

Abstract
Plant cell cultures provide an important method for production and supply of a variety of natural products, where conditions can be easily controlled, manipulated and optimized. Development and optimization of plant cell culture processes require both bioprocess engineering and metabolic engineering approaches. Cultures are generally highly heterogeneous, with significant variability amongst cells in terms of growth, metabolism and productivity of key metabolites. Taxus cultures produce the important anti-cancer agent Taxol® (i.e., paclitaxel) and have demonstrated significant variability amongst cell populations in culture with regards to paclitaxel accumulation, cell cycle participation and protein synthesis. To fully understand the link between cellular metabolism and culture behavior and to enable targeted metabolic engineering approaches, cultures need to be studied at a single cell level. This chapter describes the application of plant cell flow cytometric techniques to investigate culture heterogeneity at the single cell level, in order to optimize culture performance through targeted metabolic engineering. Flow cytometric analytical methods are described to study Taxus single cells, protoplasts and nuclei suspensions with respect to secondary metabolite accumulation, DNA content, cell size and complexity. Reproducible methods to isolate these single particle suspensions from aggregated Taxus cultures are discussed. Methods to stain both fixed and live cells for a variety of biological markers are provided to enable characterization of cell phenotypes. Fluorescence-activated cell sorting (FACS) methods are also presented to facilitate isolation of certain plant cell culture populations for both analysis and propagation of superior cell lines for use in bioprocesses.
Keywords: Plant cell culture, flow cytometry, cell sorting, FACS, Taxus, paclitaxel, culture heterogeneity, secondary metabolite, DNA content
1. Introduction
Plant cell cultures provide an important method for production of various natural products (e.g., pharmaceuticals, colors, specialty chemicals, etc.) in which environmental conditions can be easily controlled, manipulated and optimized in order to obtain high quantities of these valuable compounds (1). Development of an optimal plant cell culture process requires a successful combination of bioprocess engineering and metabolic engineering approaches. Adapting existing strategies, which are often developed using well-studied cell types (e.g., mammalian, yeast and bacteria), does not necessarily translate into productive and viable plant cell culture processes because of the inherent challenges associated with plant cells that ultimately lead to low and variable product yields. While a majority of research on plant cell culture optimization has been focused on system characterization (e.g., reactor design, medium optimization and nutrient utilization), there have been few studies emphasizing cellular metabolism and culture heterogeneity (2–6).
Cell-cell heterogeneity in culture results in unpredictable shifts in product accumulation over time and within cultures cultivated at the same time. Cell aggregation creates distinct microenvironments owing to differences in oxygen and nutrient availabilities, resulting in alterations in gene expression and metabolic function, which may be observed as phenotypic changes in culture behavior. These changes include variations in metabolic pathway participation and metabolite accumulation as well as cell subpopulation variability where certain populations of cells do not participate in growth or secondary metabolite accumulation (7). Most studies concerning metabolite production via cell culture technology rely on culture-average parameters. These measurements involve averages of culture properties over a group of cells, and are often insufficient to describe culture heterogeneity as they neglect variations at the single cell level. Analyzing cell populations with different properties, such as varying levels of metabolite accumulation, at the single cell level can significantly contribute to the understanding of the inherent molecular and metabolic differences amongst cells in culture and lead to new approaches for controlling culture heterogeneity and production variability. Techniques for investigating single cell properties, most notably flow cytometry, can provide insight into the nature of culture heterogeneity and when adapted with a sorting functionality, allow for recovery of distinct sub-populations for further analyses (e.g., gene expression profiling, metabolite profiling, DNA content and ploidy measurements, etc.).
Flow cytometry is a powerful tool to count, analyze and sort single particles (e.g., cells, protoplasts, nuclei, chromosomes, beads, etc.) in suspension. Flow cytometry allows for simultaneous multiparameter analysis of cell and/or cellular molecules, based on their light scatter and fluorescence properties, within heterogeneous populations (8). The quantitative evaluation of cellular characteristics on a per cell basis across the entire population set facilitates rapid, accurate, and sensitive analyses of culture heterogeneity. A variety of measurement techniques, including fluorescent probes, are used to identify certain cellular components to enable analysis and/or sorting. Some examples of cell and/or cell-associated properties with their respective probe/staining methods which can be measured using flow cytometry are provided in
Flow cytometry has been used extensively by scientists for the study of mammalian and microbial cell populations for biotechnological applications (20). Plant cell flow cytometry is, however, still an emerging field where many researchers adapt existing techniques developed for mammalian and microbial cells. Sorting and analysis of various plant particles/organelles (e.g., nuclei, protoplasts, chromosomes, and pollen grains) have been performed, with applications ranging from production of pharmaceuticals and specialty chemicals to crop breeding. A genomic chromosome library has been constructed through flow cytometric sorting of banana (Musa balbisiana) nuclei (21). Specific green fluorescent protein (GFP)-positive protoplasts of Arabidopsis thaliana have been isolated using fluorescence-activated cell sorting (FACS) and analyzed for gene expression (22). Chromosomes differing in DNA content have been sorted and isolated from suspension cells of Haplopappus gracilis using flow cytometry (23). Transcriptional profiling of Arabidopsis tissues was performed by obtaining highly purified pollen grains through flow cytometric sorting (24). However, because of the large size of some plant protoplasts and non-uniformity of intact single cells (i.e., those cells that contain an intact cell wall), it is challenging to use conventional methods and flow cytometers for analysis and sorting (25). Plant cells are relatively large in size (ca. 20–100 μm), limiting the use of a typical nozzle in the cytometer. The dimensions of the particles to be sorted, including intact single cells, should be compatible with the flow nozzle. It is generally assumed that for a smooth and clog-free run, the flow nozzle/orifice should be at least four times the particle size, therefore demanding special size instrument nozzles when sorting larger plant cells (26).
