

Abstract
Context:
T4-binding globulin (TBG), a protein secreted by the liver, is the main thyroid hormone (TH) transporter in human serum. TBG deficiency is characterized by reduced serum TH levels, but normal free TH and TSH and absent clinical manifestations. The inherited form of TBG deficiency is usually due to a mutation in the TBG gene located on the X-chromosome.
Objective:
Among the 75 families with X-chromosome-linked TBG deficiency identified in our laboratory, no mutations in the TBG gene were found in four families. The aim of the study was to identify the mechanism of TBG deficiency in these four families using biochemical and genetic studies.
Design:
Observational cohort, prospective.
Setting:
University research center.
Patients:
Four families with inherited TBG deficiency and no mutations in the TBG gene.
Intervention:
Clinical evaluation, thyroid function tests, and targeted resequencing of 1 Mb of the X-chromosome.
Results:
Next-generation sequencing identified a novel G to A variant 20 kb downstream of the TBG gene in all four families. In silico analysis predicted that the variant resides within a liver-specific enhancer. In vitro studies confirmed the enhancer activity of a 2.2-kb fragment of genomic DNA containing the novel variant and showed that the mutation reduces the activity of this enhancer. The affected subjects share a haplotype of 8 Mb surrounding the mutation, and the most recent common ancestor among the four families was estimated to be 19.5 generations ago (95% confidence intervals, 10.4–37).
Conclusions:
To our knowledge, the present study is the first report of an inherited endocrine disorder caused by a mutation in an enhancer region.
In humans, more than 99% of the total serum thyroid hormones (THs), T3 and its precursor T4, are bound to serum proteins: T4-binding globulin (TBG or Serpina7), transthyretin, and albumin. Of these proteins, TBG is the main TH transporter in human serum. TBG is synthesized by the liver; the mature molecule has 395 amino acids with four N-linked oligosaccharide units (1). The latter are responsible for the correct post-translational folding and the microheterogeneity of TBG when subjected to isoelectric focusing (IEF) (2, 3). The TBG promoter contains several transcription factor binding sites; among them, the hepatic nuclear factor (HNF) 1α and the HNF3α and HNF3β binding sites impart a strong liver-specific transcriptional regulation (4). Consistent with the location of the TBG gene on the long arm of the X-chromosome, familial TBG abnormalities are inherited as X-chromosome-linked traits and, based on the serum levels of TBG in hemizygous patients, manifest as complete TBG deficiency (TBG-CD), partial TBG deficiency, and TBG excess (1). Inherited TBG defects do not alter the metabolic state of the individual and do not cause thyroid disease. However, they produce alterations in total TH concentration in serum, whereas free THs remain unchanged; although the risk is low when free TH levels are measured, TBG defects may lead to inappropriate treatment and its associated complications when left unrecognized (5).
Of 75 families with TBG deficiency identified over 45 years in our laboratory, TBG gene mutations have been detected in 71 families. In four families (5%), no TBG gene alterations were identified despite sequencing all exons, introns, untranslated regions, and the promoter region of the TBG gene, covering a total of 9.2 kb, and screening for gross gene deletions and rearrangements. A plausible hypothesis is that the undetected mutations in such families could reside within a cis-regulatory element located at some distance from the TBG gene.
Enhancers often physically interact with target genes via chromatin looping at the promoter region; the latter leads to the activation of gene transcription by recruitment of transcription factors, RNA polymerase II, and by driving enhancer RNA (eRNA) synthesis (6, 7). It has been suggested that eRNAs can modulate gene expression through chromatin remodeling occurring at the promoter locus of the target gene. Accumulating evidence has recently shown that disruption of cis-regulatory regions is involved in human diseases (8).
Given the observed X-linked mode of inheritance in the four families of interest and the well-characterized candidate TBG gene, we used next-generation sequencing to analyze 1 Mb of the X-chromosome to address the potential for mutations in cis-regulatory elements of the TBG gene. In affected subjects from all four families with uncharacterized TBG deficiency, we found a G to A substitution within a 2.2-kb putative liver-specific cis-regulatory element located 20 kb downstream of the TBG gene. In vitro studies confirmed the enhancer function of this region and the reduced activity of the mutant enhancer when assayed upstream of the TBG gene promoter. To our knowledge, this is the first example that demonstrates a functional consequence for noncoding DNA variation on TBG expression.
Materials and Methods
Clinical data
The review boards of the University of Chicago approved the studies, and written consent was obtained from all subjects studied.
