

Abstract
CD93 and GAIP-interacting protein, C termius (GIPC) have been shown to interactively alter phagocytic processes of immune cells. CD93 and GIPC expression and localization during central nervous system inflammation have not yet been reported. In this study, we established a rat model of brain inflammation by lipopolysaccharide injection to the lateral ventricle. In the brain of rats with inflammation, western blots showed increased CD93 expression that decreased over time. GIPC expression was unaltered. Immunohistochemistry demonstrated extensive distribution of CD93 expression mainly in cell membranes in the cerebral cortex. After lipopolysaccharide stimulation, CD93 expression increased and then reduced, with distinct staining in the cytoplasm and nucleus. Double immunofluorescence staining in cerebral cortex of normal rats showed that CD93 and GIPC widely expressed in resting microglia and neurons. CD93 was mainly expressed in microglial and neuronal cell membranes, while GIPC was expressed in both cell membrane and cytoplasm. In the cerebral cortex at 9 hours after model establishment, CD93-immunoreactive signal diminished in microglial membrane, with cytoplasmic translocation and aggregation detected. GIPC localization was unaltered in neurons and microglia. These results are the first to demonstrate CD93 participation in pathophysiological processes of central nervous system inflammation.
Keywords: nerve regeneration, central nervous system, brain inflammation model, CD93, GIPC, neurons, microglia, expression and localization, lipopolysaccharide, intracerebroventricular injection, rats, inducible nitric oxide synthase, NSFC grants, neural regeneration
Introduction
Inflammatory reactions in the central nervous system are involved in various neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, and multiple sclerosis (Tansey and Goldberg, 2010; Alvarez et al., 2011; Xiong et al., 2011), and are also strongly associated with stroke, brain trauma, and meningitis (Bal-Price and Brown, 2003; Wyss-Coray, 2006; Block et al., 2007). A previous study found that nervous system inflammation is usually accompanied by neuronal injury induced by microglial activation and inflammatory cytokine release (Harry and Kraft, 2008; Lu et al., 2013a, b). Microglia are macrophages in the central nervous system that play a key role in maintaining homeostasis (Kettenmann et al., 2011). Under normal circumstances, microglia in the resting state monitor homeostasis, secrete neurotrophic factors, and support neuronal function. During pathological trauma and inflammation, microglia identify receptors and recognize extracellular danger signals through their surface, and convert to activated phenotypes by a series of intracellular cascades, thereby exerting immune defense functions and scavenging damaged cell debris (Akira et al., 2006; Takeuchi and Akira, 2010). However, excessively activated microglia release large amounts of proinflammatory mediators and can result in secondary nerve damage (Ulvestad et al., 1994; Van der Laan et al., 1996).
CD93 is a type I transmembrane glycoprotein (molecular weight, 126 kDa) that is expressed on the surface of platelets, and myeloid, endothelial, hematopoietic stem, and natural killer cells. CD93 is an inflammatory regulator strongly associated with immune cell phagocytosis, which regulates innate immunity and inflammation (Harhausen et al., 2010). By evaluating the effects of CD93 for examining relevant clinical results, a recent study confirmed a relationship between soluble CD93 levels and acute myocardial infarction (Youn et al., 2014). CD93 has been shown to play an important role in clearing apoptotic cells and maintaining homeostasis in normal tissue during embryonic development (Henson, 2014). In mouse models of peritonitis, soluble CD93 expression is detected, and CD93 secretion strongly associated with macrophage phagocytosis and clearance of apoptotic cells (Greenlee et al., 2009). In addition, membrane-associated CD93 regulates leukocyte migration and complement activation in a CD93−/− mouse model of peritonitis (Greenlee-Wacker et al., 2011). CD93 deletion also induces phagocyte impairment on scavenging apoptotic cells (Greenlee et al., 2009). The CD93-mediated neuroprotective mechanism of CCL21 down-regulation has been confirmed in a study of neuritis (Harhausen et al., 2010). A regulator of G protein signaling, GIPC (GAIP-interacting protein, C terminus), is expressed in the plasma membrane or within transferrin-containing vesicles located near the membrane, and a previous study found that GIPC can alter cell migration (Gao et al., 2000; Ligensa et al., 2001). Studies addressing GIPC have mainly focused on its interaction with GAIP and the G protein signaling pathway (Lou et al., 2001; Booth et al., 2002), but GIPC also interacts with myosin VI and is involved in protein transport, endocytosis, cell migration, and receptor clustering (Muders et al., 2006). Importantly, through its PDZ domain, GIPC interacts with 11 amino acids in the cytoplasmic tail of CD93 (Bohlson et al., 2005), possibly regulating phagocytosis-related membrane movement and cytoskeletal remodeling.
