

Abstract
Context:
GH-secreting pituitary adenomas exhibit heterogeneous natural history ranging from small tumors to large aggressive adenomas.
Objective:
To rigorously classify an acromegaly patient cohort defined by clinical, radiological, histopathological, and outcome characteristics.
Design:
Cross-sectional study.
Setting:
Tertiary referral pituitary center.
Patients:
Subjects were selected from a pituitary tumor research registry that includes 1178 patients with pituitary disease. Cluster analysis was performed on 338 acromegaly patients.
Interventions:
None.
Main Outcome Measures:
Biochemically active disease with elevated IGF-1 levels at follow-up.
Results:
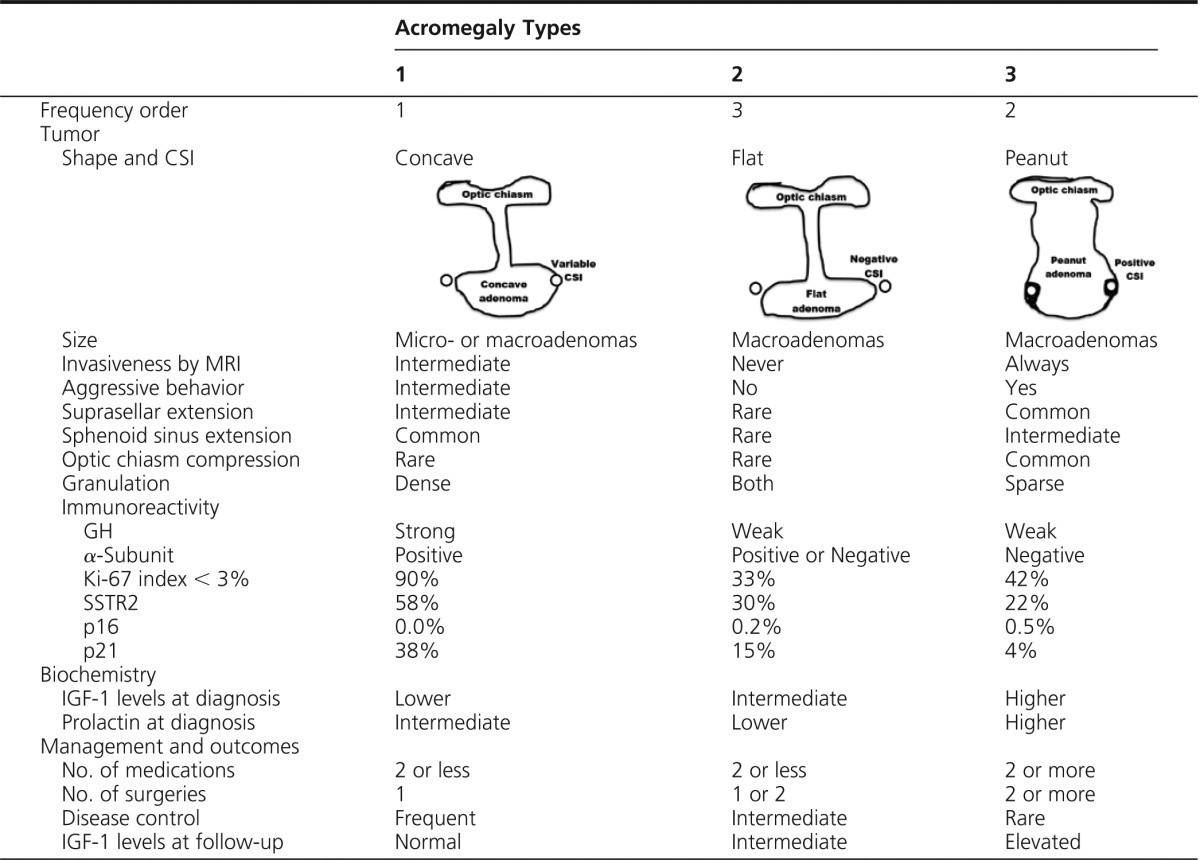
Cluster analysis of all patients yielded 292 who were rigorously classified to three acromegaly types. Type 1 (50%) comprised older patients with the longest follow-up and most favorable outcomes, characterized by densely granulated, nonaggressive microadenomas and macroadenomas. Type 1 tumors extend to the sphenoid sinus more frequently than suprasellar extension (concave tumor image) and express abundant immunoreactive p21 and somatostatin receptor 2. Type 2 (19%) comprised noninvasive, densely or sparsely granulated macroadenomas, without significant extension (flat tumor image), with intermediate biochemical outcome. Type 3 (31%) was characterized by sparsely granulated aggressive macroadenomas and comprised patients with adverse therapeutic outcomes, despite receiving more treatments. These tumors extend to both the sphenoid sinus and suprasellar regions with commonly encountered optic chiasm compression (“peanut” magnetic resonance image), with low tumor p21 and somatostatin receptor 2 expression.
Conclusions:
After validation, this classification may be useful to accurately identify acromegaly patients with distinctive patterns of disease aggressiveness and outcome, as well as to provide an accurate tool for selection criteria in clinical studies.