Intact single cells, as opposed to protoplasts, are relatively non-isotropic, which adds complexity in the application of flow cytometric methods. Moreover, large sized particles create instabilities in the flow stream, which mandates that the system flow rate and sheath pressure be maintained at low values, necessitating longer runs. Researchers have studied the physics of the cytometric sorting process to establish correlations which explain and can ultimately predict flow and droplet formation in a sorter. For instance, there have been reports of inter-dependence between parameters such as sorting efficiency, particle diameter, flow cell nozzle diameter, sheath pressure, and drive frequency (26). An extensive optimization of these parameters can lead to stable hydrodynamic flow conditions, resulting in efficient droplet formation and successful sorting of plant cells. Vacuoles, which are largely comprised of water and may constitute up to 90% of a plant cell, render plant cells fragile and shear-sensitive, thus affecting cell health and viability during sterile operations such as live cell staining and sorting. Special care (e.g., low centrifugation speeds and reduced agitation rates) must be taken during sample preparation and instrument operation to avoid any potential detrimental cellular effects. Plant cells, unlike animal and microbial cells, are non-uniform in shape, creating problems with signal detection that lead to incorrect optical measurements. To overcome this issue, a significant number of cells (~10,000 or more) should be analyzed in the cytometer, to ensure an accurate representation of the entire population set.
Another crucial difference between plant cells and other systems that limits the application of flow cytometry is the tendency of plant cells to aggregate in suspension. A first step in isolating a single particle suspension from aggregated plant cell suspensions is to induce single cell generation using enzymatic digestion to weaken the middle lamella that cements adjoining cells in an aggregate (3). Following single particle isolation, cells tend to sediment in suspension, which can complicate cytometer operation. Xanthan gum, a relatively inert material, has been used to keep large biological particles suspended during flow cytometric analysis and sorting (27).
This chapter describes the application of different flow cytometric techniques to analyze a variety of plant particles, including intact single cells. A comprehensive population analysis can be used to both optimize bioprocess conditions for growth and productivity, and to identify targets for metabolic engineering through focusing on productive cells. Here we described methods developed in Taxus plant suspensions; however, all techniques can be easily adapted to other plant culture systems. Flow cytometric methods are described to investigate heterogeneity in Taxus single cells, protoplasts and nuclei suspensions with respect to secondary metabolite accumulation, DNA content, cell size and complexity. A variety of reliable methods to isolate these single particle suspensions (i.e., single cells, protoplasts, nuclei) from aggregated Taxus suspensions are discussed. Techniques to stain cells for different types of biological molecules, that provide information about the metabolic, genomic and phenotypic state of these cultures, are also detailed. Finally, sorting of plant protoplasts and intact single cells for the isolation of distinct populations is described.
2. Materials
2.1. Cell Culture Maintenance and Methyl Jasmonate Elicitation
Gamborg’s B-5 basal medium with minimal organics (Sigma-Aldrich, St. Louis, MO) (28) is dissolved in nanopure water at 3.2 g/L (see Note 1).
Sucrose, grade I (plant cell culture tested; Sigma-Aldrich) is added to the basal medium solution at 20 g/L.
Stock solutions of α-naphthalene acetic acid (NAA) and benzyladenine (BA) (Sigma-Aldrich) are made in nanopure water, and stored at 2–8 °C for up to 6 months.
Supplemental anti-oxidants and nutrients: ascorbic acid (150 mg/L), L-glutamine (non-animal source) (6 mM) and citric acid (150 mg/L) (Sigma-Aldrich) are dissolved in nanopure water, filter sterilized through a 0.22 μm Millex® GP filter unit (Millipore) and added to the autoclaved medium. This solution is made fresh for each culture transfer (see Notes 2 and 3).
Pyrex glass shake flasks (125 ml) and Bellco semi-permeable foam closures (Vineland, NJ).
A working solution of methyl jasmonate (MJ) (95%; Sigma-Aldrich) is prepared by adding 50 μL of MJ to 450 μL of 95% (v/v) ethyl alcohol and 500 μL of nanopure water. The solution is then vortexed and filtered through a 0.2 μm Gelman acrodisc PVDF filter (Sigma-Aldrich) into a sterile container prior to culture addition.
2.2. Isolation of Single Cells and Protoplasts from Aggregated Cultures
Working solution of osmoticum is prepared by dissolving 0.5 M D-Mannitol and 0.3 % (w/v) dextran sulfate sodium salt (Sigma-Aldrich) in nanopure water. The pH is adjusted to 5.5 and the osmoticum is then autoclaved at 121 ºC for 15 minutes and stored at room temperature for up to 48 hours (see Note 4).
Cellulase from Trichoderma viride (Sigma-Aldrich) and Pectolyase Y-23 (MP Biomedicals, LLC, Solon, OH).
Miracloth® (Calbiochem, San Diego, CA) (cut into squares of 4×4 in.), 80 μm nylon mesh (Sefar American, Depew, NY) (cut into squares of 3×3 in.), aluminum foil, vacuum flask, funnel, spatula and 25 mL Erlenmeyer flasks are sterilized via autoclaving.
Phosphate buffered saline (PBS) at 0.01 M prepared in nanopure water.
Sucrose solution at 0.5 M prepared in nanopure water.
Paraformaldehyde (1 % (w/v)) (Sigma-Aldrich).
Centrifuge tubes (15 mL and 50 mL).
BD Falcon 5 mL polystyrene round-bottom tube (BD Biosciences, Bedford, MA).
BD 22G 1½ Precision Glide® needle and 1 mL syringe (BD & Co., Franklin Lakes, NJ).
Hemacytometry – V167014 light miscroscope (VWR), Leica Zoom 2000 microscope (Leica Microsystems, Buffalo, NY), or the like, hemacytometer glass slides and cover slips (VWR).
2.3. Immunofluorescent Staining for Paclitaxel
Working solutions of primary mouse monoclonal anti-paclitaxel antibody (Cardax Pharmaceuticals, Aiea, HI) and secondary goat anti-mouse phycoerythrin (PE)-conjugated antibody (Southern Biotech, Birmingham, AL) are prepared in BPT (0.25% (w/v) bovine serum albumin, 0.05% (v/v) Tween-20, 0.02% (w/v) sodium azide in 10 mM PBS). These solutions are stored at 2–8 °C until use.
Solution of TBS-T (50 mM Tris-HCl, 0.15 M sodium chloride, 0.05 % (v/v) Tween-20, 0.02 % (w/v) sodium azide), stored at 2–8 °C.
2.4. Flow Cytometric Analysis and Sorting
BD LSR II analytical flow cytometer (Becton Dickinson, San Jose, CA) with light source suitable for excitation of the fluorochromes used in the study (PE, PI and FITC) (e.g., an Argon laser tuned to 488 nm); and BD LSR II User’s Guide. The cytometer should be capable of evaluating multiparameter cellular properties (e.g., fluorescence and light scatter).
