

Abstract
Objective
Genomic analyses from blood leukocytes have concluded that mouse injury poorly reflects human trauma at the leukocyte transcriptome. Concerns have focused on the modest severity of murine injury models, differences in murine compared to human age, dissimilar circulating leukocyte populations between species, and whether similar signaling pathways are involved. We sought to examine whether the transcriptomic response to severe trauma in mice could be explained by these extrinsic factors, by utilizing an increasing severity of murine trauma and shock in young and aged mice over time, and examining the response in isolated neutrophil populations.
Design
Pre-clinical controlled in vivo laboratory study and retrospective cohort study
Setting
Laboratory of Inflammation Biology and Surgical Science and multi-institution level 1 trauma centers
Subjects
6–10 week old and 20–24 month old C57BL/6 (B6) mice and two cohorts of 167 and 244 severely traumatized (ISS >15) adult (>18 yo) patients.
Interventions
Mice underwent one of two severity polytrauma models of injury. Total blood leukocyte and neutrophil samples were collected.
Measurements and Main Results
Fold expression changes in leukocyte and neutrophil genome-wide expression analyses between healthy and injured mice (p<0.001) were compared to human total and enriched blood leukocyte expression analyses of severe trauma patients at 0.5, 1, 4, 7, 14, and 28 days after injury (Glue Grant TRDB). We found that increasing the severity of the murine trauma model only modestly improved the correlation in the transcriptomic response with humans, whereas the age of the mice did not. In addition, the genome-wide response to blood neutrophils (rather than total WBC) was also not well correlated between humans and mice. However, the expression of many individual gene families was much more strongly correlated after injury in mice and humans.
Conclusions
Although overall transcriptomic association remained weak even after adjusting for the severity of injury, age of the animals, timing, and individual leukocyte populations, there were individual signaling pathways and ontogenies that were strongly correlated between mice and humans. These genes are involved in early inflammation and innate/adaptive immunity.
Keywords: microarray, blunt trauma, shock, mouse model
Introduction
Injuries from trauma continue to be the leading cause of death for persons less than 45 years of age in the U.S. (1, 2). Despite recent advances in trauma and critical care, as well as an improved understanding of the basic pathophysiology of severe trauma and its sequelae, morbidity and mortality, especially later following trauma, remain high (3). Although it is generally accepted that murine models do not fully recapitulate the human condition in its entirety, they remain the mainstay for invasive and interventional studies in trauma and burns (4). This reliance on a single species (Mus) has raised considerable concerns about its appropriateness, given the recently published transcriptomic differences to trauma, burns and endotoxicosis between man and mouse (5), in addition to innumerable failed clinical trials based on "cures" in mice derived from monogenic approaches in assumed appropriate models of injury. Criticism of these findings has centered on potential nongenomic differences between murine and human trauma, including the severity of the injuries, the differences in comparative age between humans and mice, the differences in the baseline blood leukocyte patterns between mice and man, and the focus on the total leukocyte genome rather than on those cell-specific genes and pathways involved in inflammatory and immune responses. In the present report, we specifically examined whether more precisely matching the murine model to human trauma in terms of comparative age, severity of injury, timing of sample collection, type of circulating leukocyte population (neutrophils) and sub-analysis of combinations of these would improve the transcriptomic correlations between the two species. Of note, we have recently reported that by increasing the severity of the traditional murine trauma model (6) to one that is multi-compartmental, including hemorrhagic shock, long bone fracture, soft-tissue damage and a cecectomy, we are better able to recapitulate the early plasma inflammatory cytokine and chemokine responses as well as the phenotypic leukocyte changes following severe human trauma (7).
Materials and Methods
Human Subjects
Two populations of trauma patients were included; the first whose blood was collected and processed to conduct genome-wide expression analyses on whole blood leukocytes (1, 8), and the second who blood was processed to conduct expression analyses on isolated blood neutrophils, monocytes, and lymphocytes (data not published). The first consisted of 167 severely injured trauma patients, age 18–55 years old, who were enrolled from 2003 to 2006, along with 35 healthy age, gender, and ethnicity matched control subjects (1). This entry criteria included those who suffered from severe blunt trauma with ISS >15, systolic hypotension (<90 mmHg), or elevated base deficit ≥6 mEq/L, and requirement for blood transfusion within six hours of injury. Patients with traumatic brain injuries were excluded. All subjects were treated under study protocols utilizing standard operating procedures in order to minimize treatment variation (9) and at centers with Institutional Review Board approval. In the second set of patients which was used for the leukocyte subset analysis, 244 severely injured trauma patients ages 16–90 years (2006–2010), with the same inclusion and exclusion criteria as above were compared to 57 healthy, matched controls. Blood neutrophils, monocytes and lymphocytes were isolated as previously reported (10, 11), and genome-wide expression was determined (unpublished data). The patients were taken from the Inflammation and Host Response to Injury Large Scale Collaborative Research Program. This study was reviewed by the Institutional Review Boards at Massachusetts General Hospital and the University of Florida and was approved.