Acromegaly is a progressive disease, which, if untreated or suboptimally managed, inexorably leads to serious and incapacitating complications (1, 2). The disorder results from a pituitary somatotroph cell adenoma producing excess GH and subsequently IGF-1 (1). GH-secreting tumor behavior is heterogeneous and differs between patients; some patients may harbor small localized microadenomas, whereas others have large invasive macroadenomas (3). Although some patients seek medical attention shortly after symptoms start, most exhibit acromegaly symptoms for many years before diagnosis (4). Younger patients tend to have larger and more aggressive tumors that are diagnosed earlier, whereas older patients usually have smaller and less aggressive tumors (5). Age at diagnosis and disease duration appear to be determinants of disease outcome, likely reflecting exposure to high circulating levels of GH and IGF-1 (6–8). Magnetic resonance imaging (MRI) defines tumor size and extent of parasellar invasiveness, which is usually lateral toward the cavernous sinus and may encase the internal carotid artery (ICA). Dorsal expansion may impinge the optic chiasm, and ventral expansion extends to the sphenoid sinus (9, 10). Consequences of adenoma invasiveness render surgical procedures more difficult, resulting in adverse outcomes with higher residual tumor persistence or recurrence rates (3, 11–14).
About 50% of patients are partially or totally resistant to available somatostatin receptor ligands (SRLs) (15, 16). Somatostatin receptor (SSTR) 2 and SSTR5 receptor subtypes are usually expressed in GH-secreting adenomas, and approved SRLs bind preferentially to SSTR2 and, to a lesser extent, SSTR5 (17). Treatment resistance correlates inversely with SSTR2 abundance and may also be associated with heterogeneous SSTR type expression or signaling defects (15, 18). GH-producing pituitary adenomas are classified morphologically according to dense or sparse granulation patterns (5, 19). Larger tumors with low SSTR2a expression are more often sparsely granulated, exhibit no positivity or weak positivity for GH, and are generally more invasive (20–22). Accordingly, densely granulated adenomas, with higher SSTR2 expression, appear to exhibit a more favorable SRL response (18, 23).
GH-secreting pituitary adenoma aggressiveness is associated with dysregulated cell proliferation, particularly through the p53/p21 senescence pathway. Cyclin-dependent kinase inhibitors (CDKIs) may separately or coordinately deregulate pituitary tumor cell cycle progression (24–27). p21, a CDKI, is overexpressed in approximately 70% of GH-secreting tumors and triggers irreversible cell cycle arrest or senescence (24).
We therefore sought to classify acromegaly patients by clinical, radiological, and histopathological determinants and identified three types with significantly distinguished tumor structural-functional characteristics and treatment responsiveness. Rigorously classifying acromegaly types enables association of defined disease cohort clusters with clinical outcomes.
Patients and Methods
Study design
Subject records were reviewed from a computerized pituitary tumor research registry for which patients provided informed consent (28). The Cedars-Sinai Pituitary Tumor Research Registry comprises tertiary and quaternary referrals, including 1178 patients, 338 (29%) of whom have acromegaly (164 males, 174 females). Acromegaly diagnosis was confirmed with MRI evidence of adenoma with elevated IGF-1 or nonsuppressed GH levels in response to an oral glucose tolerance test (OGTT), or both.
Clinical characteristics before cluster analysis
Assessment of tumor size at presentation of 338 patients showed 253 (75%) macroadenomas, 54 (16%) microadenomas, and 31 (9%) not recorded. A total of 307 (91%) patients underwent surgery, including 58 (17%) undergoing multiple surgical interventions. Fifty-six (16%) subjects received radiation therapy (33 females/23 males). A total of 184 (54%) patients (88 males/96 females) were treated medically, including (% females/% males): 8.3/9.1, D2 dopamine agonists (D2DAs); 40.6/33, somatostatin analogs (SRLs); 0/1.1 pegvisomant (PEG); and 51/56.8, combined therapy. Combined therapy was recorded when two or more treatments were used concurrently.
Biochemical evaluations before cluster analysis
Fasting GH (number of subjects at diagnosis/follow-up, 204 [60%]/225 [67%]), nadir GH levels after 75-g OGTT (119 [35%]/138 [41%]), and age- and gender-adjusted IGF-1 levels (242 [71%]/274 [81%]) were established in the 338 patients. GH and IGF-1 assays used were as described (29). Control of disease activity was defined as random GH < 1 μg/L or nadir GH < 0.4 μg/L after OGTT and normal age-adjusted IGF-1 levels (30). Elevated IGF-1 was defined as increased age-adjusted IGF-1 levels at the last available evaluation. Percentages above the upper limit of normal (%ULN) for age- and gender-matched IGF-1 levels were calculated.
Imaging
Of the 338 patients, 307 radiological images were available for analysis from original reports. Tumors were classified as microadenoma (<10 mm) or macroadenoma (≥10 mm) and assessed for cavernous sinus invasion (CSI), ICA encasement, and suprasellar and sphenoid extension with MRI. Invasiveness was derived from the registry and defined using Knosp criteria (9). Suprasellar extension and sphenoid sinus extension were categorized separately. Tumor volume was calculated using the formula: 0.5 × width × length × height (mm3).