Nerve inflammation is strongly correlated with many neurological and pathological processes (Zindler and Zipp, 2010; Dong et al., 2012; Anthony and Couch, 2014). Interleukin-1β, interleukin-6, and tumor necrosis factor-α exert crucial effects on pathophysiological processes (Cekanaviciute et al., 2013; Vidal et al., 2013; Zhao et al., 2013; Almolda et al., 2014; Fenn et al., 2014). Yet involvement of CD93 and GIPC expression and localization after nerve inflammation is poorly understood. In this study, we established a rat model of brain inflammation by lipopolysaccharide injection to the lateral ventricle. To determine if CD93 and GIPC are associated with physiological and pathological processes of brain inflammation, we measured CD93 and GIPC expression by western blot assay. In addition, to determine if CD93 and GIPC colocalize in neurons and microglia, we performed double immunofluorescence staining.
Materials and Methods
Experimental animals
A total of 32 healthy male Sprague-Dawley rats weighing 180–220 g were provided by the Experimental Animal Center of Nantong University in China (Production License No. SCXK (Su) 2008-0010). All rats were housed in a specific-pathogen-free barrier environment at 23 ± 2°C with 55 ± 5% humidity and a 12-hour light/dark cycle. Rats were fed with 60Co-irradiated complete pellet feed (Suzhou Shuangshi Laboratory Animal Feed Science and Technology Co., Ltd., Suzhou, Jiangsu Province, China), and allowed free access to food and water (Animal Use License No. SYXK (Su) 2012-0030).
Experimental groups and establishment of rat model of brain inflammation
A total of 32 rats were randomly and equally assigned to sham surgery, control, and experimental groups (six time points). Rats were intraperitoneally anesthetized with sodium pentobarbital (0.2 mL/100 g), and aseptically fixed in the prone position using a Kopf brain tropism locator (Anhui Zhenghua Bio Equipment Co., Ltd., Huaibei, Anhui Province, China), ensuring the anterior fontanelle was 1 mm higher than the posterior fontanelle, according to the rat brain atlas (Paxinos and Watson, 2006). 5 μL of lipopolysaccharide (5 μg/μL; Sigma, St. Louis, MO, USA) was injected at 10 μL/5 minutes into the lateral ventricle (0.2 mm posterior to the anterior fontanelle, rostral 1.4 mm, 4.0 mm depth). In the sham surgery group, the syringe was inserted into the lateral ventricle but nothing was injected. In the control group, 5 μL of physiological saline (0.9%) was injected. The wound was rinsed, sutured with 4-0 silk suture, and covered with a sterilized dressing. Rats were then housed at room temperature and allowed free access to food. At 2, 4, 6, 9, 12, and 24 hours after lipopolysaccharide injection, 24 rats in the experimental groups and 4 rats in the control group were intraperitoneally anesthetized. Brain tissue was exposed in a dark room and the cerebral cortex dissected, placed in 1.5 μL centrifuge tubes, and stored in a −80°C ultra-low temperature freezer (Jouan, Gaithersburg, MD, USA).