Murine Models
Male C57BL/6J mice from either Jackson Laboratory (Bar Harbor, ME) (Young-6–10 weeks) or the National Institute of Aging (Old/Aged-20–24 months) were housed in pathogen-free facilities and acclimated at least one week prior to use under protocols approved by the University of Florida IACUC.
As previously described, using inhalational anesthesia, groups of mice underwent one of two models of trauma and shock (7). All mice underwent unilateral or bilateral femoral artery cannulation followed by shock induced by removing enough arterial blood to be maintained at a mean blood pressure of 30 mm Hg (± 5 mm Hg) for 90 minutes while awake. Mice were subsequently resuscitated with a crystalloid solution at four times the blood volume drawn. Next, mice were re-anesthetized and underwent either a one centimeter laparotomy to simulate the traditional model of trauma-hemorrhage (TH) or a more severe model including a one centimeter laparotomy with cecectomy combined with medial thigh dissection with femur fracture and muscle tissue damage (PT)(7). After injury, the mice were administered buprenorphrine (0.2 mg/kg BW) prior to arousal from anesthesia and every 12 hours afterward until sacrifice. Mice were euthanized after two hours and on postoperative days 1 and 3, and blood was collected for analysis. Blood samples from naïve 6–10 week old and 20–24 month old animals were used as murine controls. To determine whether old mice required a unique control group, we ran blood leukocyte genomic analyses between naïve young and old mice, and it was determined by cluster analyses that expression patterns from the two ages of mice could not be distinguished. Therefore, control samples were combined as one control group (data not shown). Four mouse replicates were used for each group as mice are expected to be genetically similar and exhibit reduced heterogeneity compared to the human population.
Gene Expression Profiling
As previously published, human total blood and enriched leukocytes were isolated, and RNA was extracted and hybridized onto either the Affymetrix HU133 Plus 2.0 or the HH/1/2/3 GeneChip™(12) according to the manufacturer’s recommendations (7, 8). For human neutrophil isolation, microfluidics were used to capture all PMNs bearing CEACAM-8, as previously described (11). Briefly, captured PMNs, were lysed and total RNA was extracted and analyzed in the same manner as described above for total leukocytes. For lymphocytes and monocytes, enriched populations of human lymphocytes and monocytes were isolated from peripheral blood samples using a two-step negative immunoselection protocol, consisting of binding antibody to unwanted cells then using density centrifugation removal of those cells, followed by enriching the remaining populations with antibody-bound bead columns (10).
Murine total blood leukocytes were isolated using the exact same procedures as for human blood samples. Murine PMNs were isolated by Ficoll-dextran isolation as previously described (11). Total cellular RNA was extracted as for the human samples and hybridized to the Affymetrix Mu430 2.0 GeneChip™ according to the manusfacturer’s recommendations.
Statistical Methods
Significant genes were selected by identifying trauma responsive genes in humans (167 patients) versus human control subjects (35) at a p-value <0.001. For genes that were represented by multiple probesets, the probeset with the highest expression value was used for analysis. Once the trauma-responsive significant genes were identified (Table 1), fold changes were calculated between mean values from human patients and control subjects. The same approach was used for the determination of the gene expression patterns from individual leukocytes from the 244 trauma patients in the second cohort. Next, the corresponding mouse orthologues to the human trauma responsive genes were identified (Table 1). Fold changes were calculated on the mouse orthologues between mice that had undergone injury and naïve control mice.
Table 1.
Number of human probe sets significant at p<0.001 and number of corresponding mouse orthologues used in analysis.
| Human Time Point (Days) |
Human Probe Sets at p<0.001 |
Mouse Orthologues |
|---|---|---|
| 0.5 | 31,847 | 11,720 |
| 1 | 31,239 | 11,585 |
| 4 | 30,643 | 11,510 |
| 7 | 28,594 | 11,029 |
| 14 | 26,218 | 11,539 |
| 21 | 21,763 | 9,475 |
| 28 | 21,492 | 10,002 |
For human total leukocyte gene expression from the 167 patients after severe trauma, an ANOVA analysis was run on discrete age ranges (16–32 versus >32–55 years, as well as 16–44 versus >44–55 years), and we found that gene expression was not different. The 244 trauma patients were divided into groups of those less than or equal to 55 years of age and those greater than 55 years of age.
Spearman’s correlations were calculated to assess the correlation between changes in gene expression in murine and human samples in multiple subgroup analyses, including age, leukocyte subset, and timing of injury. Because of the large number of correlations conducted, a conservative Bonferroni’s correction was used to adjust the alpha probability of p<0.05.
Pathway Analysis
After identifying genes whose expression changed significantly from control (p<0.001), we then performed a pathway analysis on the condition with the highest correlation at each human time point. We identified the top 100 up and down regulated genes in the humans and their corresponding mouse orthologues, and analyzed them for interactions and relationships using Ingenuity Pathway Analysis (IPA) (www.ingenuity.com). IPA was also used to identify the most over-expressed canonical pathways as well as the most highly up and down regulated genes.