BD FACSVantage SE™cell sorter (Becton Dickinson, San Jose, CA) configured with 488 nm air cooled lasers; and BD FACSVantage User’s Guide. The sorter should be equipped with a MacroSort™ option and a larger nozzle (200 μm) for sorting of large plant particles.
Software for the analysis of flow cytometric data (e.g., BD FACSDiva™ (Becton Dickinson, San Jose, CA) or FlowJo (Tree Star, Inc., Ashland, OR)).
Calibration beads for BD LSR II analytical flow cytometer - Sphero™ rainbow fluorescent particles, 3.0–3.4 μm (BD Biosciences, San Diego, CA).
Calibration beads for BD FACSVantage cell sorter - AlignFlow™ flow cytometry alignment beads, 2.5 μm for 488 nm excitation; and AlignFlow™ flow cytometry alignment beads, 2.5 μm for 633 nm excitation (Molecular Probes, Inc., Eugene, OR).
Megabead NIST Traceable Particle Size Standard, 25.0 μm (Polysciences, Inc., Warrington, PA).
Sheath buffer (sterile PBS or 0.9% sodium chloride).
Cytometer sterilization kit (10 % bleach, 70 % ethanol and distilled water) (can be prepared at the cytometer facility; follow the manufacturer’s manual).
2.5. Preparation of Nuclei Suspensions from Aggregated Cultures
Nuclei isolation buffer is prepared by dissolving MgCl2 (45 mM), 4-morpholinepropanesulfonic acid (MOPS) ≥ 99.5 % (titration) (20 mM), sodium citrate (30 mM), and Triton X-100 (0.3 % (v/v)) (Sigma-Aldrich) in nanopure water and adjusting the pH to 7. The isolation buffer is filtered through a 0.2 μm Gelman acrodisc PVDF filter and stored at −20 °C in aliquots of 10 mL. This buffer should not be refreezed once thawed.
Mini-glass scraper GSM (Allway Tools, Bronx, NY).
Polystyrene (100×15) mm. petri dishes.
2.6. Staining of Nuclear DNA
Propidium iodide (PI) stock solution of 1 mg/mL is made in nanopure water, filtered through a 0.2 μm Gelman acrodisc PVDF filter, and stored at −20 °C in 1 mL aliquots, for single use only.
Ribonuclease A (activity > 97.9 units/mg) (Fisher Scientific, Hampton, NH) stock solution of 1 mg/mL is made in nanopure water, and heated for 15 minutes at 90 °C to render any DNases inactive. The solution is stored at −20 °C in aliquots of 1 mL, for single use only.
2.7. Flow Cytometric Analysis of Stained Nuclei
Analytical flow cytometer and data analysis software (see Section 2.4).
Calibration beads, sheath buffer and sterilization kit (see Section 2.4).
3. Methods
As a first step in analyzing plant cells using flow cytometry it is crucial to develop reliable methods for isolation of plant-derived particles (i.e., nuclei, protoplasts and intact single cells). Plant cells aggregate via a middle lamella layer that cements neighboring cell walls together. While cellulose is the primary component of the plant cell wall, the middle lamella is largely composed of pectin. Enzymes such as cellulase and pectolyase Y-23 are used, at appropriately evaluated concentrations, to prepare single cells and protoplasts from aggregated cultures. Since paclitaxel is primarily stored in the cell wall, as is the case with many hydrophobic secondary metabolites (29, 30), intact isolated single cells must be used to characterize populations with regards to paclitaxel accumulation. An indirect immunofluorescent procedure is employed to identify paclitaxel-accumulating populations through flow cytometry. Protoplasts, isolated using enzymatic digestion, are useful in the study of size, complexity, and intracellular protein content. Isolated nuclei can be stained for DNA and protein content using PI and FITC, respectively, and are useful in the study of nuclear protein synthesis, ploidy levels and cell cycle participation. Here we present detailed methods for the isolation of nuclei, protoplasts and intact single cells from aggregated plant cultures, along with appropriate staining techniques, to enable population analyses via flow cytometry.
3.1. Cell Culture Maintenance and Methyl Jasmonate Elicitation
The cell lines P991, P93AF, PO93X (Taxus cuspidata) and CO93D (Taxus canadensis), obtained from US Plant Soil and Nutrition Laboratory (Ithaca, N.Y.), are maintained by subculturing every 2 weeks into fresh medium consisting of Gamborg’s B-5 basal medium (3.2 g/L) and sucrose (20 g/L). The medium is supplemented with 2.7 μM NAA and 0.1 μM BA and is adjusted to a pH of 5.5. Media is transferred into 125 mL shake flasks (40 mL media per flask), capped with Bellco foam closures, and sterilized via autoclaving at 121 ºC for 15 minutes. Media is then brought to room temperature before supplemental anti-oxidants and nutrients are added. Ten milliliters of cell culture suspension from a 14 day-old culture, with 2–3 mL of packed cell volume per 10 mL, are transferred to each of the 125 mL flasks. All cultures are maintained in gyratory shakers (125 rpm) at 23 °C in the dark.
Elicitation of the Taxus cell cultures to up-regulate the genes involved in secondary metabolite accumulation is performed with MJ 7 days post-transfer at 200 μM (31). A working stock solution of MJ is added to the culture flasks under sterile conditions.
3.2. Isolation of Single Cells and Protoplasts from Aggregated Cultures
3.2.1. Isolation of Single Cells
For isolation of single cells, an enzyme solution is prepared by adding 0.04 % (w/v) cellulase and 0.5 % (w/v) pectolyase Y-23 in osmoticum (see Notes 5 and 6). The mixture is filter sterilized through a 0.22 μm Millex® GP filter unit (Millipore) and then transferred to sterile containers (e.g., 25 mL flasks, six-well plates, etc.). The volumes of the enzyme mixture and isolated single cell suspension can be varied according to the required application (see Note 7).
Cells from late exponential phase of the culture period are vacuum-filtered using Miracloth® and added to the enzyme mixture. The ratio of cells added to the enzyme mixture is kept at a value of 1 g cells/5 mL solution. Enzymatic digestion is carried out at typical culture conditions (see Section 3.1.1) for 4 hours.
Post-digestion, the single cell suspension is centrifuged at 1000 × g for 5 minutes (see Note 8). After centrifugation, the supernatant is carefully removed with a syringe and needle (see Note 9).