Sample collection and preparation of tissue sections
Physiological saline (50 mL, 37°C) was perfused into the ascending aorta via the left ventricle, and the tissue fixed using 4% paraformaldehyde (200 mL, 4°C) for 40–60 minutes. Brains were removed and fixed in fixative for 2 hours, then immersed in 20% and 30% sucrose solutions (4°C). After tissue blocks had sunk, 8 μm thick serial frozen sections were cut, placed on polylysine-coated slides, and stored at −80°C.
Western blot assay
Brain tissue was obtained from −80°C freezer storage, weighed, and placed in a centrifuge tube. Protein lysates were prepared at 1 mL/100 mg. To rewarm lysates, centrifuge tubes were placed in an ice water mixture for 30 minutes. After homogenizing in an ice bath, lysates were incubated without stirring for 30 minutes, and then centrifuged for 15 minutes at 12,000 × g, 4°C. Protein content was measured using bicinchoninic acid protein assay kits (Beyotime, Haimen, Jiangsu Province, China). Proteins were stored separately at −80°C. Sodium dodecyl sulfate polyacrylamide gel electrophoresis and transmembrane blotting were performed next. To denature proteins, protein in 2 × sodium dodecyl sulfate loading buffer and dithiothreitol (4:5:1) was boiled in water for 5 minutes. Next, separation (10%) and stacking (5%) gels were prepared and 2 × sodium dodecyl sulfate added for every 50 μg protein. Protein molecular weight standard samples (Beyotime) were run in control wells. Electrophoresis was performed at 100 V (through the separation gel) and 150 V (through the stacking gel). Proteins were separated and electronically transferred onto polyvinylidene fluoride membrane (Roche, Vaud, Switzerland) at 300 mA for 150 minutes. Membranes were washed in Tris-buffered saline with Tween-20, and blocked with milk blocker (2.5% bovine serum albumin) at room temperature for 2 hours. Membranes were then incubated in milk blocker diluted (1:1,000) with mouse anti-CD93 monoclonal antibody (Abcam, Cambridge, UK), mouse anti-GIPC monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-inducible nitric oxide synthase monoclonal antibody (Santa Cruz Biotechnology), or mouse anti-β-actin monoclonal antibody (Sigma) at room temperature for 8 hours or 4°C overnight. After five washes in Tris-buffered saline with Tween-20 (10 minutes each), membranes were incubated with goat anti-mouse horseradish peroxidase-coupled secondary antibody diluted in Tris-buffered saline with Tween-20 containing 10% skimmed milk at room temperature for 120 minutes. After washing in Tris-buffered saline with Tween-20, membranes were incubated with Luminol Ecl Reagent (using equivalent volumes of liquids A and B) at 0.125 mL/cm2 at room temperature for 5 minutes. Membranes were exposed onto sensitive X-ray film, visualized, and fixed. Images were scanned using a scanner (Hewlett-Packard Development Company Palo Alto, CA, USA). Expression was determined by calculating the gray value of target band/gray value of internal reference, β-actin.
Immunohistochemical staining
Tissue sections stored at −80°C were re-warmed at room temperature until the slides had dried. Sections were washed three times in 0.01 mol/L PBS for 5 minutes each, then incubated in freshly prepared 3% H2O2-methanol (50 mL, 37°C) for 30 minutes, and blocked in 10% serum at room temperature for 2 hours. After removal of redundant serum, sections were incubated with rabbit anti-CD93 cytoplasmic tail C11 polyclonal primary antibody 1150 (1:50; University of California, Irvine, CA, USA). Blank control sections were incubated with antibody diluent in a wet box at room temperature for 60 minutes. Sections were washed three times with 0.01 mol/L PBS, redundant liquid removed, and then horseradish peroxidase-labeled goat anti-rabbit IgG (1:200; Sigma) added and incubated in a wet box at room temperature for 2 hours. Sections were washed with 0.01 mol/L PBS and visualized using 3,3′-diaminobenzidine. After dehydration, slides were dried at 60°C and mounted using a neutral resin. Samples were observed and photographed using a fluorescence microscope (Leica, Wetzlar, Germany). Images were analyzed and processed using ImageJ image analysis software (National Institutes of Health, Bethesda, MD, USA). Positive expression/mm2 (i.e., number of brown cells) was calculated.
