determined by real-time RT-PCR analysis of total RNA isolated from human primary thyrocytes, using GAPDH as internal standard. mRNA levels in treated cells are relative to those in vehicle-treated cells (CTRL). B, Human primary thyrocytes were solubilized and cell lysates were analyzed by Western blotting with anti BiP antibody using β-actin as a loading control. C, Densitometric quantification of human primary thyrocytes treated as described; band fluorescence intensity was assessed with ImageJ software. These results have been replicated three times. All data are shown as mean ± SEM. *, P < .001, **, P < .01 compared with CTRL cells.
AMIO-induced ER stress down-regulates Tg protein levels
Tg is a very large glycoprotein representing the major secretory product of the thyroid gland that begins to fold in the ER with the assistance of multiple molecular chaperones (20). Tg misfolding is a well-recognized cause of congenital hypothyroidism (21) in which Tg mutants are blocked in ER export and are then routed for proteasomal degradation. Because ER stress has been previously demonstrated to be involved in altered glycosylation and secretion of Tg (22), we hypothesized that AMIO-induced ER stress might lead to alterations in Tg expression. To test this hypothesis, human primary thyrocytes and ML-1 cells were treated with 5 μM AMIO for 72 hours; in both experimental systems, AMIO markedly reduced Tg protein levels (Figure 5, A–D). Interestingly, iodine treatment (10, 100, or 1000 μM) was not able to down-regulate Tg protein levels (Supplemental Figure 2).
Figure 5.

Effect of ER stress on Tg expression in human primary thyrocytes and ML-1 cells. A–D, Human primary thyrocytes and ML-1 cells were pretreated or not for 24 hours with 7.5 mM PBA and then cultured in the presence of 5 μM AMIO for 72 hours. Immunoblot analysis and quantification of total protein extracts from human primary thyrocytes (A and B) and ML-1 cells (C and D) were performed using specific antibodies against Tg and β-actin. E, Real-time RT-PCR analysis from human primary thyrocytes and ML-1 cells vehicle-treated (CTRL) or treated with 5 μM AMIO for 72 hours. Tg mRNA levels in treated cells are relative to those in control cells. GAPDH was used as internal standard. Experiments were repeated three times, and values are mean ± SEM. *, P < .001, ***, P < .001 compared with CTRL cells.
Next, we sought to establish the causal relationship between the observed decrease of Tg- and AMIO-induced ER stress analyzing Tg protein in cells that were treated with AMIO in the presence of PBA. PBA prevented AMIO-dependent reduction of Tg protein level, suggesting that reversing ER stress may promote Tg-correct folding in thyroid cells. A number of studies indicate that ER stress plays an essential role in modulating cellular protein levels by fine-tuning gene transcription (23) and protein degradation (24). To test whether AMIO-induced down-regulation of Tg was due to suppression of Tg transcription or due to an up-regulation of Tg protein degradation, we investigated the effect of AMIO-induced ER stress on the mRNA levels of Tg. Real-time RT-PCR analysis revealed that AMIO treatment had a significant but very small effect on Tg mRNA levels (Figure 5E), both in primary human thyrocytes and ML-1 cells, suggesting that protein degradation may play a major functional role in AMIO-induced Tg down-regulation.
AMIO-induced ER stress down-regulates Tg protein levels by a proteasome-dependent mechanism
The proteasome-dependent pathway represents one of the molecular mechanisms by which ER stress induces protein degradation. Improperly folded proteins are retained in the ER and delivered for proteasomal degradation after retrotranslocation into the cytosol. This tightly controlled process is called ER-associated degradation (ERAD) (25). To determine whether the proteasome mechanistically participates in ER stress-related Tg degradation, we treated primary thyrocytes and ML-1 cells with AMIO in presence of MG132, a proteasome-specific inhibitor. As shown in Figure 6, in both cellular systems preincubation with MG132 prevented the AMIO-induced down-regulation of Tg, supporting our hypothesis that ER stress triggered by AMIO leads to Tg degradation via a proteasome-mediated pathway.