Thyroid function tests
Blood was collected locally and shipped for analysis in our laboratory. Total T4 (TT4), total T3 (TT3), total rT3, TSH, thyroglobulin, and antibodies to thyroglobulin and thyroperoxidase were measured as previously described (9). Free T4 (FT4) was measured by equilibrium dialysis (10) and TBG by an immunometric assay with a sensitivity of 3.0 to 3.5 μg/mL (0.055 to 0.065 μmol/L).
Isoelectric focusing
Samples mixed with buffer containing [125I]-T4 were electrophoresed at 5°C on ampholine-containing gels. To validate the presence of TBG in serum of hemizygous patients from families 1 and 2, excess unlabeled T4 was added to [125I]-T4.
DNA mutation analysis
DNA was isolated from peripheral blood leukocytes using QIAamp DNA Mini Kit (QIAGEN), followed by amplification of genomic DNA (gDNA) by PCR and direct sequencing.
X-chromosome sequencing of the surrounding region of the TBG gene
Target enrichment probes were designed to include the TBG locus, including all RefSeq annotated exons on the X-chromosome. To capture long-range regulatory elements of the TBG gene, the 740-kb region spanning the gene was tiled (build 36, chrX: 105007703-105750097). Barcoded sequencing libraries were constructed according to the Agilent Sure Select protocol. Libraries were sequenced on a HiSeq 2000 at the University of Chicago High Throughput Genome Analysis Core facility. Genotypes were called using the GATK Unified Genotyper. A custom annotation pipeline was used to define for each variant, type, derived allele frequency, and evolutionary sequence constraint. In a second analysis, variants were identified using a haplotype-aware Bayesian model implemented in FreeBayes (version 0.9.12) (11). Samples were separated according to gender, and variant calling was performed in each set separately to explicitly account for gender differences in ploidy. Reads with a mapping quality < 3 were excluded, and only bases with a base quality > 1 were considered. Finally, all sites that did not have strong evidence of being polymorphic were excluded.
Reporter and expression plasmids
A 105-bp fragment corresponding to the minimal promoter of the human TBG gene was amplified from gDNA (4) and cloned in a pGL3basic reporter vector, thus having the luciferase (Luc) driven by the minimal TBG promoter (MP-Luc construct).
To generate the plasmid carrying the wild-type enhancer upstream of the minimal TBG promoter in the MP-Luc, a 2.2-kb region corresponding to the human TBG gene enhancer was amplified from gDNA of a normal subject and cloned upstream of the minimal TBG promoter (EnWT-MP-Luc construct).
The plasmid carrying the mutant enhancer upstream of the MP-Luc (EnMut-MP-Luc construct) was similarly generated, the mutant enhancer being amplified from the gDNA of a hemizygous patient (I-1, family 1; Figure 1).
Figure 1.

Pedigrees of the four families with X-linked TBG deficiency. Results of thyroid function tests are aligned below each individual. Low values are marked in blue and high values in red. #, not measured. The slightly elevated TSH in subject III-7 of family 2 may represent mild hypothyroidism.
Cell culture, transfection, and Luc assay
Human hepatoblastoma-derived (HepG2) cells were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco).
Transient transfection was performed using Fugene 6 (Promega). Cells were harvested 48 hours after transfection, and the relative luciferase activity was measured using a Dual-Luciferase Assay System (Promega).
The same experiments were performed using Cos-7 and HeLa cells. Functional analysis was also performed in the presence of in vitro synthesized HNF1α.
Electrophoretic mobility shift assay
EMSA experiments were performed using the DIG Gel Shift Kit, second generation (Roche), and nuclear extracts from HepG2 cells. In vitro translated HNF1α was prepared according to the manufacturer's instructions for T7 TnT Quick Coupled Transcription/Translation System (Promega). Three different probes were designed, and the gel mobility shifts of the respective mutant probes were compared with that of the wild-type probe.
Expression of eRNA in HepG2 cells
RNA was isolated from HepG2 cells using TRIzol reagent (Invitrogen, Life Technologies). RNA samples were treated with recombinant DNase I, RNAse-free (Roche) and reverse transcribed to cDNA. To measure eRNA expression for this TBG enhancer (eTBG), primers were designed to amplify a 182-bp product including the mutation locus from the cDNA sample.
Silencing eRNA in HepG2 cells
Four RNAse H active antisense oligonucleotides (IDT Integrated DNA Technologies) were designed to silence the activity of eRNA in HepG2 cells. They were transfected with the antisense oligonucleotides using Lipofectamine 2000 (Life Technologies). Cells exposed to Lipofectamine without antisense oligonucleotides served as negative controls. Isolated RNA was reverse transcribed to cDNA, and TBG mRNA was measured by quantitative PCR.