Enzyme-free osmoticum is added to the cells to increase the total volume to the required working volume. Single cells are then purified by filtering twice through 80 μm nylon mesh into new centrifuge tubes. Filtration is performed by pouring/transferring (using a cut 1 mL pipette tip) the cell suspension through the nylon mesh (held over an open centrifuge tube) and collecting the filtrate in the tubes.
Cell density is then measured using hemacytometry. A suitable working range of cell density for flow cytometry and sorting is (0.5 – 1.0) × 106 cells/mL. The cell suspension should be diluted, if necessary, with PBS to achieve this cell density. Care should be taken to account for the loss of cells during cell counting and subsequent staining and filtering steps, especially when using small volumes (i.e., less than 2 mL) of solution (see Notes 9 and 10).
The isolated single cells can be utilized for several types of analysis. Cells may be fixed and stained for paclitaxel accumulation (see Section 3.3) and analyzed in the BD LSR II flow cytometer (see Section 3.4.1) or used for cell sorting through the BD FACSVantage cell sorter (see Section 3.4.2). In order to avoid any loss of important metabolic information in the isolated single cells, samples should be analyzed and/or sorted in the cytometer immediately following preparation (see Note 11).
3.2.2. Isolation of Protoplasts
For formation of protoplasts, the enzyme solution is prepared at a different concentration than that required for single cell generation. 1 % (w/v) cellulase and 0.1 % (w/v) pectolyase Y-23 is dissolved in osmoticum (see Note 5). The mixture is filter sterilized through a 0.22 μm Millex® GP filter unit (Millipore) into a sterile container.
Cells are added to the enzyme mixture and digestion is carried out as described in Section 3.2.1.2.
Post-digestion, the suspension is centrifuged at 300 × g for 5 minutes (see Note 9). The supernatant is carefully removed using a syringe and needle (see Note 9).
Enzyme-free osmoticum is added to the sample to increase the volume to the required working volume. The sample is then filtered through 80 μm nylon mesh into sterile centrifuge tubes (see Section 3.2.1.4).
Approximately one-third volume of 0.5 M sucrose is added to the sample and mixed by inversion. The sample is allowed to sit for 20–30 min, during which time the sucrose and osmoticum separate due to density differences, and the protoplasts settle in a noticeable ring at the liquid-liquid interface. The protoplasts are then carefully removed using a 1 mL cut pipette tip.
3.3. Immunofluorescent Staining for Paclitaxel
The isolated single cells (as described in Section 3.2.1.6) are fixed with 1 % (w/v) paraformaldehyde at 4 °C for 1 h. The cells are then washed two times with an equal volume of PBS to remove any remaining fixative. Washing includes the following steps: (i) centrifuge at 400 × g for 1 minute; (ii) remove the supernatant using a syringe and needle (see Note 9); (iii) add PBS; and (iv) repeat starting at step (i).
The fixed cells are then divided into two equal volumes and transferred to suitable containers. The first sample serves as the fluorescence control and is treated only with the secondary antibody. The second sample is incubated with both primary and secondary antibody to stain specifically for paclitaxel.
To stain for paclitaxel, the sample is incubated with the primary antibody at a dilution ratio of 1:500 (see Notes 12 and 13) at typical culture conditions (125 rpm, 23 °C and dark) for 1 h. After incubation, the sample is washed two times with one-half volume of TBS-T, in a similar manner as described in step 1 above. Both the stained sample and the control sample are then incubated with secondary antibody at a dilution ratio of 1:500 for 1 h at typical culture conditions (see Notes 12 and 13). Thereafter, each sample is washed two times with TBS-T as described above.
Both the control and stained samples are filtered through 80 μm mesh (see Section 3.2.1.4), re-suspended in equal volume of PBS and transferred to 5 mL round-bottom tubes for flow cytometric analysis (see Note 14).
3.4. Flow Cytometric Analysis and Sorting
3.4.1. Flow Cytometric Analysis of Paclitaxel Accumulation
The BD LSR II is used for the analysis of paclitaxel accumulation in stained single cells. The cytometer setup is performed according to the instructions in the BD LSR II User’s Guide. Typical steps in the setup procedure include starting the computer and the cytometer, setting up the optical filters and mirrors, preparing sheath and waste containers, and configuring the fluidics system. A cytometer quality control (QC) procedure is performed prior to sample analysis. To optimize laser and fluidics performance, QC calibration beads (Sphero™ rainbow fluorescent particles) are run first, results recorded and setup parameters are adjusted based on bead performance. Refer to the BD LSR II User’s Guide for more details.
Once the cytometer setup is complete, the settings for the sample type and fluorochromes used are selected (i.e., selection of filter sets to be used to collect data) before running a sample. The BD LSR II is generally equipped with 488 nm and 633 nm fixed alignment lasers. The primary blue 488 nm laser can detect 5 fluorochromes (including the PE fluorochrome conjugated to the antibody used in the immunostaining for paclitaxel), while the red 633 nm laser can detect 3 fluorochromes.
The control cell sample (see Section 3.3.3) is run first through the BD LSR II cytometer (see Note 15). Typically, data collected for immunofluorescent analysis includes forward scatter (FSC), side scatter (SSC), and PE-fluorescence. Data are visualized real-time using scatter dot plots (FSC and SSC) and histograms (PE). Voltage settings on the photomultiplier tubes (PMT) for each detector must be adjusted so that measurements are scaled properly to spread across the dot plot; otherwise data points may be grouped at the maximum or minimum of the relevant axis. Additionally, the PE detector voltage should be set so that the control histogram falls at the low end of the fluorescent range, since positively stained samples will have a higher fluorescence and must not reach the maximum of the range. Once voltage settings are adjusted, they remain unchanged for analysis of subsequent samples so that different samples can be directly compared to each other (see Note 16).
Subsequently, data for 10,000 control cells are recorded. If necessary, the FSC threshold can be adjusted and/or a manual gate drawn to exclude debris from the population of interest and defined as population P1 (Fig. 3.1a).
The stained cell sample is then run on the flow cytometer and data for 10,000 cells are recorded. The stained sample histogram is analyzed and the non-overlapping part, using the same relative fluorescence cutoff, is obtained. The cells in this region are the PE-positive cells and represent the cells in the culture that are positively stained for paclitaxel (Figs. 3.1b and 3.1c). Cells having fluorescence which exceeds 99.5 % of that of the control cells are defined as positively stained cells (8). [Fig. 3.1 near here]
Fig 3.1.