Immunohistochemistry
Pituitary tissue was obtained after written consent was given by registry patients (IRB nos. 2873 and 21964). Pathology review used one or two cores (1.2 mm) collected per tissue block. Control tissues included normal pituitaries, tubulovillous colon adenomas, non-small cell lung cancer, lung adenocarcinoma, brain and spinal melanoma, pheochromocytoma, metastatic carcinoid, breast fibroadenoma, and cancer. After deparaffinization and antigen retrieval, tissues were incubated with 3% hydrogen peroxide and blocked. For GH staining, slides were pretreated on Ventana Benchmark Ultra boards at high pH, followed by incubation with GH antibody (Predilute; Ventana Medical Systems, Inc) and detection with Ventana Ultraview DAB Kit. To detect SSTR2, we used antibodies from Epitomics (1:300; catalog no. 3582-1), and to detect p16, the monoclonal antibody from CINTec (Ventana Predilute, clone E6H4) or anti-p21 (1:50; Cell Signaling, catalog no. 2947) at 4°C, blocked, and incubated with antirabbit HRP (Dako Envision) and DAB. Secondary antibody (MACH; Biocare Medical) was followed by visualization with chromogen (Vector Labs) and counterstained with Mayer's hematoxylin. Adenomas (n = 292; 86%) were categorized as sparsely (n = 118) or densely (n = 174) granulated GH-expressing tumors using CAM5.2 cytokeratin immunostaining (5). Seventy-seven percent of patients were naive to SRL treatment, with equal distribution between groups (P = .87). Slides were digitized by high-resolution Leica SCN400NF whole-slide scanner (Translational Research Core). Acquired color images were sequentially analyzed on an automated image analysis platform, and OptTMA software (Leica) was used to locate and circle TMA cores subjected to image segmentation (Tissue IA Optimizer; Leica) to identify single cell nuclei, cytoplasm, and cell membranes. Chromogen images (hematoxylin, FastRed, and DAB) deconvoluted from color images were used for biomarker quantification. Integrated compartmentalized signal intensity was thresholded to determine biomarker positivity. Respective percentages of negative and positive cells were exported for statistical analysis.
Statistical analyses
Normally distributed data were expressed as means ± standard deviation, and skewed variables were expressed using median and interquartile ranges. To identify acromegaly types, two-step model-based cluster analysis was performed. Baseline clinical, histopathological, and radiological characteristics of all acromegaly patients in the registry were assessed (Supplemental Figure 1). Schwarz's Bayesian Information Criterion was used as a likelihood-based measure to indicate which clustering model and number of clusters yielded a maximized fit with a minimal number of parameters (<5). Discriminant analysis tested the accurate classification of patients and overlap between groups. χ2, Mann-Whitney U, or Kruskal-Wallis tests were used as indicated. Kaplan-Meier analyses evaluated activity of acromegaly and elevated IGF-1 levels at follow-up between types. Log-rank test was used for statistical comparison. P value ≤.05 was considered significant using SPSS version 20.0 software (IBM Corp).
Results
Clinical, histopathological, and radiological characteristics of acromegaly patients in the registry were subjected to cluster analysis (Supplemental Figure 1). Tumor size (micro- or macroadenomas), invasiveness (CSI and internal carotid encasement by MRI), and granulation pattern (dense or sparse) were identified for the maximum significant likelihood for classifying 338 patients into three different groups (Table 1). Cluster analysis excluded 46 patients because of incomplete data for one or more of such variables (Supplemental Figure 1). The analyses subsequently selected patients' cluster membership and saved it as a new variable (“types”). Discriminant analysis confirmed that 91% of the original grouped cases were correctly classified, with 9% (n = 26) of cases overlapping (Figure 1A). The most common acromegaly type was number 1 (n = 147; 50%).
Table 1.
Characteristics of Three Acromegaly Types as Determined by Cluster Analysis
| Acromegaly Types |
P Value | |||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| Prevalence | 147 (50) | 55 (19) | 90 (31) | |
| Parameters used by cluster analysis | ||||
| Granulation pattern* | ||||
| Densely granulated | 147 (100) | 27 (49) | 0 (0.0) | <.001a |
| Sparsely granulated | 0 (0.0) | 28 (51) | 90 (100) | <.001a |
| Tumor size** | ||||
| Microadenoma | 55 (37) | 0 (0.0) | 0 (0.0) | <.001a |
| Macroadenoma | 92 (63) | 55 (100) | 90 (100) | <.001a |
| Invasiveness*** | ||||
| Negative | 92 (63) | 55 (100) | 0 (0.0) | <.001a |
| Positive | 55 (37) | 0 (0.0) | 90 (100) | <.001a |
| Clinical and biochemical features | ||||
| Age at diagnosis, y (means ± SD) | 53.2 ± 12 | 40.6 ± 8.9 | 31.9 ± 7.8 | .001b |
| Year of diagnosis (means ± SD) | 1996 ± 9 | 1998 ± 9 | 1996 ± 7 | .20b |
| Gender | ||||
| Males | 76 (52) | 25 (45) | 45 (50) | .86a |
| Females | 71 (48) | 30 (55) | 45 (50) | |
| Disease duration, y | ||||
| Before diagnosis | 7.0 (2.8–14.2) | 7.2 (2.3–10.8) | 3.7 (0.7–7.1) | <.0001c |
| After diagnosis | 4.9 (1.7–11.1) | 5.3 (2.5–8.2) | 4.5 (2.0–12.4) | .865c |
| Disease duration | 15.5 (8.9–25.0) | 13.0 (10.5–18.5) | 9.6 (4.5–17.3) | .001c |
| Diagnosis (serum) | ||||
| GH, ng/mL | 13.0 (7.7–29.4) | 15.6 (7.3–46.1) | 17.5 (7.2–36.3) | .83c |
| Nadir GH after OGTT | 12.0 (6.8–24.2) | 8.6 (4.8–30.8) | 13.8 (5.1–32.1) | .58c |
| IGF-1, ng/mL | 638 (385–876) | 683 (381–952) | 823 (491–1027) | <.0001c |
| IGF-1, %ULN | 297 (250–349) | 307 (264–349) | 395 (332–458) | .01c |
| Prolactin, ng/mL | 13.0 (7.5–24.1) | 13.2 (8.5–24.0) | 22.0 (10.7–54.1) | .01c |
| Calcium, mg/dL | 9.6 (9.3–10.0) | 9.6 (9.5–9.9) | 9.5 (9.4–9.7) | .78c |
Data are expressed as number (percent) or median (interquartile range), unless described otherwise. %ULN represents the upper normal limit for age and gender IGF-1 levels.