Haplotype analysis
Thirteen short tandem repeats (STRs) spanning a total of 20 Mb surrounding the enhancer locus on the X-chromosome were identified using the Marshfield, Genethon, and deCODE genetic maps. DNA samples from four hemizygous males, one from each family, were used to directly sequence these regions and generate haplotypes. The shared haplotype was also examined by in silico analysis of the high throughput sequencing data.
Estimating the age of the mutation
The genetic length of the ancestral haplotype shared between the hemizygous males was used to estimate the time since their most recent common ancestor (MRCA). The Gamma method was applied using the high-density single nucleotide polymorphism (SNP) data surrounding the mutation (12).
Statistics
All results are expressed as mean ± SEM. Statistical analysis was performed using two-tailed Student's t test for unpaired observations. P ≥ .05 was considered to be nonsignificant.
Results
Thyroid function tests
The pedigrees and results of thyroid function tests for the four families are shown in Figure 1.
Family 1
The propositus (I-1), a 50-year-old man of Arab (Palestinian) origin, had an apparent phenotype of TBG-CD. His two daughters (II-3 and II-4) had half-normal TBG levels, compatible with a heterozygous state.
Family 2
The family was of Arab (Lebanese) origin. The serum TT4, TT3, and total rT3 concentrations of the propositus (II-2) and his brother (II-3) were low, whereas their TSH and FT4 levels were normal, and TBG was undetectable. The three daughters of the propositus also had low serum TT4 and TT3 concentrations and half-normal TBG levels.
Family 3
This family of Arab (Lebanese) origin was previously reported as having TBG deficiency without TBG gene mutation (13). The propositus (II-1) and his brother (II-2) had thyroid function tests typical of TBG-CD. His two sisters (II-3 and II-4) had half-normal TBG levels, indicating a heterozygous state. Interestingly, his mother (I-1) displayed a phenotype similar to that of her two affected sons, suggesting that she was homozygous for the TBG defect. At the time her blood sample was obtained, she was taking L-T4, explaining the low normal serum TSH level.
Family 4
This family was also previously reported (14). When investigated at the University of Chicago, the proposita (I-1) was on L-T4 150 μg/d. She presented with low serum TT4, TT3, and TBG levels but had high FT4 and suppressed TSH levels due to L-T4 overtreatment. Her identical twin sons (II-1 and II-2) also had low serum TT4 and TT3 and a TBG < 3 μg/mL (0.055 μmol/L), but normal serum TSH levels. The family lives in the United Kingdom, but the ethnic origin of the female proband was not reported to the authors.
Isoelectric focusing
Using IEF, a technique able to detect as little as 1% of serum TBG, samples from hemizygous affected individuals from families 1 and 2 were found to contain very small amounts of TBG with normal isoforms when compared to a patient known to have TBG-CD (Figure 2). To confirm that the bands represented labeled T4 bound to TBG, the IEF was repeated in the presence of excess unlabeled T4, which displaced the labeled T4, leading to the disappearance of the TBG bands.
Figure 2.
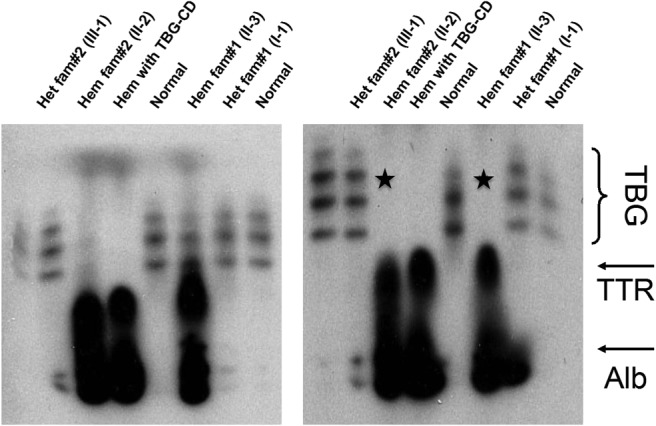
IEF gels showing the presence of small amounts of TBG with normal isoforms in hemizygous males. A, Note the presence of faint TBG isoform bands in serum of the hemizygotes of families 2 and 1 (subjects II-2 and II-3), compared to the total absence of TBG bands in the subject with TBG-CD. The excess labeled T4 not bound to TBG in the affected males is associated with transthyretin (TTR) and albumin (Alb). B, Addition of unlabeled T4 cleared the bands corresponding to the TBG isoforms in the samples from affected individuals of families 2 and 1 (lanes 3 and 6 counting from the left) and indicated by an asterisk, corroborating that the faint bands do correspond to TBG. Het, heterozygous; Hem, hemizygous.