Double immunofluorescence staining
Sections were obtained from −20°C freezer storage, re-warmed for 1 hour, and blocked in 0.01 mol/L serum (same animal source as the secondary antibody; Shanghai Yikesai Biological Products Co., Ltd., Shanghai, China) at room temperature for 2 hours. After removal of redundant serum, a primary antibody mixture was added. Sections were incubated with a combination of mouse cell marker antibody CD11b monoclonal antibody (1:100; AbD Serotec, Kidlington, UK), NeuN monoclonal antibody (1:600; Abcam), rabbit anti-CD93 polyclonal antibody (1:100; Abcam), and rabbit anti-GIPC polyclonal antibody (1:200; Santa Cruz Biotechnology) at room temperature for 2 hours and then 4°C for 16–18 hours. Sections were washed three times in 0.01 mol/L PBS (10 minutes each), and redundant fluid removed. Sections were incubated with FITC-labeled goat anti-mouse secondary antibody IgG (1:100; Abcam) and Cy3-labeled donkey anti-rabbit secondary antibody IgG (1:2,000; BBI, Oakland, CA, USA) at 4°C for 2 hours, washed with 0.01 mol/L PBS, mounted using buffered glycerol, and stored in the dark in a wet box at 4°C. Samples were observed and photographed using a fluorescence microscope (Leica). FITC has an excitation wavelength of 488 nm and Cy3 of 568 nm.
Statistical analysis
Stata 7.0 (Stata Corp, College Station, TX, USA), Sigma Plot 10.0 (Sigma), and Adobe Photoshop 11.0 (Adobe, San Jose, CA, USA) were used for statistics and mapping. Measurement data were expressed as the mean ± SEM. Differences among groups were compared using one-way analysis of variance and the least significant difference tests. P values < 0.05 were considered statistically significant. Experiments were repeated in at least triplicate.
Results
CD93 and GIPC protein expression in a rat model of brain inflammation
Inducible nitric oxide synthase is a rate-limiting enzyme that catalyzes nitric oxide production. Inducible nitric oxide synthase expression is low in normal brain tissue. Activated inducible nitric oxide synthase induces large amounts of nitric oxide, increases vascular permeability, leads to interstitial edema, causes lipid peroxidation in cell membranes, and increases cell apoptosis (Sheng et al., 1998). Therefore, expression of inducible nitric oxide synthase can be used to determine if our rat model of brain inflammation has been successfully established. Accordingly, our western blot results show that inducible nitric oxide synthase expression increased over time, peaking at 12 hours after lipopolysaccharide injection (P < 0.01), and thereby demonstrating successful establishment of our model. We used a well characterized CD93 extracellular domain antibody (ab78859) to detect CD93 expression in our rat model of brain inflammation. Western blot results show that CD93 expression significantly increased at 2 hours after lipopolysaccharide injection, and then decreased (P < 0.05). No significant difference in GIPC expression was observed (Figure 1A, B). These results show that in our rat model of brain inflammation, CD93 protein expression increases and then decreases with time, but GIPC protein expression is unaltered.
Figure 1.
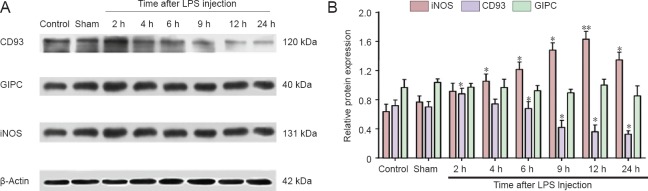
Time course of CD93 and GIPC expression in a rat model of brain inflammation.