GH-positive granules (5).
Microadenoma (<10 mm) and macroadenoma (≥10 mm).
CSI and/or ICA encasement observed on MRI using Knosp criteria (9).
P value from linear by linear association χ2.
One-way ANOVA test.
Kruskal-Wallis H test; P value <.05; Mann-Whitney U test between type 1 or 2 vs type 3.
Figure 1.

A, Discriminant canonical function scatter plot of acromegaly types showing 95% of patients classified without overlap between groups. B–G, GH-secreting adenoma invasiveness, extension, and size evaluated in patients with acromegaly types 1 (white), 2 (gray), and 3 (black). *, P < .01; **, P < .001. H–L, Adenoma histopathological characteristics evaluated in patients with acromegaly types 1 (white), 2 (gray), and 3 (black). Ki-67, n = 40; p16, p21, and SSTR2 expression, n = 27. Percentage represents (number of positive/number of total cells) × 100. *, P < .05; **, P < .001. M and N, p21 immunoreactivity in type 1 (M) and type 3 (N). O and P, SSTR2 immunoreactivity in type 1 (O) and type 3 (P). Error bars represent 95% confidence interval.
Clinical and biochemical parameters in clustered patients
Gender distribution was similar between groups. Year of diagnosis was similar in all groups; however, age at diagnosis was significantly higher in type 1 vs type 3 (P = .001; Table 1). Types 1 and 2 exhibited longer disease duration before diagnosis, suggesting less symptomatology (P < .0001). After diagnosis was established, acromegaly types showed similar disease duration (Table 1). Baseline and nadir GH levels were similar between groups at diagnosis. IGF-1 levels at diagnosis were significantly higher in type 3 (P < .0001). Prolactin was significantly higher (P = .01) in type 3 (Table 1).
Tumor size in clustered patients
Of the 292 clustered patients, 237 (81%) harbored macroadenomas, and 55 (19%) harbored microadenomas. All patients with microadenoma were classified as type 1, with a mean tumor size of 6 ± 2.5 mm. Fifty percent, 19%, and 31% of macroadenomas were distributed in types 1, 2, and 3, respectively (P < .001; Table 1). With micro- and macroadenomas considered together, tumor volumes were significantly smaller in type 1. Also, macroadenomas were statistically larger in type 2 > type 3 > type 1 (all P < .01; Figure 1B).
Tumor invasiveness in clustered patients
No type 2 patients harbored invasive macroadenomas (P < .001; Table 1). In contrast, all type 3 tumors were invasive macroadenomas (P < .001), whereas 37% of type 1 patients had invasive tumors. Distribution of CSI and ICA encasement is depicted in Figure 1, C and D. Type 1 comprises fewer aggressive tumors than type 3, with a higher proportion of patients without CSI (n = 44 [52%] vs n = 0 [0%]; P < .0001) and without ICA encasement (n = 54 [74%] vs n = 10 [24%]; P < .001).
Suprasellar extension and optic chiasm compression were more common in type 3 at diagnosis and follow-up (all P < .001; Figure 1, E and F). Sphenoid sinus extension at diagnosis was significantly more prevalent in types 1 and 3 (P = .004; Figure 1G). Compared to type 3, residual sphenoid sinus extension at follow-up was more common in type 1 (Figure 1G). Cases without suprasellar (Figure 1E) and sphenoid sinus extension (Figure 1G) were encountered particularly frequently in type 2 patients.
Histopathological characteristics in clustered patients
All tumors classified as type 1 acromegaly were densely granulated, and all type 3 tumors were sparsely granulated (Table 1). Type 2 was characterized by a similar proportion of densely and sparsely granulated tumors (Table 1). Positive GH (n = 147 [100%]; P < .0001; Figure 1H) and positive α-subunit (n = 42 [51%]; P = .01; Figure 1I) immunoreactivity were frequent type 1 findings. The only two cases with undetected GH immunoreactivity were encountered in type 3. No significant difference was identified for prolactin immunoreactivity between groups. A linear association was found with Ki-67 proliferation index between groups. Ki-67 proliferation index (n = 40) was <3% in 89% (n = 17) of type 1 adenomas (Figure 1J). In contrast, 67 and 58% of type 2 and type 3 patients, respectively, showed Ki-67 index > 3% (P for trend = .004; Figure 1J).
Immunohistochemistry for SSTR2, p21, and p16
p16 immunoreactivity (n = 27) was undetectable or low in all acromegaly types (median [interquartile range] of the proportion of positive cells in acromegaly types 1, 2, and 3 were 0.0 [0–0.9], 0.2 [0–1.5], and 0.5% [0.1–1.4], respectively; P = .45). p21 (n = 27), however, was expressed in all acromegaly tumor types but was significantly more abundant in type 1 than in the other two types (38% [29–51], 15% [5–59], and 4% [2–48], respectively; P = .02; Figure 1, K–N). Type 1 showed the highest proportion of cells with positive SSTR2 staining (n = 27) (58% [34–90], 30% [10–52], and 22% [5.9–46.6], respectively; P = .04; Figure 1, L–P).
Treatments in clustered patients
Surgery
A total of 277 patients underwent at least one surgery to resectable GH-producing adenoma. In 15 cases, operative information was not available. In the 277 surgeries performed, 80% of patients were operated on once, 17% twice, and 3% three or more times. Eighty-eight percent of type 1 patients required one surgery, whereas all subjects who required three or more surgeries were classified as type 2 or 3 (P = .006; Table 2).
Table 2.