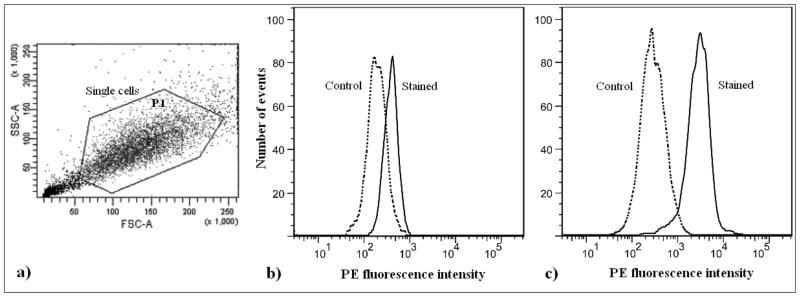
(a) Flow cytometric scatter dot plot (FSC vs. SSC) of Taxus cuspidata P991 single cells, stained for paclitaxel (see Section 3.3). The figure shows a method for gating the population of interest while recording the fluorescent data. A manual polygon-shaped gate, excluding the debris and aggregates, is drawn in the dot plot before recording the data. Debris, which is characterized by low FSC and low SSC values in the dot plot, is largely comprised of cellular fragments and undesirable sub-cellular particles. While these are often counted as individual events by the cytometer, they are not intact single cells and do not accurately represent the correct metabolic information pertaining to the stained cells. Aggregates and larger particles, which are formed by fusion of two or more cells, are also not included in the analysis, owing to their incorrect optical measurements. (b and c) Flow cytometric histograms of control (secondary antibody only) and stained (primary and secondary antibody) samples of Taxus cuspidata P991 single cells: (b) cells were not elicited with methyl jasmonate (−MJ), i.e., “low/nonpaclitaxel-accumulating” cells; (c) cells were elicited with methyl jasmonate (+MJ), i.e., “paclitaxel-accumulating cells”. Both elicited and nonelicited cells were stained under identical conditions (see Section 3.3), and can thus be compared on the basis of their fluorescent histograms. Percentage positive is defined as the percentage of stained cells above the threshold set by 99.5 % of control cells (8). In this experiment, nonelicited cells were 2 % positive, while elicited cells were 89 % positive, indicating that significantly more elicited cells accumulate paclitaxel. The relative fluorescent intensity, defined as the difference between the mean fluorescence of stained and unstained cells, can also be used to compare samples. In this experiment, the relative fluorescent intensities of nonelicited and elicited cells were 882 and 2895, respectively, also indicating that elicited cells accumulate more paclitaxel than nonelicited cells. These data indicate cell-cell heterogeneity with regards to secondary metabolite accumulation, as demonstrated by the distribution in paclitaxel-related fluorescence in the samples. This type of analysis can be used to understand secondary metabolite heterogeneity in cell culture populations to ultimately suggest strategies for optimizing accumulation and stability.
3.4.2. Sorting of Single Cells and Protoplasts
A BD FACSVantage, a special order system equipped with a 200 μm nozzle and a MacroSort™ option, is used for sorting of single cells and protoplasts. The sorter setup is performed according to the instructions in the BD FACSVantage User’s Guide, which includes, starting up the instrument, instrument optimization and quality control, and sample processing. QC calibration beads (AlignFlow™ flow cytometry alignment beads, 2.5 μm for 488 nm excitation) are run and the data are used to align the 488 nm laser. The signal from this primary laser is optimized for consistency by adjusting controls such as nozzle alignment, objective lens, FSC obscuration bar, beam focus, fluorescence focus, etc. If using a two-color sorting, the signals from the secondary laser (633 nm) should also be optimized using the AlignFlow™ flow cytometry alignment beads, 2.5 μm for 633 nm excitation. Refer to the BD FACSVantage User’s Guide for more details.
Before recording data, instrument settings (FSC and SSC voltages, FSC threshold, and fluorescence PMT voltages) are optimized by running large beads (Megabead NIST Traceable Particle Size Standard, 25.0 μm), which represent an average plant cell size. These beads are sorted based on scatter properties, and the configuration settings corresponding to high sort purity (> 90%) are recorded. The instrument is subsequently kept at the same settings to sort plant cells and protoplasts (see Note 17). Refer to the BD FACSVantage User’s Guide for more details.
Isolated single cells (see Section 3.2.1) and protoplasts (see Section 3.2.2) are then separately run through the cell sorter. The data are acquired on a FSC vs. SSC scatter dot plot for a minimum of 10,000 events. The events, corresponding to the cells to be sorted, are selected from the dot plot based on high FSC and SSC properties, and gated as P1 (Figs. 3.2a and 3.2c). These cells/protoplasts are then sorted in the FACSVantage and collected in tubes containing sterile 50:50 medium (see Note 11).
The sorted cells/protoplasts are then analyzed for sort purity by running the recovered populations through the sorter. The scatter data corresponding to the sorted cells/protoplasts are recorded and the number of events lying inside the gated P1 region is determined (Figs. 3.2b and 3.2d). Sort purity is defined as the percentage of events in the post-sorting plot that fall within the P1 region. [Fig. 3.2 near here]
Fig 3.2.

Flow cytometric scatter dot plots of sorting of single cells (a and b) and protoplasts (c and d) by size: (a) single cells before sorting, 52 % (P1) of the entire parent events were selected for sorting based on high FSC and SSC values; (b) single cells after sorting, 91 % of the selected events fall within P1 in the sorted cells plot; (c) protoplasts before sorting, 62 % (P1) of the entire parent events were selected for sorting based on high FSC and SSC values; and (d) protoplasts after sorting, 93 % of the selected events fall within P1 in the sorted protoplasts plot. Single cells and protoplasts were isolated from Taxus cuspidata cell line P991 (see Section 3.2) and were run through a BD FACSVantage (see Section 3.4.2). High sort purities (91 % for single cells and 93 % for protoplasts) were obtained for sorting based on size. These results demonstrate the feasibility of sorting both intact Taxus cells and protoplasts. These distinct populations differing in size and complexity can be isolated and further analyzed to investigate culture heterogeneity.
3.5. Preparation of Nuclei Suspensions from Aggregated Cultures
For preparation of nuclei suspensions, cells from day 12 of the culture (note: here cells were day 12, but cells could be isolated from any culture time) are vacuum-filtered through Miracloth® and approximately 0.5 g of cells are transferred to the center of a plastic Petri dish.