Figure 6.

Proteasome activation is involved in ER-stress induced Tg protein down-regulation. A–D, Human primary thyrocytes and ML-1 cells were grown on glass coverslips for 24 hours and subsequently pretreated or not overnight with 0.8 μM MG132 followed by 72 hours incubation in medium with 5 μM AMIO. Human primary thyrocytes (A) and ML-1 cells (C) were stained with a specific antibody against Tg (green). Nuclei were counterstained with DAPI (blue). B, Quantification of fluorescence intensity for Tg in human primary thyrocytes and cultured ML-1 cells (D) treated as described. The data in this figure are representative of four replicate experiments. Asterisks indicate statistically significant differences (*, P < .05, ***, P < .001 compared with CTRL cells).
Discussion
AMIO, a biiodinated benzofuran derivative, is a compound with unique pharmacokinetics and electrophysiological properties. It represents one of the most used and effective antiarrhythmic drugs, beneficial for both atrial and ventricular arrhythmias (26, 27). However, AMIO has been associated with multiple serious adverse effects, including hypothyroidism and hyperthyroidism (9). The abnormal thyroid functions are especially challenging because patients receiving AMIO usually have severe heart disease associated with serious arrhythmias, both of which can be exacerbated by hyper- or hypothyroidism. The AMIO side effect profile is a function of dose as well as the duration of therapy, but the molecular mechanisms have not been fully investigated. In the present study, we identify, for the first time, ER stress as a key molecular link between AMIO treatment and destructive thyroiditis and a potential therapeutic target.
The ER is the cellular compartment in which proteins are synthesized and acquire their correct three-dimensional folding to become biologically active. The protein-folding machinery in the ER is particularly challenged in professional secretory cells owing to their high demand for protein synthesis, which represents a constant source of stress (18). Recently it has been proposed that ER-stressed cells could eventually die or produce neoautoantigens, triggering autoimmunity in genetically susceptible individuals (28). Dysregulation of the UPR may contribute to autoimmunity through four potential different mechanisms: 1) misfolded proteins may themselves act as autoantigens; known examples are ankylosing spondylitis and inclusion body myositis (29, 30); 2) UPR-related genes may themselves act as autoantigens, and this mechanism has been suggested in patients with rheumatoid arthritis and in a mouse model of Sjogren's syndrome (31, 32); 3) defective UPR pathways in nonimmune cells overwhelm normal mechanisms of immune tolerance, causing development of colitis (33), atherosclerosis (34), and experimental autoimmune encephalomyelitis (35); and 4) the up-regulation of the ERAD pathway may allow autoreacitve cells to escape UPR-mediated apoptosis or confer a survival advantage; such a mechanism has been proposed in a mouse model of inflammatory arthritis (36).
Thyroid cells are specialized secretory cells synthesizing and secreting thyroid hormones, T4 and T3 as well as their precursor Tg. Indeed, thyrocytes are very sensitive to disruptions in ER homeostasis. Here we show that AMIO therapy can induce chronic ER stress with the activation of the UPR in human thyrocytes and that this could represent a central and integrating mechanism underlying AMIO-induced thyroid toxicity. To cope with ER stress, cells turn on an adaptive cellular reaction, the UPR, a coordinated program that enhances the ER folding capacity to buffer fluctuations in unfolded protein load. The UPR is a unique self-protective mechanism tailored to the needs of the cell, determining its fate by acting as a switch between survival and death. If the adaptive response fails, cells execute apoptosis (37). Our data unveiled a key role for UPR signaling in regulating thyrocytes survival under AMIO-induced ER stress as demonstrated by increased levels of phospho-eIF2α, CHOP, and spliced XBP-1 in ML-1 cells treated with AMIO. The UPR has been shown to play a central role in several human diseases. For example, the UPR can serve as an apoptotic executor that kills beneficial cells causing pathological conditions such as Fabry's disease, cystic fibrosis, and type 2 diabetes mellitus. On the other hand, UPR can also act as a cytoprotector in other diseases such as osteogenesis imperfecta, Parkinson's disease, and congenital hypothyroid goiter (38). Interestingly, subtoxic doses of AMIO were recently reported to interfere with protein folding in the ER in glioma cells (39), supporting our findings and indicating that this response is occurring in other cell types.