Treatment Administered to Acromegaly Patients Classified by Types
| Acromegaly Types |
P Value | |||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| Surgery, n | 137 | 51 | 89 | .006a |
| 1 | 121 (88) | 38 (74.5) | 63 (71) | |
| 2 | 16 (12) | 12 (23.5) | 18 (20) | |
| 3 or more | 0 (0) | 1 (2) | 8 (9) | |
| Radiotherapy, n | 139 | 53 | 87 | .02a |
| Irradiated cases | 23 (16.5) | 9 (17) | 20 (23) | |
| Medical treatments, n | 89 | 32 | 62 | |
| None | 4 (4.5) | 1 (3.1) | 1 (1.6) | .32b |
| Somatostatin analogs (SRLs) | 71 (80) | 27 (84) | 58 (93.5) | .02b |
| D2DAs | 47 (53) | 12 (37.5) | 36 (58) | .62b |
| PEG | 11 (12) | 6 (19) | 17 (27) | .02b |
| Combined | 53 (59) | 16 (50) | 47 (77) | .01b |
| SRLs, D2DA, PEG1 | .006b | |||
| 1 group medication | 46 (52) | 18 (56) | 23 (37) | |
| 2 groups medications | 34 (38) | 12 (37.5) | 26 (42) | |
| 3 groups medications | 5 (6) | 1 (3) | 12 (19) | |
| No. of combined treatments received2 | 145 | 55 | 89 | <.0001a |
| 1 | 56 (39) | 20 (36) | 22 (25) | |
| 2 | 38 (26) | 15 (27) | 17 (19) | |
| 3 | 30 (21) | 12 (22) | 21 (24) | |
| 4 | 14 (10) | 7 (13) | 18 (20) | |
| 5 or more | 7 (5) | 1 (2) | 11 (12) | |
| Follow-up | ||||
| GH, ng/mL | 2.2 (1.0–5.1) | 1.8 (0.6–4.3) | 3.7 (1.1–8.6) | .02c |
| Nadir GH after OGTT, ng/mL | 1.0 (0.2–2.9) | 0.8 (0.4–2.6) | 1.5 (0.6–5.0) | .11c |
| GH after OGTT, n | 65 | 30 | 31 | .04e |
| >0.4 ng/mL | 44 (68) | 20 (67) | 27 (87) | |
| IGF-1 (ng/mL) | 221 (134–324)d | 234 (145–369)d | 370 (193–637) | <.0001c |
| IGF-1 levels (%ULN) | 99 (81–117) | 93 (78–107) | 137 (94–180) | .04b |
| IGF-1 levels, n | 129 | 53 | 81 | .004a |
| Not elevated | 88 (68) | 33 (62) | 39 (48) | |
Data are expressed as number (percent) or median (interquartile range), unless described otherwise.
Linear by linear association χ2.
Fisher exact χ2 test.
Kruskal-Wallis H test.
P value <.05, Mann-Whitney U test between type 1 or 2 vs type 3.
P value from Fisher's χ2 exact test.
Number of medical treatments used throughout follow-up, not necessarily together (SRLs and/or D2DAs and/or PEG).
Number of treatments used throughout follow-up (SRLs, and/or D2DAs, and/or PEG, and/or number of surgeries, and/or radiotherapy).
Radiotherapy
Fifty-two clustered patients received radiotherapy (18%), 31 conventional radiotherapy, and 21 radiosurgery (16 stereotactic and five gamma knife). All receiving gamma knife radiosurgery had macroadenomas, except for one case. Radiotherapy was most frequently administered to type 3 patients (P = .02; Table 2).
Medical treatment
Sixty-three percent of patients (n = 183) received medications to control acromegaly activity. Octreotide, octreotide LAR, or lanreotide autogel/depot was used in 156 (85%) patients, bromocriptine in 66 (36%), cabergoline in 43 (23.5%), PEG in 34 (19%), and octreotide infusion in one (1%). D2DAs were used as monotherapy only in type 1 patients. PEG was rarely used as monotherapy, and combined treatment was more commonly employed in type 3 patients (77%; P = .01; Table 2). Medication resistance was more commonly encountered in type 3, and although medications were not necessarily administered together, these patients received two or three different groups of medications (P = .006; Table 2).
Number of combined treatments received
Type 1 (86%) and type 2 (85%) patients received three or fewer treatment modalities in comparison to type 3 patients, who required three or more treatments in more than half of the cases (P < .0001; Table 2). Two patients received six treatment modalities, and one patient required seven, all categorized as type 3.
Acromegaly types and outcomes
The hazard ratio for active disease at follow-up and the hazard ratio of elevated age-adjusted serum IGF-1 levels at follow-up were analyzed using estimated duration of disease. Type 1 patients exhibited more favorable outcomes with controlled disease and serum IGF-1 levels at follow-up (Figure 2 and Table 3). In contrast, type 3 patients showed less favorable outcomes with an increased hazard for active disease and elevated IGF-1 levels at follow-up, despite more treatment modalities administrated in such patients. Type 1 patients had a median of 16 years with controlled disease, whereas type 2 and type 3 patients had a median of 13 and 9.7 years, respectively (P < .0001; Tables 3 and 4). We analyzed the independent role of disease duration with the probability for active disease or elevated IGF-1 levels at follow-up. Logistic regression analysis showed that disease duration per se was not independently associated with primary outcomes (Supplemental Table 1).
Figure 2.

A–D, Cumulative survival probability and hazard ratio for active disease (A and B) and elevated IGF-1 levels (C and D) at last documented follow-up in patients with acromegaly types 1 (black line), 2 (gray line), and 3 (dotted line).
Table 3.