Mutation identification and genotyping of members of the four families
The X-linked pattern of inheritance is noted in Figure 1. gDNA sequencing of all affected members of the four families failed to show any DNA alterations within 9.2 kb encompassing the entire coding regions of the TBG gene, all introns, a 2.7-kb region upstream of the ATG translation start codon and containing the TBG gene promoter, and 1 kb downstream of the TGA translation stop codon and containing the two polyadenylation sites (Supplemental Figure 1) (4, 15). The absence of mutations in the TBG gene, the X-linked transmission of TBG deficiency, and the presence in serum of small amounts of normal TBG prompted us to sequence approximately 1 Mb of the X-chromosome, including all annotated exons and the 740-kb region spanning the TBG locus. A total of 10 subjects (four affected males, four affected females, and two unaffected relatives) were sequenced (Supplemental Table 1). A total of 2363 SNPs and microindels were called; 23 rare variants were found to segregate with the phenotype in at least one family, and four variants (Supplemental Table 2) were concordant in all four families. Of these four variants, the G to A change located on chromosome-X:105138624 (GRCh36/hg18), here denoted as Xq22G>A, was not reported in public databases and was the only one with functional relevance (analyzed in the following section). In addition, it was not present in random DNA samples from subjects of Arab origin (150 alleles) and 189 alleles from six other ethnic groups. Sequencing of all 27 subjects belonging to the four families confirmed that the Xq22G>A change was present in all affected individuals and segregated with the phenotype (Figure 3).
Figure 3.

Genotyping of all four families for the G to A substitution in the TBG enhancer. Note that two females (I-1 Fam 3 and I-1 Fam 4) are apparently homozygous for the mutation.
Identification of a long-range enhancer in the mutant region
The Xq22G>A mutation is located 20 kb downstream of the TBG gene and 50 kb upstream of the Nik-related Kinase (NRK) gene (NG_021425). Several lines of evidence from ENCODE suggested that the region surrounding the mutation acts as a liver-specific enhancer, including: 1) the mutation resides within a 2.2-kb sequence predicted to be a “strong enhancer” by chromHMM (16) (Supplemental Figure 2); 2) the mutation overlaps a DNAse I hypersensitive region identified in HepG2 and Huh-7 cells; 3) the mutation overlaps ChIP-seq binding sites for nine factors in HepG2 cells; 4) the region is significantly constrained across vertebrates; and 5) the region is modestly transcribed in HepG2 cells. Additionally, data from ENCODE and GTEx demonstrate that the gene expression pattern of TBG and the putative enhancer are both liver specific; furthermore, the putative enhancer and the TBG gene are flanked by two predicted insulators.
Effect of the Xq22G>A mutation on enhancer activity
To assess whether the cis-regulatory region functions as an enhancer of the TBG gene, we transfected EnWT-MP-Luc and EnMut-MP-Luc into liver HepG2 cells, which express TBG and in which the putative enhancer is uniquely active (Figure 4). In HepG2 cells, the EnWT-MP-Luc construct increased the luciferase activity by 42-fold compared to the MP-Luc vector (P < .001). The EnMut-MP-Luc construct showed a 70% decrease (P < .001) compared to the EnWT-MP-Luc construct (Figure 4). These results confirm that the cis-regulatory region has the predicted enhancer properties and that the mutation reduces its activity on the TBG gene promoter.
Figure 4.