The frozen nuclei isolation buffer is thawed at room temperature (approximately 30 minutes) and allowed to liquefy completely. Approximately 1–3 mL of the cold nuclei isolation buffer is added to the Petri dish (32).
Cells are immersed in the buffer and chopped immediately with the mini-glass scraper for five minutes (32) (see Note 18). The resultant sample is mixed properly with the buffer several times by tilting the Petri dish to ensure uniform chopping of the entire sample. If necessary, additional buffer can be added to facilitate chopping (Fig. 3.3a).
The chopped sample is then transferred to a 15 mL centrifuge tube and PBS is added to make up the volume to 10 mL.
The nuclei suspension is then filtered through 80 μm mesh (see Section 3.2.1.4) and transferred into sterile 15 mL centrifuge tubes for staining of DNA.
Fig 3.3.

(a) Preparation of Taxus cuspidata P991 nuclei suspensions: cells were placed in a Petri dish with the isolation buffer and chopped with a mini glass scrapper (see Section 3.5); and (b) histogram of PI fluorescence intensity as a measurement of relative 2C nuclear DNA content obtained after PI staining and subsequent flow cytometric analysis of the isolated nuclei suspension (see Sections 3.6 and 3.7). Note that there are two peaks in the histogram. A nucleus in the first peak has two copies of the unreplicated genome and has a relative DNA content of 2C. Similarly, a nucleus in the second peak has 4C relative DNA content. The two peaks can be gated and their mean fluorescence intensities determined which can be used to estimate the absolute DNA content using known standards. This method can be used to explore culture variability by determining the DNA content/genome size variation across different cells lines and under different culture conditions.
3.6. Staining of Nuclear DNA
For staining of DNA, both PI and Ribonuclease A are added to the nuclei suspension (isolated from nonelicited cultures as described above) at final concentrations of 50 μg/mL each (see Note 19). The sample is mixed by inverting the tubes several times. The sample is then incubated on ice for 15 minutes.
The PI stained sample is filtered through 80 μm mesh and transferred to 5 mL round-bottom tubes for flow cytometric analysis.
3.7. Flow Cytometric Analysis of Stained Nuclei
The BD LSR II analytical flow cytometer is used to measure the fluorescence of the stained nuclei suspensions. The fluorochrome used is PI, which can be detected using the 488 nm laser under the PI detector. Refer to Section 3.4.1 for cytometry setup and other related procedures.
The PI stained sample is run in the flow cytometer and data for 5,000 events are acquired in a histogram chart containing PI fluorescence intensity as a measure of relative nuclear DNA content. The scatter dot plot is gated manually to eliminate background noise arising from debris or aggregates (see Notes 20 and 21) (Fig. 3.3b). [Fig. 3.3 near here]
Minimize interruptions during operation of the flow cytometer (see Notes 22 and 23)
Table 1.1.
Examples of some important flow cytometric-measurable parameters and measurement techniques and/or fluorescent probes.
| Measurable variables | Measurement techniques and/or associated probe(s) | Examples |
|---|---|---|
| Cell count and size | Scatter dot plots | Nicotiana tabacum (9) |
| DNA, ploidy, cell cycle | DAPI, mithramycin, propidium iodide | Dioscorea (10) |
| Chlorophyll content | Autofluorescence | Nicotiana tabacum (11) |
| Cell viability | PI and fluorescein diacetate (FDA) | Brassica napus (12) |
| Cell-wall components | Specific antibodies, Calcofluor white | Brassica napus (13) |
| Apoptosis and related studies | Annexin-V | Nicotiana plumbaginifolia (14) |
| Membrane fluidity | Diphenylhexatriene (DPH) | Lupinus albus L. (15) |
| Membrane potential | Rhodamine 123 | Solanum tuberosum L. (16) |
| Intracellular Ca2+ | Indo-1, fura-2 | Barley aleurone (17) |
| Gene expression | GFP or other reporter genes | Arabidopsis (18) |
| Intracellular molecules | Specific dyes, antibodies tagged with fluorescence after cell fixation and permeabilization | Coptis japonica (19) |
Acknowledgments
The authors would like to acknowledge support for this work by the National Science Foundation (CBET 0730779 and CBET 0521223) and the National Institutes of Health (R01 GM070852-01). The authors would like to thank Dr. Donna Gibson of the US Plant Soil and Nutrition Laboratory of the USDA for the Taxus cell cultures.
Footnotes
Nanopure water used to prepare solutions should have a resistivity greater than 18 MΩ-cm.
All culture medium preparation and cultivation steps should be carried out in a laminar flow hood under sterile conditions.
Supplemental plant anti-oxidants and nutrients should be made fresh and added to the autoclaved media under sterile conditions. These compounds are labile and degrade upon autoclaving.
Osmoticum should be prepared a day before single cell and protoplast isolation to enable potential equilibrium, and stored at room temperature.
Cell wall digesting enzymes are labile and will denature upon heating, so ensure that enzyme solutions are not heated to facilitate dissolution. Also, do not vortex enzymes, as proteins are more readily denatured at air-water interfaces, particularly at high speeds. Dissolve the enzymes by adding small amounts in succession with intermittent mixing/shaking through inversion by hand.
Specific enzyme concentrations to efficiently dissociate aggregates into single cells and protoplasts must be determined for different plant species and cell types. The single cell yield (SCY), which represents the percentage of cell clusters that contain only a single cell, serves as an indicator of effective disaggregation and can be obtained using hemacytometry (3). Working with flow cytometry necessitates the use of single cell suspension, and therefore, obtaining a high SCY is critical to ensure accurate representation of aggregated cultures.
The amounts of isolated single cells and protoplasts depend on the relevant application. For instance, relatively greater volumes will be required for a cell sorting application as compared to analytical flow cytometric evaluation. Typical reaction containers used are 15 mL flasks, six-well plates, and 1.5 mL centrifuge tubes.
Isolated single cells and protoplasts should be handled with special care to avoid any breakage. Do not centrifuge for either longer periods or at higher speeds than indicated.
Minimize sample/cell loss during washing and other steps by removing the supernatant carefully with a syringe and needle, instead of either decanting or using a pipette tip.
When diluting the isolated cell suspension to the required level, always consider to account for cell loss during subsequent steps (e.g., staining, washing, etc.). It is advisable to maintain a cell density slightly above the recommended levels to account for any potential loss during manipulation.