Controlled Disease Duration and Normal IGF-1 Levels Grouped by Acromegaly Types
| Type | Disease Duration, y |
|||
|---|---|---|---|---|
| n (%) |
After Diagnosis, Median (95% Confidence Interval) |
|||
| Controlled | Normal IGF-1 | Controlled | Normal IGF-1 | |
| 1 | 11 (13) | 79 (74) | 16 (13–19) | 39 (33–44) |
| 2 | 2 (5) | 26 (61) | 13 (11–16) | 28 (10–46) |
| 3 | 1 (2) | 36 (49) | 9.7 (7–13) | 18 (12–24) |
| Overall | 14 | 141 | 14 (12–16) | 30 (25–35) |
| P value | <.001 | <.001 | ||
Table 4.
Summary of Acromegaly Classification by Types
Discussion
Pituitary tumors exhibit heterogeneous behavior ranging from very small tumors, which may be difficult to detect and have limited biochemical activity, to large, very aggressive, highly active adenomas (3). Importantly, GH-secreting tumors are almost invariably benign (31). Cell cycle disruption leads to pituitary tumor growth, and several mechanisms may underlie the constraint of pituitary adenoma growth from malignant transformation (24, 32). Aneuploidy and DNA damage lead to p53 pathway induction, and in pituitary adenomas, p53 induces CDKIs such as p21 (24), which leads to irreversible cell cycle arrest (33). In GH-secreting tumors, abundant p21 expression is lost in rarely encountered carcinomas (24). Acromegaly clinical outcome markers include GH or IGF-1 levels at follow-up and mortality (6, 8, 34, 35). Other factors associated with outcomes include age at diagnosis; disease duration; prior radiotherapy administration (6, 7, 34); radiological parameters such as tumor size, CSI, and ICA encasement (9, 10); and histopathological factors including granulation and SSTR2 expression (18, 20, 21). Such characteristics have been associated with differing treatment responsiveness with impacts on survival (35).
Clinical, radiological, and histopathological characteristics derived from our registry (28) were used to classify three acromegaly types distinguished by tumor aggressiveness and treatment responsiveness, expression profile of somatotroph surface receptors and markers of cell senescence, and disease outcomes. However, these patients were selected from a tertiary referral database, likely comprising more complex patients than usually encountered in a primary endocrinology clinic, but reflective of similar analysis of pituitary tumor natural history for treatment outcomes in a tertiary center (14). Nevertheless, this classification must be validated in other clinical settings.
Type 1 patients comprised older patients with the longest follow-up, with densely granulated, nonaggressive micro- and macroadenomas. Tumors extended to the sphenoid sinus more frequently than the suprasellar region, and when suprasellar extension occurred, optic chiasmic compression was rarely encountered (Figure 1F). Therefore, we propose that this tumor type be identified as a concave MRI shape (Table 4). Because tumor extension occurs mainly to the sphenoid sinus, these tumors are more accessible for debulking and likely explain why only one surgical procedure was needed in most of these patients (Table 2). IGF-1 levels at diagnosis are lower, and symptoms are more discrete. Mass effects are less commonly seen, resulting in longer disease duration from symptom onset before a biochemical diagnosis is made. p16 expression was very low in all three types, consistent with previous reports showing specific lineage expression of p16 in gonadotroph cells (25). p21 immunoreactivity, however, was particularly high in type 1 adenomas, suggesting cell senescence features in these tumors that explain, at least in part, less aggressive tumor behavior of type 1 patients (24). A higher proportion of type 1 patients have Ki-67 index <3%, indicating lower proliferative activity (36). Expression of SSTR2 and treatment responsiveness were higher in type 1, and outcomes were more favorable in terms of lower hazard ratio for active disease at follow-up (Figure 2) and higher median number of years with normal IGF-1 levels (Table 3).
Type 2 patients comprised densely or sparsely granulated macroadenomas with no invasive features. Densely granulated adenomas in this group responded less effectively to treatments than type 1 patients. Interestingly, sparsely granulated tumors in acromegaly type 2 are not invasive. Although these tumors seem less aggressive, these patients have higher IGF-1 levels at diagnosis, and required more treatments than did type 1 patients. Based on our observations, we propose that these tumors may be identified with a flat MRI shape (Table 4). Abundance of SSTR2 and p21 immunoreactivity was intermediate, as were clinical outcomes. Thus, patients with acromegaly type 2 adenomas exhibit a particular phenotypic behavior not necessarily determined exclusively by granulation pattern.
Type 3 comprised more aggressive and sparsely granulated macroadenomas. These tumors extend to both the sphenoid sinus and suprasellar regions with commonly encountered optic chiasm compression. Therefore, we propose that these tumors be identified on MRI as a “peanut” or round shape (Table 4). Symptoms and mass effects are more severe, likely because type 3 patients exhibited shorter disease duration before biochemical diagnosis (3.7 y [0.7–7.1]) (Table 1). Prolactin levels were increased, but not tumor prolactin immunoreactivity, suggesting a consequence of stalk-section rather than a mixed somatotroph/lactotroph tumor component. The lower expression of α-subunit, p21, and SSTR2 may also reflect reversion to a less differentiated somatotroph cell, without cycle arrest and more aggressive tumor growth. Overall follow-up was also of shortest duration with controlled disease (Table 3). Mortality was not evaluated because of the inherent challenges of a cross-sectional study to accurately ascertain cause of death. However, tumor aggressiveness, enhanced disease activity, and the short time before and after diagnosis suggest that these patients experience an adverse survival. High tumor aggressiveness correlated with very low p21 expression (<10% of cells), and decreased SSTR2 expression likely led to attenuated treatment responsiveness (18).