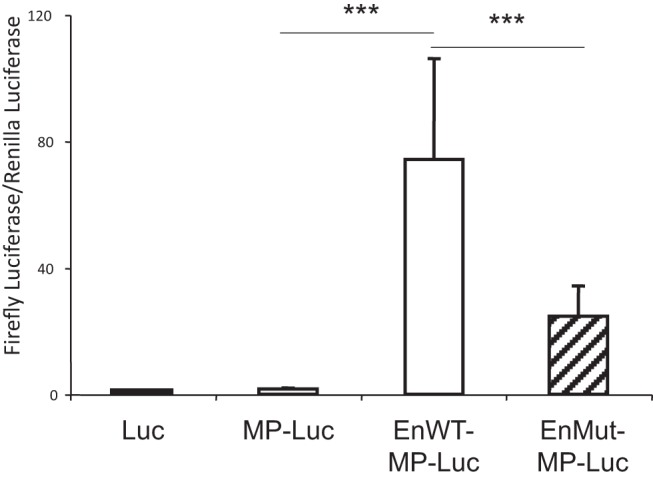
In vitro functional analysis of the mutant enhancer. HepG2 cells were transfected with the empty vector containing only luciferase (Luc), the minimal TBG promoter upstream of Luc (MP-Luc), and a 2-Kb sequence of the enhancer cloned upstream of the minimal promoter (MP). The latter constructs contained either the wild-type (EnWT-MP-Luc) or mutant (EnMut-MP-Luc) enhancers. Note the 42-fold increase in luciferase activity (normalized with renilla) stimulated by the wild-type enhancer and the attenuated stimulation by the mutant enhancer. Data are represented as the mean ± SEM. ***, P < .001.
Functional studies were also carried out in Cos-7 and HeLa cells transfected with the vectors described above. As previously demonstrated in non-liver-derived CV1 and CHO cells (4), transfection of MP-Luc increased the basal activity of the empty Luc vector by only 1- to 2-fold. This activity was increased one more fold by coexpressing HNF1α, presumably through the presence of a HNF1α binding site in the minimal promoter of the MP-Luc vector (4). However, the vectors containing the wild-type or mutant enhancers (EnWT-MP-Luc, EnMut-MP-Luc) showed no difference in Luc activity compared to the corresponding vectors containing the minimal promoter alone (MP-Luc) in the absence or presence of HNF1α (data not shown), indicating that additional liver-specific factors are responsible for the effect observed in HepG2 cells.
EMSA results
To further characterize the mechanism by which the mutation decreases the enhancer activity on TBG gene expression, we examined the effect of the mutation on the binding of HNF1α and HNF3β using probes which include their putative binding sites near the enhancer mutation, 55 bp downstream and 35 bp upstream, respectively. No consistent differences in the shifted bands formed with wild-type and mutant probes were observed using the HepG2 nuclear extracts or the in vitro synthesized HNF1α (data not shown).
eRNA expression and silencing in HepG2 cells
We next determined whether eRNA is transcribed from the enhancer DNA region of interest in HepG2 cells. We showed that eRNA is present in HepG2 cells, whereas no amplification was evident when the reaction was carried out in the absence of reverse transcriptase (Figure 5). We then blocked eRNA action by transfecting HepG2 cells with antisense oligonucleotides and quantified the expression of TBG gene by quantitative PCR. Silencing the eRNA did not appear to decrease TBG gene expression (data not shown).
Figure 5.
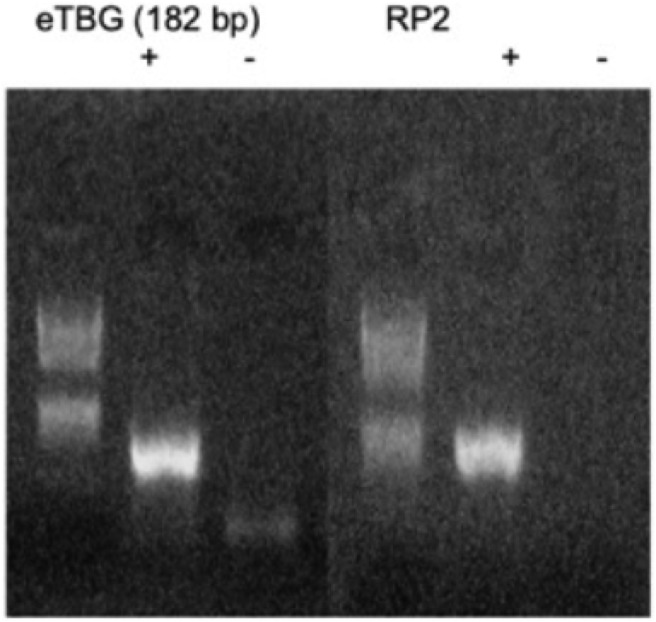
Expression of eRNA in the mutation locus in HepG2 cells. Representative image of a 3% agarose gel showing the expression of eRNA in the mutation locus in HepG2 cells (abbreviated eTBG). RNA was collected from HepG2 cells, treated with DNAse, precipitated with ethanol, and reverse transcribed to cDNA. A total of 200 ng of cDNA were used for the eTBG and 80ng for the RP2 (housekeeping gene) PCR. Plus sign denotes the presence and minus sign denotes the absence of reverse transcriptase during reverse transcription to control for gDNA contamination.