Results
Injury severity, but not sample timing or age, improves gene expression correlations between murine and human trauma
For human trauma, we used existing data from 167 trauma patients obtained from the Inflammation and Host Response to Injury, Large Scale Collaborative Research Program (Glue Grant) who had total blood leukocyte genome-wide expression determined (8). Human trauma patients had blunt trauma without brain injury, an Injury Severity Score (ISS) > 15, systolic hypotension (<90 mm Hg), or elevated base deficit ≥ 6 mEq/L, and requirement for blood transfusion within six hours of injury. Blood samples were collected in the first 12 hours and at days 1, 4, 7, 14, 21, and 28 after trauma, during hospitalization. In comparison, we used two murine trauma models: the traditional murine trauma model used in earlier studies where C57BL/6 (B6) mice underwent 90 minutes of shock (mean arterial blood pressure of 30 mmHg) and resuscitation via femoral artery cannulation followed by a midline abdominal laparotomy (TH) (13, 14), and a more recent model of polytrauma where the TH model was supplemented with a cecectomy and femur fracture with muscle tissue damage (PT) (7). Blood samples were collected at two hours and one and three days. Genome-wide expression analysis was performed on total leukocytes and enriched blood neutrophil populations. Murine trauma was induced in both 6–10 week old animals (equivalent to a 14–20 year old human) and in 20–24 month old animals (equivalent to 55 –70 year old humans) (15).
We first examined the human probe sets whose expression changed at p < 0.001 at individual time points (between 21,492 and 31,847), and subsequently identified their mouse gene orthologues (between 9,475 and 11,720) (Table 1). Focusing on these genes, we compared the changes in gene expression of total blood leukocytes in a traditional TH model of murine trauma to human blunt trauma, and we see weak correlations, regardless of the timing of the measurements (Figure 1). These findings confirm those of Seok et al (5) demonstrating sporadically significant correlations between human trauma and murine TH, and in some cases significant inverse correlations. However, because of the large number of genes included in the correlation, the biological significance of statistically significant correlations ranging from 0.05 – 0.1 is essentially zero. These weak correlations are independent of the age of the mouse, employing either a juvenile mouse (6–10 week), or an older mouse (20–24 months). We also distinguished gene expression between adults under the age of 45 years versus those 45 –55 years of age from the initial data set (8), and found no improvement in the genome-wide correlation in gene expression with either juvenile or aged mice (data not shown).
Figure 1. Increasing murine model severity improves gene expression correlations between murine and human trauma.
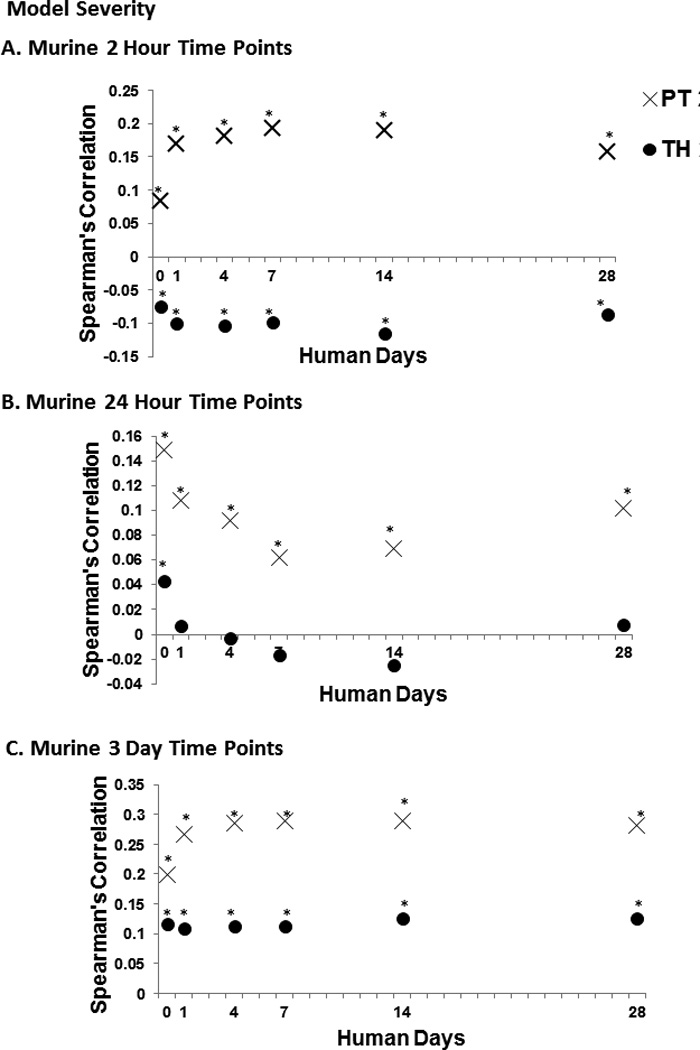
Graphs illustrating genome wide expression correlations of total blood leukocytes between human trauma patients and murine trauma models: TH, a traditional model of murine trauma and shock, PT, a more complex model of trauma and shock with injuries to multiple compartments and an Injury Severity Score >15. Asterisks (*) indicate statistically significant correlations (p<0.05 after Bonferroni’s correction). This was performed at each murine time point (A, B, and C). By increasing model severity, we significantly improve correlations at all human time points.