Given the protective role of UPR-mediated ER chaperone response in metabolic disorders, small molecules with chaperone-like activity have drawn attention as candidates for novel therapies. Preclinical and clinical studies have recently demonstrated the therapeutic potential of PBA and TUDCA in several metabolic diseases caused by ER stress. The beneficial properties of PBA and TUDCA were discovered and explored in cystic fibrosis (40), metabolic syndrome (41), obesity (42), and atherosclerosis (43). Our results in ML-1 cells and in human primary thyrocytes suggest that PBA and TUDCA may be beneficial in AIT type 2, as shown by the prevention of BiP up-regulation by PBA and TUDCA in our system.
As a result of AMIO-induced ER stress, the Tg protein level was significantly reduced. Moreover, we found that the deleterious consequences of AMIO treatment on Tg protein were fully prevented by the chemical chaperone PBA (Figure 5). Therefore, we postulated that AMIO causes Tg misfolding in human thyrocytes, triggering its ERAD. Supporting this hypothesis, pretreatment with MG132, a proteasome-specific inhibitor, reversed the AMIO-induced down-regulation of Tg protein levels (Figure 6). These findings meet the ERAD criteria of misfolded secretory glycoproteins, suggesting an attractive new molecular mechanism underlying AMIO-induced toxicity. The importance of ERAD-mediated degradation of misfolded Tg has been confirmed previously by Kim et al (44), who demonstrated that a Tg R19K missense mutation induces local misfolding in its amino-terminal domain and has global effects on thyroid hormonogenesis. In their model, Tg failed to be secreted and was instead disposed intracellularly via proteasomal digestion without arriving in the Golgi complex (44).
AMIO-induced thyroid toxicity has been classically ascribed to its iodine content (3), especially in iodine-deficient patients or in subjects with previously undiagnosed goiter. A study by Chiovato et al (45) has shown that thyroid cytotoxicity caused by AMIO is mainly due to its direct effect on thyroid cells, albeit excess iodine released from the drug may further contribute to AMIO toxic action. Therefore, we examined whether iodine might be responsible for AMIO-induced ER stress in human thyrocytes. Remarkably, our findings indicated that iodine, even at very high superphysiological levels, was not involved in UPR up-regulation in thyroid cells, demonstrating that AMIO induces ER stress in thyroid cells in an iodine-independent manner. These data provide a molecular mechanism that could account for AMIO toxicity in subjects that are iodine sufficient and with normal thyroid glands. Our data are consistent with previous studies suggesting an iodine-independent thyrotoxicity of AMIO, leading to follicular damage in the gland (46).
Our findings point to a key role for ER stress in AMIO-induced thyroid dysfunction, but the exact mechanisms by which AMIO triggers ER stress in thyroid cells is not yet known. Previous studies performed in various cell types indicated that AMIO increases intracellular calcium levels (47), which is known to trigger ER stress. Therefore, perturbation of the ER calcium homeostasis is likely involved in the AMIO-induced ER stress and activation of UPR in thyroid cells.
In an attempt to reduce the AMIO-related adverse effects, a noniodinated derivative, dronedarone, was introduced as an alternative antiarrhythmic drug in 2009. However, recent studies demonstrated that dronedarone is significantly less effective than AMIO for the maintenance of sinus rhythm (48). Moreover, dronedarone has also been associated with increased mortality in patients with heart failure (49). In view of the failure of dronedarone to replace AMIO as a safe and effective antiarrhythmic agent, there is a need for a better understanding of AMIO-induced toxicity to develop new therapeutic modalities that prevent AMIO toxicity and enable its continued use as an effective antiarrhythmic agent.