Haplotype analysis and estimation of the age of the mutation
Because all four families shared the identical Xq22G>A mutation and three of them had confirmed Arab origin, we hypothesized that they may have a common ancestor. Using the copy number of 13 STRs on the X-chromosome covering a total of 20 Mb surrounding the mutation, we generated the haplotype of the hemizygous males from each family and found that they share a DNA segment of 8 Mb (build g36, chrX: 100392537-108595208) (Figure 6). We also took advantage of SNP data available from deep sequencing to confirm the shared haplotype with in silico analysis. In addition, the mothers of the two hemizygous males from families 3 and 4 (I-1 in both pedigrees) presented with a clinical phenotype suggesting a homozygous state. We sequenced the STRs of the shared haplotype and inspected the SNP data from deep sequencing and found that they appear to have a stretch of homozygous calls.
Figure 6.
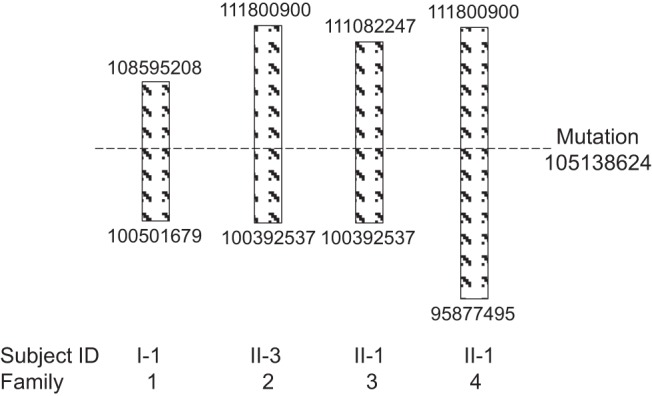
Shared haplotype common between four hemizygous males, one from each family (I-1 Fam 1, II-3 Fam 2, II-1 Fam 3, II-1 Fam 4). Numbers indicate left and right end positions of the shared haplotype in the physical map, in base pairs.
We used a recently described method of dating rare mutations, the Gamma method (12), which can be applied to small samples like ours and utilizes high-density SNP data to estimate the age of the MRCA. Assuming an independent genealogy, the MRCA of the four families was estimated to be 19.5 generations in the past [95% confidence interval, 10.4–37]. Assuming a correlated genealogy, ie, a tree-like genealogy, the MRCA and the propositi were estimated to be 17.8 generations apart (95% confidence interval, 7.7–42.8). Considering the length of a generation to be 20–25 years, it seems that the four families originated from a common ancestor 400–500 years ago.
Discussion
Inherited TBG deficiency is a well-characterized condition caused by mutations in the TBG gene. We present, herein, four families with a novel mutation causing X-linked TBG deficiency that resides not in the TBG gene, but rather in a region with enhancer properties. Although serum TBG was undetectable when measured by routine laboratory technique, using an assay with a detection limit of 3 μg/mL (0.055 μmol/L) cannot confirm complete TBG deficiency. By definition, complete TBG deficiency requires that serum TBG concentration in an affected hemizygous male be less than 0.005 μg/mL (0.0009 μmol/L) or 0.003% the average normal (17). Thus, we sought a more sensitive method to determine whether a small amount of TBG was present in serum. In fact, IEF was able to detect small amounts of TBG in the affected hemizygous males, which contained normal isoforms. This confirmed that the cause of TBG deficiency in these families does not involve the structure of the TBG gene, but rather represents a quantitative defect of TBG synthesis, pointing to an alteration in the control of gene expression. This rationale led to the deep sequencing of the 740-kb region encompassing the TBG locus in search of a defect in a regulatory element. We also sequenced all RefSeq annotated exons of the X-chromosome to assess for the possibility of a mutation in a protein located on the X-chromosome that is involved in the regulation of TBG synthesis.
Enhancers modulate their target gene promoters via chromatin remodeling and DNA looping. Still, many questions remain regarding their functional architecture. Valuable information has been gained from projects, such as the ENCODE, which has provided transcription factor binding data. For mammalian enhancers, the “enhanceosome” model has been proposed. In this model, the enhancers have several transcription factor binding sites and contain both activating and repressing domains. Appropriate spatial assembly of the recruited proteins is a condition sine qua non for enhancer activation to ensue, rendering these regions susceptible to mutations (18).