We then examined the effect of model severity on how well the human and mouse genomic changes correlate following severe trauma. We found that by increasing the model severity alone, we were able to improve the correlation in genome-wide expression between murine PT and human trauma, as evidenced by improved Spearman’s correlations (Figure 1), however, the improvements were modest.
We then examined the effects of age and timing on the correlations using the more severe PT model and found that there was no consistent improvement in the correlation between mouse and human genome-wide expression based on the timing of the blood sampling nor were the genome-wide correlations in expression improved by inducing severe polytrauma in aged mice (data not shown).
Analyzing and comparing circulating human leukocyte subsets to murine samples improves gene expression correlations
It is well known that the dissimilarities in the cellular composition of the circulating leukocytes between mice and humans may underlie some of the differences seen when comparing the murine and human responses to trauma (Figure 2A) (5, 16, 17). To explore whether exploiting these differences would improve the correlation between human trauma and murine models, we performed subgroup analysis comparing murine total leukocytes to enriched human T-cells, monocytes, and PMNs. The human patients in this previously-unpublished analysis consisted of 244 blunt trauma patients with the same inclusion criteria listed previously in whom neutrophils, monocytes and T-cells were isolated, and genome-wide expression was performed on enriched cell populations. In this second cohort obtained from the Glue Grant between 2006 – 2010, we compared the expression of murine total leukocytes to each human isolated leukocyte as well as performed a direct comparison of murine and human circulating neutrophils. This subgroup analysis was able to improve the correlation by three-fold as compared to the best value for the TH model for the majority of the correlations, and these best correlations with their optimal adjustments illustrated in Figure 3.
Figure 2. Murine to human age equivalents and circulating leukocyte populations.
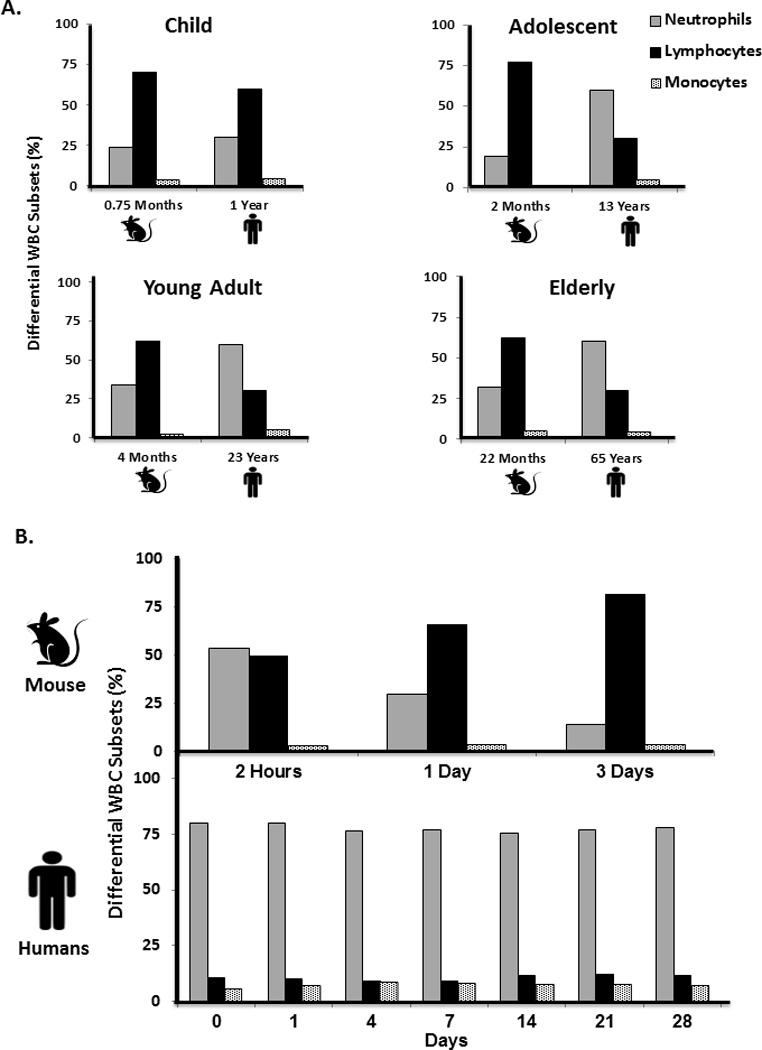
A. Diagram representing the mouse age of C57BL/6J mice in months as is equivalent to human age in years (modified from Jackson Laboratories http://research.jax.org/faculty/harrison/ger1vLifespan1.html) and (20). Bars represent WBC differential counts of neutrophils, lymphocytes, and monocytes at each age. (Human differential data obtained from http://www.childrensmn.org/manuals/lab/hematology/018981.asp). B. Murine and human differential percentages of neutrophils, lymphocytes, and monocytes following severe trauma in humans and PT in mice (n=4 per time point).