In conclusion, our data suggest that ER stress is a key mechanism of AMIO-associated thyroid dysfunction, causing ERAD of Tg protein. Our findings have significant translational implications, given the excellent safety profiles of PBA and TUDCA in humans. Indeed, the Food and Drug Administration has recently approved PBA and TUDCA for the treatment of urea-cycle disorders and primary biliary cirrhosis, respectively (50). Both chemical chaperones appear to be safe and have few side effects, including weight gain, hair loss, epigastric discomfort, and diarrhea (51, 52). Therefore, it is possible that these compounds may be candidates for future trials as a novel therapy for the treatment of AMIO-induced destructive thyroiditis by restoring Tg protein folding homeostasis.
Acknowledgments
We thank Christoph Buettner and Derek LeRoith for their experimental help.
This work was supported by NIH Grants DK061659, DK067555, and DK073681 from the National Institute of Diabetes and Digestive and Kidney Diseases In addition, this material is based upon work supported in part by the Department of Veterans Affairs.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AIT
- AMIO-induced thyrotoxicosis
- AMIO
- amiodarone
- BiP
- Ig heavy chain-binding protein
- CHOP
- CCAAT/enhancer-binding protein homologous protein
- DAPI
- 4′,6′-diamino-2-phenylindole
- eIF2α
- eukaryotic translation initiation factor 2α
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- IRE1
- inositol requiring enzyme 1
- NaI
- iodine
- PBA
- 4-phenylbutyric acid
- Tg
- thyroglobulin
- THAP
- thapsigargin
- TUDCA
- tauroursodeoxycholic acid
- UPR
- unfolded protein response
- XBP
- X-box binding protein-1.
References
- 1. Fuster V, Ryden LE, Cannom DS, et al. 2011 ACCF/AHA/HRS focused updates incorporated into the ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines developed in partnership with the European Society of Cardiology and in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. J Am Coll Cardiol. 2011;57:e101–e198. [DOI] [PubMed] [Google Scholar]
- 2. Vassallo P, Trohman RG. Prescribing amiodarone: an evidence-based review of clinical indications. JAMA. 2007;298:1312–1322. [DOI] [PubMed] [Google Scholar]
- 3. Han TS, Williams GR, Vanderpump MP. Benzofuran derivatives and the thyroid. Clin Endocrinol (Oxf). 2009;70:2–13. [DOI] [PubMed] [Google Scholar]
- 4. Ahmed S, Van Gelder IC, Wiesfeld AC, Van Veldhuisen DJ, Links TP. Determinants and outcome of amiodarone-associated thyroid dysfunction. Clin Endocrinol (Oxf). 2011;75:388–394. [DOI] [PubMed] [Google Scholar]
- 5. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Amiodarone Trials Meta-Analysis Investigators. Lancet. 1997;350:1417–1424. [PubMed] [Google Scholar]
- 6. Yamazaki K, Mitsuhashi T, Yamada E, et al. Amiodarone reversibly decreases sodium-iodide symporter mRNA expression at therapeutic concentrations and induces antioxidant responses at supraphysiological concentrations in cultured human thyroid follicles. Thyroid. 2007;17:1189–1200. [DOI] [PubMed] [Google Scholar]
- 7. Bogazzi F, Bartalena L, Gasperi M, Braverman LE, Martino E. The various effects of amiodarone on thyroid function. Thyroid. 2001;11:511–519. [DOI] [PubMed] [Google Scholar]
- 8. Bogazzi F, Tomisti L, Bartalena L, Aghini-Lombardi F, Martino E. Amiodarone and the thyroid: a 2012 update. J Endocrinol Invest. 2012;35:340–348. [DOI] [PubMed] [Google Scholar]
- 9. Franklyn JA, Gammage MD. Treatment of amiodarone-associated thyrotoxicosis. Nat Clin Pract Endocrinol Metab. 2007;3:662–666. [DOI] [PubMed] [Google Scholar]
- 10. Osman F, Franklyn JA, Sheppard MC, Gammage MD. Successful treatment of amiodarone-induced thyrotoxicosis. Circulation. 2002;105:1275–1277. [PubMed] [Google Scholar]