The in vitro assay in HepG2 cells provided strong evidence that the cis-regulatory region does have enhancer properties and that the mutation results in a significant decrease of the enhancer activity. Trying to elucidate the underlying mechanism, and based on the current knowledge of the function of enhancers, we speculated that the mutation might disrupt binding sites for transcription factors and thus decrease the activity of the TBG gene promoter. However, EMSA experiments were inconclusive. Considering the in vitro setup of EMSA, this result is not surprising. It is possible that in vivo, the architecture of the gDNA looping needed to bring the enhancer near the promoter and the sequential binding of the proteins required for this tethering may be affected.
An alternative hypothesis involves the possible role of noncoding eRNA on the TBG gene promoter activity. eRNAs have been shown to participate in the formation or stabilization of chromatin loops between enhancers and their target genes; their loss can significantly decrease target gene expression. However, this may occur without necessarily modifying chromatin looping (7, 19, 20). Although we demonstrated the expression of eRNA from the enhancer region in HepG2 cells, silencing it did not seem to decrease TBG gene expression, at least in HepG2 cells. Despite this finding, it would be interesting to fully characterize the eRNA transcribed from this region and understand its relevance in the enhancer complex.
To unveil the underlying mechanism that affects the activity of the Xq22G>A mutation, a future project could involve direct mutagenesis of the liver-specific HepG2 cell line, in which the single nucleotide substitution G>A would be introduced through the CRISPR-Cas9 system (21). This would allow precise characterization of the properties of the mutant knock-in cell line with regard to TBG gene expression, helping to identify the transcription factors and mechanisms involved in modulating TBG gene expression.
It was surprising that all four families with TBG deficiency, out of a total of 75 families evaluated over a period of 45 years, were found to harbor the same mutation in the liver-specific enhancer. Three families had confirmed Arab descent, whereas the ethnic origin of the fourth family (from the maternal side) remains unknown. Family 1 lives in the United States, family 2 in Germany, family 3 in Austria, and family 4 in the United Kingdom. Although there was no indication of a relation between them and no additional genealogical information was available, it was strongly suspected that these families may share a common ancestor, and the common Arab descent in three of them reinforced this possibility; based on the Palestinian and Lebanese origin of the three families, their MRCA would most likely be geographically located in the Middle East. Using STRs and SNP data from deep sequencing, the shared haplotype common among the four families was estimated to be 8 Mb flanking the mutation. Furthermore, the propositi and their MRCAs were estimated to be approximately 20 generations, ie, 400–500 years, apart. However, if one considers an earlier age of reproduction in the Arab population and thus a shorter generation length of approximately 15 years, then the MRCAs should be 300–375 years in the past.
Estimating the age of the mutation was complicated for several reasons. The mutation is novel and rare, the sample size is small, and genetic data for estimation of recombination rates on the X-chromosome is insufficient. This precludes the use of methods based on the frequency of the mutation, gene trees, and the linkage disequilibrium around the mutation for dating. The recently described Gamma method assumes that the mutation carriers will have inherited the mutation as a segment of an ancestral chromosome and that the length of this segment decreases over time. It also makes effective use of high-density SNP data, making it suitable in the present situation (12). One limitation that should be addressed, though, is that available SNP data in this study were not derived from whole genome sequencing and thus were not continuous in covering the entire X-chromosome. Another interesting point is that two mothers, in families 3 and 4, were homozygous for the Xq22G>A mutation with no indication of deletion of the respective region in one allele by sequencing and deep sequencing, which suggests some degree of inbreeding.
In conclusion, we report a novel mutation in four families with X-linked TBG deficiency, which resides not in the TBG gene, but rather in a cis-regulatory element with enhancer properties. To the best of our knowledge, this is the first demonstration of a congenital endocrine disorder caused by an enhancer mutation. Fully elucidating the mechanism of action of the novel mutation on TBG gene expression represents a very interesting area of future research.
Acknowledgments
This work was supported by Grants R37DK15070 and UL1TR000430 from the National Institutes of Health and the Seymour J. Abrams, Rabbi Morris Esformes, and Tivy Katz funds for thyroid research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes And Digestive And Kidney Diseases or the National Institutes of Health.
A.M.D. was supported by National Institutes of Health Grant F32 DK 91016.
Current address for R.E.W.: Department of Medicine, University of Miami Miller School of Medicine, 1120 NW 14th Street, R-761, Room 310, Miami, FL 33136.
Current address for A.M.F.: Istituto Oncologico Veneto (IOV-IRCCS), Via Gattamelata, 64 – Padova, Italy 35128.