Figure 3. Analyzing and comparing circulating human leukocyte subsets to murine samples improves gene expression correlations.
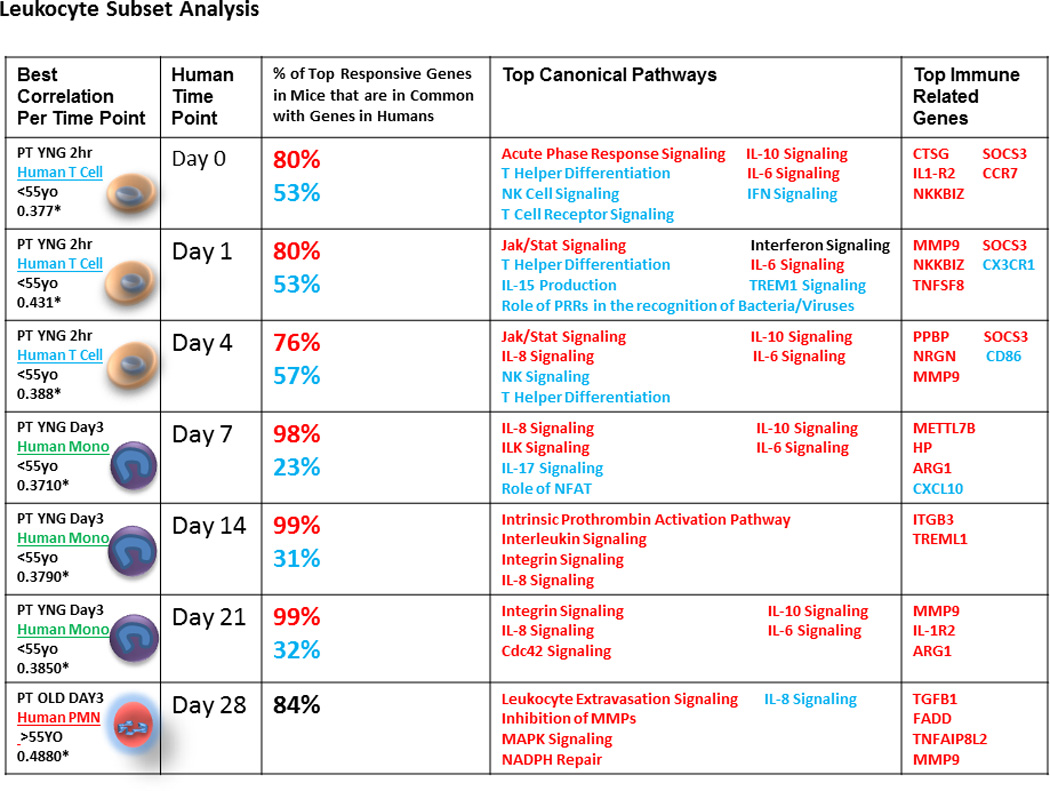
From left to right, columns one and two show the best correlation between murine PT models and human trauma at each human time point. The murine correlations were chosen as the best correlation of total mouse leukocytes from either the 2 hour, 1 day, or 3 day time points and either young or old mice. Asterisks (*) indicate statistically significant correlations (p<0.05 after Bonferroni’s correction). The best human correlations were chosen with regards to patient age (<55 years old or >55 years old) and leukocyte subset (lymphocytes, monocytes, or PMNs). Column three shows the percentage of up-regulated and down-regulated genes in mice that are in common with the top 100 up and down regulated genes in humans at each time point. Column four shows immune related pathways of interest chosen from the top 30 canonical pathways from an IPA analysis of the top 100 up and top 100 down regulated genes in humans. Column five shows the top up and down regulated genes from the same IPA analysis. (Red=up regulated, Blue=down regulated, Black=both up and down regulated).
Performing a more restrictive analysis on matching enriched cell populations improves Spearman’s correlations between human trauma and murine trauma models
We then compared the expression of enriched murine neutrophils following the PT model to human neutrophils at the same time points, and there was no improvement in the correlations over the comparison between mixed leukocyte populations (Supplemental Table 1). However, when a more restrictive analysis was performed examining only those genes whose expression had a greater than 1.5 fold change, we were able to significantly improve the best Spearman correlations between neutrophil enriched populations (0.21 to 0.43, respectively) (Supplemental Table 1).