- 11. Bogazzi F, Bartalena L, Brogioni S, et al. Color flow Doppler sonography rapidly differentiates type I and type II amiodarone-induced thyrotoxicosis. Thyroid. 1997;7:541–545. [DOI] [PubMed] [Google Scholar]
- 12. Bogazzi F, Bartalena L, Dell'Unto E, et al. Proportion of type 1 and type 2 amiodarone-induced thyrotoxicosis has changed over a 27-year period in Italy. Clin Endocrinol (Oxf). 2007;67:533–537. [DOI] [PubMed] [Google Scholar]
- 13. Schonberger J, Bauer J, Spruss T, et al. Establishment and characterization of the follicular thyroid carcinoma cell line ML-1. J Mol Med (Berl). 2000;78:102–110. [DOI] [PubMed] [Google Scholar]
- 14. Huber AK, Finkelman FD, Li CW, et al. Genetically driven target tissue overexpression of CD40: a novel mechanism in autoimmune disease. J Immunol. 2012;189:3043–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lombardi A, Ulianich L, Treglia AS, et al. Increased hexosamine biosynthetic pathway flux dedifferentiates INS-1E cells and murine islets by an extracellular signal-regulated kinase (ERK)1/2-mediated signal transmission pathway. Diabetologia. 2012;55:141–153. [DOI] [PubMed] [Google Scholar]
- 16. Kuznetsov G, Bush KT, Zhang PL, Nigam SK. Perturbations in maturation of secretory proteins and their association with endoplasmic reticulum chaperones in a cell culture model for epithelial ischemia. Proc Natl Acad Sci USA. 1996;93:8584–8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. [DOI] [PubMed] [Google Scholar]
- 19. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. [DOI] [PubMed] [Google Scholar]
- 20. Kim PS, Bole D, Arvan P. Transient aggregation of nascent thyroglobulin in the endoplasmic reticulum: relationship to the molecular chaperone, BiP. J Cell Biol. 1992;118:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Medeiros-Neto G, Kim PS, Yoo SE, et al. Congenital hypothyroid goiter with deficient thyroglobulin. Identification of an endoplasmic reticulum storage disease with induction of molecular chaperones. J Clin Invest. 1996;98:2838–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Di Jeso B, Formisano S, Ulianich L. Perturbation of cellular calcium delays the secretion and alters the glycosylation of thyroglobulin in FRTL-5 cells. Biochem Biophys Res Commun. 1997;234:133–136. [DOI] [PubMed] [Google Scholar]
- 23. Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658. [DOI] [PubMed] [Google Scholar]
- 24. Werner ED, Brodsky JL, McCracken AA. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc Natl Acad Sci USA. 1996;93:13797–13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. [DOI] [PubMed] [Google Scholar]
- 26. D'Ascia SL, D'Ascia C, Marino V, et al. Cardiac resynchronisation therapy response predicts occurrence of atrial fibrillation in non-ischaemic dilated cardiomyopathy. Int J Clin Pract. 2011;65:1149–1155. [DOI] [PubMed] [Google Scholar]
- 27. Cairns JA. Antiarrhythmic therapy in the post-infarction setting: update from major amiodarone studies. Int J Clin Pract. 1998;52:422–424. [PubMed] [Google Scholar]
- 28. Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8:663–674. [DOI] [PubMed] [Google Scholar]
- 29. Nagaraju K, Raben N, Loeffler L, et al. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci USA. 2000;97:9209–9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mear JP, Schreiber KL, Munz C, et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol. 1999;163:6665–6670. [PubMed] [Google Scholar]