* A.M.F. and T.P. contributed equally to this work.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- eRNA
- enhancer RNA
- eTBG
- enhancer TBG
- FT4
- free T4
- gDNA
- genomic DNA
- HNF
- hepatic nuclear factor
- IEF
- isoelectric focusing
- MRCA
- most recent common ancestor
- SNP
- single nucleotide polymorphism
- STR
- short tandem repeat
- TBG
- T4-binding globulin
- TBG-CD
- complete TBG deficiency
- TH
- thyroid hormone
- TT3
- total T3
- TT4
- total T4.
References
- 1. Refetoff S, Murata Y, Mori Y, Janssen OE, Takeda K, Hayashi Y. Thyroxine-binding globulin: organization of the gene and variants. Horm Res. 1996;45:128–138. [DOI] [PubMed] [Google Scholar]
- 2. Murata Y, Magner JA, Refetoff S. The role of glycosylation in the molecular conformation and secretion of thyroxine-binding globulin. Endocrinology. 1986;118:1614–1621. [DOI] [PubMed] [Google Scholar]
- 3. Grimaldi S, Bartalena L, Ramacciotti C, Robbins J. Polymorphism of human thyroxine-binding globulin. J Clin Endocrinol Metab. 1983;57:1186–1192. [DOI] [PubMed] [Google Scholar]
- 4. Hayashi Y, Mori Y, Janssen OE, et al. Human thyroxine-binding globulin gene: complete sequence and transcriptional regulation. Mol Endocrinol. 1993;7:1049–1060. [DOI] [PubMed] [Google Scholar]
- 5. Refetoff S, Selenkow HA. Familial thyroxine-binding globulin deficiency in a patient with Turner's syndrome (XO). Genetic study of a kindred. N Engl J Med. 1968;278:1081–1087. [DOI] [PubMed] [Google Scholar]
- 6. Marsman J, Horsfield JA. Long distance relationships: enhancer-promoter communication and dynamic gene transcription. Biochim Biophys Acta. 2012;1819:1217–1227. [DOI] [PubMed] [Google Scholar]
- 7. Plank JL, Dean A. Enhancer function: mechanistic and genome-wide insights come together. Mol Cell. 2014;55,:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Epstein DJ. Cis-regulatory mutations in human disease. Brief Funct Genomic Proteomic. 2009;8:310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ferrara AM, Onigata K, Ercan O, Woodhead H, Weiss RE, Refetoff S. Homozygous thyroid hormone receptor β-gene mutations in resistance to thyroid hormone: three new cases and review of the literature. J Clin Endocrinol Metab. 2012;97:1328–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nelson JC, Tomei RT. Direct determination of free thyroxin in undiluted serum by equilibrium dialysis/radioimmunoassay. Clin Chem. 1988;34:1737–1744. [PubMed] [Google Scholar]
- 11. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv. 2012;1207.3907 Available at: arxiv.org/pdf/1207.3907.pdf.
- 12. Gandolfo LC, Bahlo M, Speed TP. Dating rare mutations from small samples with dense marker data. Genetics. 2014;197:1315–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reutrakul S, Dumitrescu A, Macchia PE, Moll GW, Jr, Vierhapper H, Refetoff S. Complete thyroxine-binding globulin (TBG) deficiency in two families without mutations in coding or promoter regions of the TBG genes: in vitro demonstration of exon skipping. J Clin Endocrinol Metab. 2002;87:1045–1051. [DOI] [PubMed] [Google Scholar]
- 14. Narendran P, Lado-Abeal J, Moeller LC, Refetoff S. Partial thyroxine-binding globulin (TBG) deficiency in a family with no detectable mutation of the TBG gene. Clin Endocrinol (Oxf). 2003;59:824–825. [DOI] [PubMed] [Google Scholar]
- 15. Kambe F, Seo H, Murata Y, Matsui N. Cloning of a complementary deoxyribonucleic acid coding for human thyroxine-binding globulin (TBG): existence of two TBG messenger ribonucleic acid species possessing different 3′-untranslated regions. Mol Endocrinol. 1988;2:181–185. [DOI] [PubMed] [Google Scholar]
- 16. Hoffman MM, Ernst J, Wilder SP, et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41:827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Refetoff S. Inherited thyroxine-binding globulin abnormalities in man. Endocr Rev. 1989;10:275–293. [DOI] [PubMed] [Google Scholar]
- 18. Thanos D, Maniatis T. Virus induction of human IFN β gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091–1100. [DOI] [PubMed] [Google Scholar]
- 19. Lai F, Orom UA, Cesaroni M, et al. Activating RNAs associate with mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013;23:1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]