Although overall genome-wide expression between murine models of trauma and humans may be modest, functional analysis reveals that selected individual gene or pathway responses are more similar
A general criticism of using a broad, genomic approach may be that it has limited significance if the investigator has a specific interest in individual genes or pathways. Even in the absence of overall correlations in gene expression, the expression of selected individual gene or pathway responses may be very similar. For example, when examining the top 100 responsive genes to trauma in humans over time, we observed that 80–99% of the same genes were also upregulated following murine PT (Figure 3), thus indicating that the majority of top responsive genes are behaving similarly in both mice and humans after injury. Not surprisingly, many of these genes were involved in early inflammatory processes and innate immunity. This was confirmed by functional analysis using Ingenuity Pathway Analysis (IPA) which revealed that the majority of these pathways were involved in early inflammatory responses and activation of innate immunity (Figure 3). They included up-regulation of NF-κB, MAPK, JAK/STAT signaling pathways associated with cytokine signaling, and down-regulation of T-helper cell differentiation pathways.
Discussion
Although there has been recent criticism about the validity of murine models of trauma and burns (5), most investigators in the field recognize the critical role they have played. Despite this persistent question regarding the appropriateness of using murine models to study human trauma and burns, there remains considerable evidence that murine models have proven useful for evaluating other inflammatory processes in humans, most notably in the development of anti-inflammatory and anti-immunity drugs for rheumatoid arthritis (e.g. etanercept, anakinra, abatacept, tocilizumab, tofacitinib) (18). However, rheumatoid arthritis is a chronic low grade human inflammatory condition caused by autoimmunity, and not the "genomic storm" of severe injury (8), which may also help to explain the failure to translate these identical therapies in the acute injury setting.
Understanding the limitations of murine studies is essential as the majority of new drugs target specific genes and pathways, and differences between mice and humans can lead to inconsequential or incorrect conclusions. Similar to Soek et al, we have illustrated that the overall total murine leukocyte transcriptomic response to severe injury and shock is dissimilar to man (5) when utilizing current rodent models of trauma appropriately limited because of animal welfare concerns. Only modest improvements can be made by humanely increasing the severity of the injury, and specifically looking at certain times post injury as well as focusing on individual human leukocyte populations.
Undoubtedly, the one determinant that can be best improved upon is the severity of the traumatic injury, as moving from a simple trauma hemorrhage model to a model of polytrauma significantly improves the overall genome-wide correlations. Regardless, overall comparison of total blood leukocytes is still suboptimal and recognizing the limitations of these models is essential. However, the results are not all gloom and doom. Focusing on specific genes or cell signaling pathways can still prove valuable in predicting what the human response would be under similar conditions. For example, in Figure 3, we list immune related pathways and genes of interest garnered from an analysis comparing the top 100 up and top 100 down-regulated genes in humans to their murine ortholgues. Here we show that canonical pathways including acute phase response signaling, IL-10, IL-6, and Jak/Stat signaling all behave in a similar manner in both mice and humans following trauma. Similarly, we list genes involved in early inflammatory responses and activation of innate immunity such as ARG1, MMP9, and SOCS3 that behave in the same manner. Efforts to validate the comparative responses of individual genes or pathways in these models might improve their acceptance by the scientific and medical communities.
At the genome-wide response in total circulating leukocytes under the best of circumstances, less than 10% of the variation in human gene expression could be predicted by changes in the mouse genome. Mouse models may fail to surpass this level for multiple reasons, including, but not limited to, species differences, differences in homogenous versus heterogeneous responses, and differences in the severity of their injuries. For example, the murine PT is a nonlethal model and the genomic response is returning to baseline by three days (data not shown). SInce there was no consistent difference in the correlation between mouse and human genome-wide expression based on the timing of the blood sample collection, the fact that the correlations of PT day three mice did not significantly decline when compared to later time points post human trauma sampling may indicate that earlier time points in murine models may still reflect sub-acute and chronic ICU patients at some level. Alternatively, in the human studies, approximately one-third of the cohort had an uncomplicated outcome and were discharged from the ICU within five days, while roughly, another third either were hospitalized for organ failure for longer than 14 days or died (1, 8). Thus, the lack of time dependence may reflect the heterogeneity of the human population.
However, this was not necessarily true for specific gene pathways and ontologies. In fact, of those genes that changed the most in the human blood leukocytes after trauma, 88% – 100% of the murine orthologues changed in a similar direction. Similarly, the most over-represented pathways and ontologies were those involved in cytokine signaling and inflammation in humans after trauma and were generally recapitulated in the mouse, as listed in Figure 3.
It is not clear as to why gene expression from total murine leukocytes at certain time points better reflect gene expression in specific human leukocyte populations in the early and later phases after trauma (Figure 3). An overall pattern emerged that the genomic response of murine total circulating leukocytes best reflects the human T-cell response in the early phases after severe injury, while later time points in both mice and humans better correlate to expression by human myeloid cells (data not shown). This may be a reflection of the myeloid response associated with sepsis and trauma that is associated with the persistent inflammation immunosuppression catabolism syndrome (PICS) (19). Regardless, the transcriptomic differences or similarities between mice and humans cannot be easily explained by differences or similarities in the prevalent type of circulating leukocyte after injury (Figure 2B). In general, the genome-wide correlations between mice and humans are improved after accounting for the severity of the injury model, sampling intervals and differences in leukocyte populations with time, although they remain modest at best (Figure 4).