- 31. Purcell AW, Todd A, Kinoshita G, et al. Association of stress proteins with autoantigens: a possible mechanism for triggering autoimmunity? Clin Exp Immunol. 2003;132:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blass S, Union A, Raymackers J, et al. The stress protein BiP is overexpressed and is a major B and T cell target in rheumatoid arthritis. Arthritis Rheum. 2001;44:761–771. [DOI] [PubMed] [Google Scholar]
- 33. Bertolotti A, Wang X, Novoa I, et al. Increased sensitivity to dextran sodium sulfate colitis in IRE1β-deficient mice. J Clin Invest. 2001;107:585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seimon TA, Wang Y, Han S, et al. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Invest. 2009;119:886–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin W, Bailey SL, Ho H, et al. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest. 2007;117:448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamasaki S, Yagishita N, Tsuchimochi K, Nishioka K, Nakajima T. Rheumatoid arthritis as a hyper-endoplasmic-reticulum-associated degradation disease. Arthritis Res Ther. 2005;7:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–719. [DOI] [PubMed] [Google Scholar]
- 38. Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007;87:1377–1408. [DOI] [PubMed] [Google Scholar]
- 39. Kim IY, Kang YJ, Yoon MJ, et al. Amiodarone sensitizes human glioma cells but not astrocytes to TRAIL-induced apoptosis via CHOP-mediated DR5 upregulation. Neuro Oncol. 2011;13:267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant α1-antitrypsin (α1-AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in α1-AT deficiency. Proc Natl Acad Sci USA. 2000;97:1796–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ozcan L, Ergin AS, Lu A, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51. [DOI] [PubMed] [Google Scholar]
- 42. Xiao C, Giacca A, Lewis GF. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and β-cell dysfunction in humans. Diabetes. 2011;60:918–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Erbay E, Babaev VR, Mayers JR, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med. 2009;15:1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim PS, Lee J, Jongsamak P, et al. Defective protein folding and intracellular retention of thyroglobulin-R19K mutant as a cause of human congenital goiter. Mol Endocrinol. 2008;22:477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chiovato L, Martino E, Tonacchera M, et al. Studies on the in vitro cytotoxic effect of amiodarone. Endocrinology. 1994;134:2277–2282. [DOI] [PubMed] [Google Scholar]
- 46. Di Matola T, D'Ascoli F, Fenzi G, et al. Amiodarone induces cytochrome c release and apoptosis through an iodine-independent mechanism. J Clin Endocrinol Metab. 2000;85:4323–4330. [DOI] [PubMed] [Google Scholar]
- 47. Gupta SS, Ton VK, Beaudry V, Rulli S, Cunningham K, Rao R. Antifungal activity of amiodarone is mediated by disruption of calcium homeostasis. J Biol Chem. 2003;278:28831–28839. [DOI] [PubMed] [Google Scholar]
- 48. Iannone P, Haupt E, Flego G, et al. Dronedarone for atrial fibrillation: the limited reliability of clinical practice guidelines. JAMA Intern Med. 2014;174:625–629. [DOI] [PubMed] [Google Scholar]
- 49. Kober L, Torp-Pedersen C, McMurray JJ, et al. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med. 2008;358:2678–2687. [DOI] [PubMed] [Google Scholar]
- 50. Cao SS, Kaufman RJ. Targeting endoplasmic reticulum stress in metabolic disease. Expert Opin Ther Targets. 2013;17:437–448. [DOI] [PubMed] [Google Scholar]
- 51. Collins AF, Pearson HA, Giardina P, McDonagh KT, Brusilow SW, Dover GJ. Oral sodium phenylbutyrate therapy in homozygous beta thalassemia: a clinical trial. Blood. 1995;85:43–49. [PubMed] [Google Scholar]
- 52. Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–1273. [DOI] [PubMed] [Google Scholar]