It is advisable to analyze/sort samples in the flow cytometer immediately after preparation. However, if required, isolated single cells can be stored overnight in a tailor-made 50:50 medium (50 % fresh medium + 50 % conditioned medium from day 12 of the culture period) without affecting viability or SCY.
In order to avoid any fluorescence loss, protect the cells from light while incubating with the fluorophore-attached secondary antibody by wrapping containers with metal foil or keeping the containers inside a larger opaque container.
Optimal antibody concentrations must be determined in a titration experiment, in which increasing concentrations of antibodies are used until fluorescence saturation is reached. In the case of abundant molecules present in high concentrations, for which saturation is not possible, a constant antibody concentration may be used, in which case relative fluorescence shifts can be compared between different samples.
Single cells can be filtered a final time through 80 μm mesh prior to flow cytometric analysis, if any aggregation is observed in the sample. This is particularly important for cell sorting applications as aggregates can complicate the creation of a stable hydrodynamic flow in the instrument.
Gently invert the round-bottom tubes containing cells to be analyzed several times to properly suspend cells, and then run enough cells (~2,000, provided sufficient cells are available) in the BD LSR II cytometer before recording data. This will help stabilize the sample cell counting rate.
Once the appropriate PMT voltage settings for the control are obtained, do not change them. Both the control and the stained sample should be run at the same voltage settings. Since the control sample is used to set the level of background fluorescence, it is advisable to check for any possible non-specific binding of the secondary antibody. Ensure that the background fluorescence (i.e., control) is on the order of endogenous fluorescence associated with unstained (i.e., no secondary antibody) cells.
Sorting of single cells and protoplasts should be performed at optimized flow and sorting conditions for the instrument (26). Parameters may vary with the type of instrument and sample; therefore, an extensive optimization to obtain a suitable working range for the following parameters should be carried out – sheath pressure, drop drive frequency, break-off point, drop delay range, and sample differential.
The amount of cells and buffer as well as chopping time should be determined according to the species to enable effective nuclei isolation. Note that some cells may be more resistant to breakage. The razor should be sharp and used only once. Do not chop vigorously or significant cell damage will occur.
As PI also binds to dsRNA, add RNase simultaneously with PI to the nuclei suspension. Also, heat the RNase solution at 90 °C for 15 minutes to deactivate any DNase activity.
In order to obtain PI peaks without undesirable low channel signals, choose an appropriate PMT voltage threshold to minimize off signals coming from cell debris and autofluorescent compounds. The scatter dot plots may be gated to exclude any debris and aggregates as described previously.
In case of significant debris and aggregates in the isolated nuclei suspension, the concentration of the nonionic detergent (i.e., Triton X-100) can be increased in the isolation buffer. Increasing concentrations aid in the release and cleaning of nuclei solutions, by decreasing the aggregation affinity of nuclei and debris without affecting the fluorescence properties of the dye molecule (33). Besides, there are several possible reasons (e.g., improper staining protocol, vigorous chopping, blunt razor blade, incorrect instrument operation and data acquisition, negative cytosolic effects, recalcitrant tissues, etc.) for broad peaks and large amount of debris background that should be appropriately addressed (34).
Avoid air bubbles in the flow cytometer system by minimizing sample mixing and tube changes during operation.
If there is a clog in the flow chamber or line tubing, or the sheath pressure and/or the event rate drops unexpectedly, pause the sorting, clean the flow chamber, tubing, in-line filters, and inspect for leaks. Cleaning is performed using the cytometer sterilization kit (10 % bleach, 70 % ethanol and distilled water) as mentioned in the User’s Guide for the instrument.
References
- 1.Kolewe ME, Gaurav V, Roberts SC. Pharmaceutically active natural product synthesis and supply via plant cell culture technology. Molecular Pharmaceutics. 2008;5(2):243–256. doi: 10.1021/mp7001494. [DOI] [PubMed] [Google Scholar]
- 2.Roberts SC, Shuler ML. Large-scale plant cell culture. Current Opinion in Biotechnology. 1997;8:154–159. doi: 10.1016/s0958-1669(97)80094-8. [DOI] [PubMed] [Google Scholar]
- 3.Naill MC, Roberts SC. Preparation of single cells from aggregated Taxus suspension cultures for population analysis. Biotechnology and Bioengineering. 2004;87(6):817–826. doi: 10.1002/bit.20083. [DOI] [PubMed] [Google Scholar]
- 4.Naill MC, Roberts SC. Flow cytometric identification of paclitaxel-accumulating subpopulations. Biotechnology Progress. 2005;21:978–983. doi: 10.1021/bp049544l. [DOI] [PubMed] [Google Scholar]
- 5.Naill MC, Roberts SC. Cell cycle analysis of Taxus suspension cultures at the single cell level as an indicator of culture heterogeneity. Biotechnology and Bioengineering. 2005;90(4):491–500. doi: 10.1002/bit.20446. [DOI] [PubMed] [Google Scholar]
- 6.Naill MC, Roberts SC. Flow cytometric analysis of protein content in Taxus protoplasts and single cells as compared to aggregated suspension cultures. Plant Cell Reports. 2005;23:528–533. doi: 10.1007/s00299-004-0875-y. [DOI] [PubMed] [Google Scholar]
- 7.Yanpaisan W, King N, Doran P. Flow cytometry of plant cells with applications in large-scale bioprocessing. Biotechnology Advances. 1999;58:515–528. doi: 10.1016/s0734-9750(98)00014-7. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro HM. Practical flow cytometry. 4. John Wiley & Sons, Inc; Hoboken, NJ: 2003. [Google Scholar]