Figure 4. Although genome-wide correlations between mice and humans are modest at best, when optimizing the model, age, and leukocyte subset correlations between murine and human gene expression can be significantly improved.
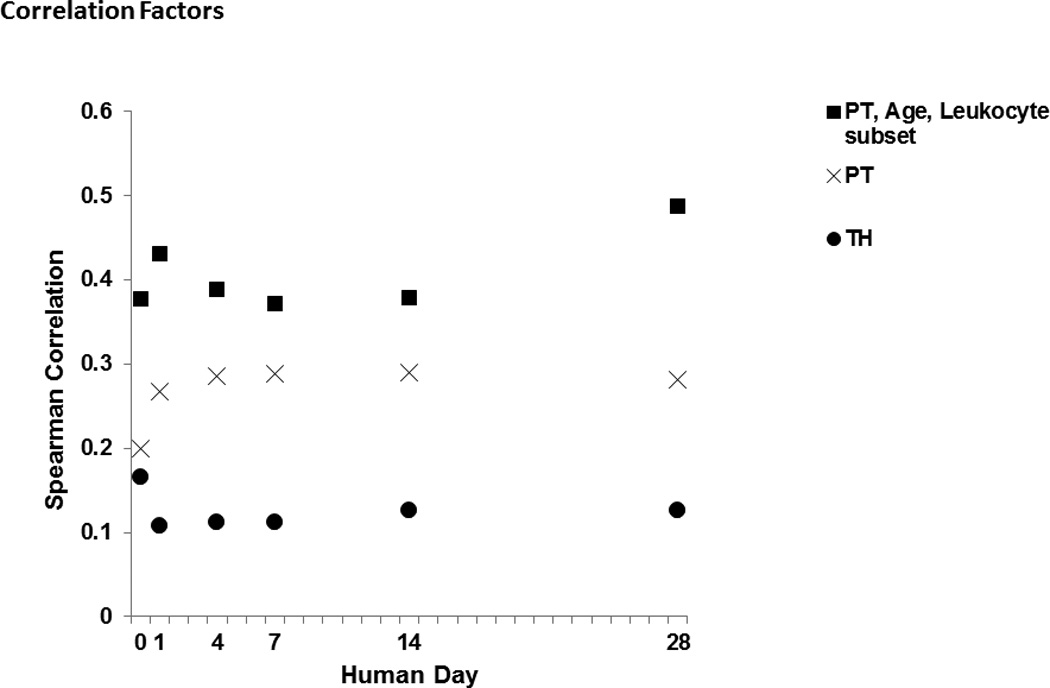
Graph shows best murine correlation for each human time point for the analysis of the TH model alone, the PT model alone (increased model severity), and an analysis including all factors (model severity, age, and overall best leukocyte subset) compared to each human time point. All reported correlations are statistically significant correlations (p<0.05 after Bonferroni’s correction). The correlations are improved at each time point as the number of factors considered increases.
Despite more precisely matching the murine model of injury to the injury sustained in human trauma patients, another limitation remains: the fact that severely injured human trauma patients receive continued ICU care including, but not limited to, mechanical ventilation, blood transfusions, placement of invasive central lines and Foley catheters, whereas the mice are merely resuscitated and then receive ongoing pain management.
Indeed while blood leukocytes are undoubtedly important cells in the immune response, they most certainly are not the sole cell type to help orchestrate the immune response. Many other immune tissues and cell types are involved in a septic or traumatic insult. These may be equally important as blood leukocytes, and may even show stronger correlations to humans; hence the investigation into appropriate murine models that reflect the human condition should continue.
Conclusions
In conclusion, the findings suggest murine biology cannot be assumed to accurately represent the entire complexity of the human response, and thus, murine research should not be used indiscriminately without validation a priori. Investigators must strive within sufficient capacity to develop rodent models that best mimic human insults and pathology. However, mouse models can suitably reflect specific components of the human response, and these physiologic or pathophysiogic functions can be examined, assuming there is an appropriate understanding of the limitations of the data obtained, as well as reasonable validity of the model under investigation.
Supplementary Material
Acknowledgements
AGC and LFG were supported by a T32 training grant (T32 GM-008721-13) in burns and trauma from the NIGMS. Other support was provided by R01 GM-40586-24 and R01 GM-80576-06, awarded by the NIGMS. Support was also provided by the Claude D. Pepper Older Americans Independence Center (NIH/NIA P30AG028740).
This work was also supported by a contract (U54 GM- 062119-10) awarded by the National Institute of General Medical Sciences (NIGMS) to Dr. Ronald Tompkins, Massachusetts General Hospital, and represents a secondary use of the Trauma Related Database developed by that Program. However, the results and conclusions contained in the manuscript have not necessarily been reviewed by the Program, and do not necessarily represent the views of the Glue Grant Program or the NIGMS.
None
Footnotes
Conflict of Interest Statement: No conflict of or competing interests have been declared.