Study limitations include the cross-sectional design, which allows describing associations and not cause-and-effect relationships. Also, follow-up information was not available for some parameters. Patients in the registry with insufficient documentation of tumor size, invasiveness, and/or granulation pattern could not be rigorously classified by cluster analysis (Supplemental Figure 1). Immunohistochemistry of small tissue samples may also not be wholly representative of the adenoma, and immunoreactivity may not be sufficiently sensitive to detect low-level gene expression. Because some markers (p16, p21, SSTR2, Ki-67) were not available for all subjects, these results require confirmation in larger groups. Strengths of the study include: the number of patients evaluated for this rare disorder in a single center, the robust statistical power and strong differences between groups observed, and also the multiple clinical parameters used to distinguish the types—in particular, the strong association observed after comparing the hazard ratio for active disease at follow-up (Figure 2). The significance of these distinguishing differences was also supported by immunohistochemistry results, suggesting unique pathophysiological mechanisms underlying the variation in aggressiveness and treatment responsiveness between groups.
This classification enables identification of acromegaly patients with distinctive patterns of disease aggressiveness and outcome and provides an accurate tool for selection criteria in clinical studies. Validation in other centers will be important.
Acknowledgments
D.C.-R. thanks the Department of Endocrinology, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico City, Mexico, for their support. The authors thank Grace Labrado for help with manuscript preparation.
This work was supported by National Institutes of Health (NIH) Grants CA 75979 (to S.M.) and DK085148 (to O.C.), the Doris Factor Molecular Endocrinology Laboratory, and the National Center of Advancing Translational Sciences Grant UL1TR000124. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Disclosure Summary: J.D.C. has consulted for Novartis, Ipsen, Chiasma, and Genentech. S.M. has consulted for Chiasma, Novartis, Pfizer, Ipsen, and Genentech. V.S.B. has consulted for Pfizer, Ipsen, and Chiasma. D.C.-R., O.C., A.G., and A.N.M. have nothing to disclose.
Footnotes
- CDKI
- cyclin-dependent kinase inhibitor
- CSI
- cavernous sinus invasion
- D2DA
- D2 dopamine agonist
- ICA
- internal carotid artery
- MRI
- magnetic resonance imaging
- OGTT
- oral glucose tolerance test
- PEG
- pegvisomant
- SRL
- somatostatin receptor ligand
- SSTR
- somatostatin receptor
- %ULN
- percentage above the upper limit of normal.
References
- 1. Melmed S. Medical progress: acromegaly. N Engl J Med. 2006;355:2558–2573. [DOI] [PubMed] [Google Scholar]
- 2. Annamalai AK, Webb A, Kandasamy N, et al. A comprehensive study of clinical, biochemical, radiological, vascular, cardiac, and sleep parameters in an unselected cohort of patients with acromegaly undergoing presurgical somatostatin receptor ligand therapy. J Clin Endocrinol Metab. 2013;98:1040–1050. [DOI] [PubMed] [Google Scholar]
- 3. Jane JA, Jr, Starke RM, Elzoghby MA, et al. Endoscopic transsphenoidal surgery for acromegaly: remission using modern criteria, complications, and predictors of outcome. J Clin Endocrinol Metab. 2011;96:2732–2740. [DOI] [PubMed] [Google Scholar]
- 4. Reid TJ, Post KD, Bruce JN, Nabi Kanibir M, Reyes-Vidal CM, Freda PU. Features at diagnosis of 324 patients with acromegaly did not change from 1981 to 2006: acromegaly remains under-recognized and under-diagnosed. Clin Endocrinol (Oxf). 2010;72:203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Melmed S, Braunstein GD, Horvath E, Ezrin C, Kovacs K. Pathophysiology of acromegaly. Endocr Rev. 1983;4:271–290. [DOI] [PubMed] [Google Scholar]
- 6. Holdaway IM, Rajasoorya RC, Gamble GD. Factors influencing mortality in acromegaly. J Clin Endocrinol Metab. 2004;89:667–674. [DOI] [PubMed] [Google Scholar]
- 7. Ayuk J, Clayton RN, Holder G, Sheppard MC, Stewart PM, Bates AS. Growth hormone and pituitary radiotherapy, but not serum insulin-like growth factor-I concentrations, predict excess mortality in patients with acromegaly. J Clin Endocrinol Metab. 2004;89:1613–1617. [DOI] [PubMed] [Google Scholar]
- 8. Biermasz NR, Dekker FW, Pereira AM, et al. Determinants of survival in treated acromegaly in a single center: predictive value of serial insulin-like growth factor I measurements. J Clin Endocrinol Metab. 2004;89:2789–2796. [DOI] [PubMed] [Google Scholar]
- 9. Knosp E, Steiner E, Kitz K, Matula C. Pituitary adenomas with invasion of the cavernous sinus space: a magnetic resonance imaging classification compared with surgical findings. Neurosurgery. 1993;33:610–617; discussion 617–618. [DOI] [PubMed] [Google Scholar]
- 10. Albarel F, Castinetti F, Morange I, et al. Outcome of multimodal therapy in operated acromegalic patients, a study in 115 patients. Clin Endocrinol (Oxf). 2013;78:263–270. [DOI] [PubMed] [Google Scholar]