- 9.Nicoloso FT, Val J, van der Keur M, van Iren F, Kijne JW. Flow-cytometric cell counting and DNA estimation for the study of plant cell population dynamics. Plant cell, tissue and organ culture. 1994;39:251–259. [Google Scholar]
- 10.Obidiegwu JE, Rodriguez E, Ene-obong EE, Loureiro J, Muoneke CO, Santos C, Kolesnikova-Allen M, Asiedu R. Estimation of the nuclear DNA content in some representative of genus Dioscorea. Scientific Research and Essay. 2009;4(5):448–452. [Google Scholar]
- 11.Galbraith DW, Harkins KR, Jefferson RA. Flow cytometric characterization of the chlorophyll contents and size distributions of plant protoplasts. Cytometry. 1988;9(1):75–83. doi: 10.1002/cyto.990090112. [DOI] [PubMed] [Google Scholar]
- 12.Schulze D, Pauls KP. Flow cytometric characterization of embryogenic and gametophytic development in Brassica napus microspore cultures. Plant and Cell Physiology. 1998;39(2):226–234. [Google Scholar]
- 13.Schulze D, Pauls KP. Flow cytometric analysis of cellulose tracks development of embryogenic Brassica cells in microspore cultures. New Phytologist. 2002;154(1):249–254. [Google Scholar]
- 14.O’Brien IEW, Reutelingsperger CPM, Holdaway KM. Annexin-V and TUNEL use in monitoring the progression of apoptosis in plants. Cytometry. 1997;29:28–33. [PubMed] [Google Scholar]
- 15.Gantet P, Hubac C, Brown SC. Flow cytometric fluorescence anisotropy of lipophilic probes in epidermal and mesophyll protoplasts from water-stressed Lupinus albus L. Plant Physiology. 1990;94:729–737. doi: 10.1104/pp.94.2.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petit PX. Flow cytometric analysis of Rhodamine 123 fluorescence during modulation of the membrane potential in plant mitochondria. Plant Physiology. 1992;98:279–286. doi: 10.1104/pp.98.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bush DS, Jones RL. Measurement of cytoplasmic calcium in aleurone protoplasts using indo- 1 and fura-2. Cell Calcium. 1988;8:455–472. doi: 10.1016/0143-4160(87)90029-7. [DOI] [PubMed] [Google Scholar]
- 18.Birnbaum K, Shasha DE, Wang JY, Jung JY, Lambert GM, Galbraith DW, Benfey PN. A gene expression map of the Arabidopsis root. Science. 2003;302(5652):1956–1960. doi: 10.1126/science.1090022. [DOI] [PubMed] [Google Scholar]
- 19.Hara Y, Yamagata H, Morimoto T, Hiratsuka J, Yoshioka T, Fujita Y, Yamada Y. Flow cytometric analysis of cellular berberine contents in high- and low-producing cell lines of Coptis japonica obtained by repeated selection. Planta Medica. 1989;55(2):151–154. doi: 10.1055/s-2006-961910. [DOI] [PubMed] [Google Scholar]
- 20.Sklar LA. Flow cytometry for biotechnology. 1. Oxford University Press, Inc; New York, NY: 2005. [Google Scholar]
- 21.Safar J, Noa-Carrazana JC, Vrana J, Bartos J, Alkhimova O, Lheureux F, Simkova H, Caruana ML, Dolezel J, Piffanelli P. Creation of a BAC resource to study the structure and evolution of the banana (Musa balbisiana) genome. Genome. 2004;47:1182–1191. doi: 10.1139/g04-062. [DOI] [PubMed] [Google Scholar]
- 22.Birnbaum K, Jung JW, Wang JY, Lambert GM, Hirst JA, Galbraith DW, Benfey PN. Cell type-specific expression profiling in plants via cell sorting of protoplasts from fluorescent reporter lines. Nature Methods. 2005;2:615–619. doi: 10.1038/nmeth0805-615. [DOI] [PubMed] [Google Scholar]
- 23.de Laat AMM, Blaas J. Flow-cytometric characterization and sorting of plant chromosomes. TAG Theoretical and Applied Genetics. 1984;67(5):463–467. doi: 10.1007/BF00263414. [DOI] [PubMed] [Google Scholar]
- 24.Becker JD, Boavida LC, Carneiro J, Haury M, Feijo JA. Transcriptional profiling of Arabidopsis tissues reveals the unique characteristics of the pollen transcriptome. Plant Physiology. 2003;133:713–725. doi: 10.1104/pp.103.028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dolezel J, Greilhuber J, Suda J, editors. Flow cytometry with plant cells. Wiley-VCH; Weinheim, Baden-Württemberg, Germany: 2007. (illustrated ed.) [Google Scholar]
- 26.Harkins KR, Galbraith DW. Factors governing the flow cytometric analysis and sorting of large biological particles. Cytometry. 1987;8:60–70. doi: 10.1002/cyto.990080110. [DOI] [PubMed] [Google Scholar]
- 27.Freyer JP, Fillak D, Jett JH. Use of xantham gum to suspend large particles during flow cytometric analysis and sorting. Cytometry. 1989;10:803–806. doi: 10.1002/cyto.990100620. [DOI] [PubMed] [Google Scholar]
- 28.Gamborg OL, Miller RA, Ojima K. Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res. 1968;50:151–158. doi: 10.1016/0014-4827(68)90403-5. [DOI] [PubMed] [Google Scholar]
- 29.Roberts SC, Naill M, Gibson DM, Shuler ML. A simple method for enhancing paclitaxel release from Taxus canadensis cell suspension cultures utilizing cell wall digesting enzymes. Plant Cell Reports. 2003;21(12):1217–1220. doi: 10.1007/s00299-003-0575-z. [DOI] [PubMed] [Google Scholar]
- 30.Aoyagi H, DiCosmo F, Tanaka H. Efficient paclitaxel production using protoplasts isolated from cultured cells of Taxus cuspidata. Planta Med. 2002;68(5):420–424. doi: 10.1055/s-2002-32082. [DOI] [PubMed] [Google Scholar]
- 31.Ketchum REB, Gibson DM, Croteau RB, Shuler ML. The kinetics of taxoid accumulation in cell suspension cultures of Taxus following elicitation with methyl jasmonate. Biotechnology and Bioengineering. 1999;62:97–105. doi: 10.1002/(sici)1097-0290(19990105)62:1<97::aid-bit11>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 32.Galbraith DW, Harkins KR, Maddox JM, Ayres NM, Sharma DP, Firoozabady E. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science. 1983;220(4601):1049–1051. doi: 10.1126/science.220.4601.1049. [DOI] [PubMed] [Google Scholar]
- 33.Kapuscinski J. DAPI: a DNA-specific fluorescent probe. Biotech Histochem. 1995;70(5):220–233. doi: 10.3109/10520299509108199. [DOI] [PubMed] [Google Scholar]
- 34.Dolezel J, Greilhuber J, Suda J. Estimation of nuclear DNA content in plants using flow cytometry. Nature Protocols. 2007;2:2233–2244. doi: 10.1038/nprot.2007.310. [DOI] [PubMed] [Google Scholar]