Author Contributions
LFG contributed to the analysis of data, conception, design, composition, drafting, and editing of the manuscript. DCN performed animal experiments, data collection, and contributed to the editing of the manuscript. MCP contributed to the analysis of data and editing of the manuscript. EV, AGC, BAM, FAM, and AC contributed to the manuscript editing. RU contributed animal experiments and laboratory data collection as well as to the editing of the manuscript. TOB contributed to the editing of the manuscript. AB and CL contributed to the design and editing of the manuscript. JC and RVM contributed to the conception and design of the manuscript, and the editing of the manuscript. HVB and LLM contributed to the conception and design of the manuscript, and the composition/editing of the manuscript. PAE contributed to the analysis of data, conception, design, composition, drafting, and editing of the manuscript.
References
- 1.Cuenca AG, Gentile LF, Lopez MC, et al. Development of a Genomic Metric That Can Be Rapidly Used to Predict Clinical Outcome in Severely Injured Trauma Patients. Crit Care Med. 2013 doi: 10.1097/CCM.0b013e318277131c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Probst C, Pape HC, Hildebrand F, et al. 30 years of polytrauma care: An analysis of the change in strategies and results of 4849 cases treated at a single institution. Injury. 2009;40(1):77–83. doi: 10.1016/j.injury.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Davidson GH, Hamlat CA, Rivara FP, et al. Long-term survival of adult trauma patients. JAMA. 2011;305(10):1001–1007. doi: 10.1001/jama.2011.259. [DOI] [PubMed] [Google Scholar]
- 4.Dyson A, Singer M. Animal models of sepsis: why does preclinical efficacy fail to translate to the clinical setting? Crit Care Med. 2009;37(1 Suppl):S30–S37. doi: 10.1097/CCM.0b013e3181922bd3. [DOI] [PubMed] [Google Scholar]
- 5.Seok J, Warren HS, Cuenca AG, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu HP, Hwang TL, Hsieh PW, et al. Role of estrogen receptor-dependent upregulation of P38 MAPK/heme oxygenase 1 in resveratrol-mediated attenuation of intestinal injury after trauma-hemorrhage. Shock. 2011;35(5):517–523. doi: 10.1097/SHK.0b013e318209e931. [DOI] [PubMed] [Google Scholar]
- 7.Gentile LF, Nacionales DC, Cuenca AG, et al. Identification and Description of a Novel Murine Model for Polytrauma and Shock. Crit Care Med. 2013 doi: 10.1097/CCM.0b013e318275d1f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao W, Mindrinos MN, Seok J, et al. A genomic storm in critically injured humans. The Journal of experimental medicine. 2011;208(13):2581–2590. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuschieri J, Johnson JL, Sperry J, et al. Benchmarking outcomes in the critically injured trauma patient and the effect of implementing standard operating procedures. Ann Surg. 2012;255(5):993–999. doi: 10.1097/SLA.0b013e31824f1ebc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laudanski K, Miller-Graziano C, Xiao W, et al. Cell-specific expression and pathway analyses reveal alterations in trauma-related human T cell and monocyte pathways. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(42):15564–15569. doi: 10.1073/pnas.0607028103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kotz KT, Xiao W, Miller-Graziano C, et al. Clinical microfluidics for neutrophil genomics and proteomics. Nat Med. 2010;16(9):1042–1047. doi: 10.1038/nm.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W, Seok J, Mindrinos MN, et al. Human transcriptome array for high-throughput clinical studies. Proc Natl Acad Sci U S A. 2011;108(9):3707–3712. doi: 10.1073/pnas.1019753108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang P, Ba ZF, Burkhardt J, et al. Trauma-hemorrhage and resuscitation in the mouse: effects on cardiac output and organ blood flow. The American journal of physiology. 1993;264(4 Pt 2):H1166–H1173. doi: 10.1152/ajpheart.1993.264.4.H1166. [DOI] [PubMed] [Google Scholar]
- 14.Gill R, Ruan X, Menzel CL, et al. Systemic inflammation and liver injury following hemorrhagic shock and peripheral tissue trauma involve functional TLR9 signaling on bone marrow-derived cells and parenchymal cells. Shock. 2011;35(2):164–170. doi: 10.1097/SHK.0b013e3181eddcab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison DE. Lifespan as a Biomarker. 2011 In. [Google Scholar]
- 16.von Vietinghoff S, Ley K. Homeostatic regulation of blood neutrophil counts. J Immunol. 2008;181(8):5183–5188. doi: 10.4049/jimmunol.181.8.5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 18.Miller AV, Ranatunga SK. Immunotherapies in rheumatologic disorders. The Medical clinics of North America. 2012;96(3):475–496. ix–x. doi: 10.1016/j.mcna.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Gentile LF, Cuenca AG, Efron PA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. The journal of trauma and acute care surgery. 2012;72(6):1491–1501. doi: 10.1097/TA.0b013e318256e000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turnbull IR, Wlzorek JJ, Osborne D, et al. Effects of age on mortality and antibiotic efficacy in cecal ligation and puncture. Shock. 2003;19(4):310–313. doi: 10.1097/00024382-200304000-00003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.