- 11. Mamelak AN, Carmichael J, Bonert VH, Cooper O, Melmed S. Single-surgeon fully endoscopic endonasal transsphenoidal surgery: outcomes in three-hundred consecutive cases. Pituitary. 2013;16:393–401. [DOI] [PubMed] [Google Scholar]
- 12. Kreutzer J, Vance ML, Lopes MB, Laws ER., Jr Surgical management of GH-secreting pituitary adenomas: an outcome study using modern remission criteria. J Clin Endocrinol Metab. 2001;86:4072–4077. [DOI] [PubMed] [Google Scholar]
- 13. Starke RM, Raper DM, Payne SC, Vance ML, Oldfield EH, Jane JA., Jr Endoscopic vs microsurgical transsphenoidal surgery for acromegaly: outcomes in a concurrent series of patients using modern criteria for remission. J Clin Endocrinol Metab. 2013;98:3190–3198. [DOI] [PubMed] [Google Scholar]
- 14. Zada G, Woodmansee WW, Ramkissoon S, Amadio J, Nose V, Laws ER., Jr Atypical pituitary adenomas: incidence, clinical characteristics, and implications. J Neurosurg. 2011;114:336–344. [DOI] [PubMed] [Google Scholar]
- 15. Colao A, Auriemma RS, Lombardi G, Pivonello R. Resistance to somatostatin analogs in acromegaly. Endocr Rev. 2011;32:247–271. [DOI] [PubMed] [Google Scholar]
- 16. Kleinberg DL. Primary therapy for acromegaly with somatostatin analogs and a discussion of novel peptide analogs. Rev Endocr Metab Disord. 2005;6:29–37. [DOI] [PubMed] [Google Scholar]
- 17. Ben-Shlomo A, Melmed S. Pituitary somatostatin receptor signaling. Trends Endocrinol Metab. 2010;21:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brzana J, Yedinak CG, Gultekin SH, Delashaw JB, Fleseriu M. Growth hormone granulation pattern and somatostatin receptor subtype 2A correlate with postoperative somatostatin receptor ligand response in acromegaly: a large single center experience. Pituitary. 2013;16:490–498. [DOI] [PubMed] [Google Scholar]
- 19. Kiseljak-Vassiliades K, Shafi S, Kerr JM, Phang TL, Kleinschmidt-DeMasters BK, Wierman ME. Clinical implications of growth hormone-secreting tumor subtypes. Endocrine. 2012;42:18–28. [DOI] [PubMed] [Google Scholar]
- 20. Horvath E, Kovacs K. Pathology of acromegaly. Neuroendocrinology. 2006;83:161–165. [DOI] [PubMed] [Google Scholar]
- 21. Larkin S, Reddy R, Karavitaki N, Cudlip S, Wass J, Ansorge O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naive patients with somatotroph adenomas. Eur J Endocrinol. 2013;168:491–499. [DOI] [PubMed] [Google Scholar]
- 22. Mayr B, Buslei R, Theodoropoulou M, Stalla GK, Buchfelder M, Schöfl C. Molecular and functional properties of densely and sparsely granulated GH-producing pituitary adenomas. Eur J Endocrinol. 2013;169:391–400. [DOI] [PubMed] [Google Scholar]
- 23. Fougner SL, Casar-Borota O, Heck A, Berg JP, Bollerslev J. Adenoma granulation pattern correlates with clinical variables and effect of somatostatin analogue treatment in a large series of patients with acromegaly. Clin Endocrinol (Oxf). 2012;76:96–102. [DOI] [PubMed] [Google Scholar]
- 24. Chesnokova V, Zonis S, Kovacs K, et al. p21(Cip1) restrains pituitary tumor growth. Proc Natl Acad Sci USA. 2008;105:17498–17503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chesnokova V, Zonis S, Zhou C, et al. Lineage-specific restraint of pituitary gonadotroph cell adenoma growth. PLoS One. 2011;6:e17924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quereda V, Martinalbo J, Dubus P, Carnero A, Malumbres M. Genetic cooperation between p21Cip1 and INK4 inhibitors in cellular senescence and tumor suppression. Oncogene. 2007;26:7665–7674. [DOI] [PubMed] [Google Scholar]
- 27. Woloschak M, Yu A, Xiao J, Post KD. Frequent loss of the P16INK4a gene product in human pituitary tumors. Cancer Res. 1996;56:2493–2496. [PubMed] [Google Scholar]
- 28. Drange MR, Fram NR, Herman-Bonert V, Melmed S. Pituitary tumor registry: a novel clinical resource. J Clin Endocrinol Metab. 2000;85:168–174. [DOI] [PubMed] [Google Scholar]
- 29. Carmichael JD, Bonert VS, Mirocha JM, Melmed S. The utility of oral glucose tolerance testing for diagnosis and assessment of treatment outcomes in 166 patients with acromegaly. J Clin Endocrinol Metab. 2009;94:523–527. [DOI] [PubMed] [Google Scholar]
- 30. Giustina A, Chanson P, Kleinberg D, et al. Expert consensus document: a consensus on the medical treatment of acromegaly. Nat Rev Endocrinol. 2014;10:243–248. [DOI] [PubMed] [Google Scholar]
- 31. Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009;119:3189–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol. 2011;7:257–266. [DOI] [PubMed] [Google Scholar]
- 33. Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. [DOI] [PubMed] [Google Scholar]
- 34. Holdaway IM, Bolland MJ, Gamble GD. A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. Eur J Endocrinol. 2008;159:89–95. [DOI] [PubMed] [Google Scholar]
- 35. Dekkers OM, Biermasz NR, Pereira AM, Romijn JA, Vandenbroucke JP. Mortality in acromegaly: a metaanalysis. J Clin Endocrinol Metab. 2008;93:61–67. [DOI] [PubMed] [Google Scholar]
- 36. Jaffrain-Rea ML, Di Stefano D, Minniti G, et al. A critical reappraisal of MIB-1 labelling index significance in a large series of pituitary tumours: secreting versus non-secreting adenomas. Endocr Relat Cancer. 2002;9:103–113. [DOI] [PubMed] [Google Scholar]