(A) Protein expression profiles of CD93, GIPC, iNOS, and the internal reference β-actin in sham surgery, control and experimental groups (at 2, 4, 6, 9, 12, and 24 hours). (B) Statistical results of A: each column represents the ratio of target protein to β-actin at various time points. *P < 0.05, **P < 0.01, vs. sham surgery group (sham). Data are expressed as the mean ± SEM with four rats in each group; one-way analysis of variance and the least significant difference tests were performed. iNOS expression was significantly increased at 4, 6, 9, and 24 hours, and peaked at 12 hours. CD93 immunoreactivity was significantly increased at 2 hours, and then gradually reduced (remaining significant at 9, 12, and 24 hours). GIPC expression was not altered at any time point. GIPC: GAIP-interacting protein, C terminus; iNOS: inducible nitric oxide synthase; LPS: Lipopolysaccharide; h: hours.
CD93 immunoreactivity and distribution in the cerebral cortex in a rat model of brain inflammation
Immunohistochemical staining revealed extensive CD93 distribution throughout the cerebral cortex, mainly in cell membranes (Figure 2A). CD93 immunoreactivity was low in control and sham surgery groups, but at 6 hours after lipopolysaccharide injection, intracellular CD93 immunoreactivity began to increase, peaking at 9 hours before decreasing at 12–24 hours (Figure 2B). This result is not completely consistent with our western blot results, but suggests that the CD93 cytoplasmic tail may play a specific biological role in intracellular function during the immune response. Immunohistochemical staining also demonstrated a significant increase in cytoplasmic CD93-immunoreactive signal at 9 hours after injection (P < 0.01), indicating that with increased inflammatory reaction, CD93 cytoplasmic tail expressed in the cell membrane may migrate towards the cytoplasm or nucleus (Figure 2A c, d).
Figure 2.
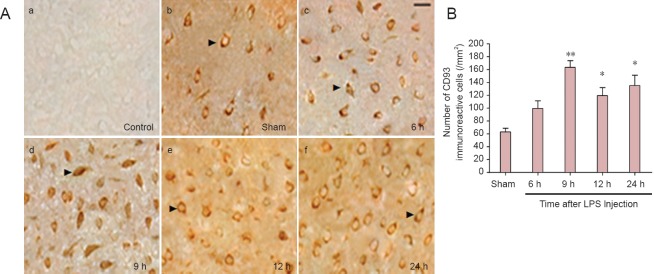
CD93 immunoreactivity and localization in the cerebral cortex of rats with brain inflammation.
(A) Immunohistochemical staining using CD93 antibody in the cerebral cortex of rats with brain inflammation (light microscope). (a) control group; (b) sham surgery (sham) group; (c–f): staining results at 6, 9, 12, and 24 hours after lipopolysaccharide injection. Arrows show CD93-immunoreactive cells. (a) no CD93-immunoreactive signal; (b, e, f) CD93 mainly expressed in cell membrane; (c, d) CD93 expressed in cytoplasm and nuclei. Scare bar: 40 μm. (B) Statistical results of A: each column represents the number of CD93-immunoreactive cells per mm2 at various time points. Compared with the sham group, the number of CD93-immunoreactive cells is higher in experimental groups at various time points. Significant differences are visible at 9, 12, and 24 hours. The number of CD93-immunoreactive cells is greatest at 9 hours. *P < 0.05, **P < 0.01, vs. sham group. Data are expressed as the mean ± SEM with four rats in each group; one-way analysis of variance and the least significant difference tests were performed. LPS: Lipopolysaccharide; h: hours.
Localization of CD93 and GIPC during the early stage of central nervous system inflammation
Immunohistochemical staining showed that cytoplasmic CD93 immunoreactivity increased at 9 hours, suggesting that CD93 may play a specific role in pathophysiological processes during the early stage of central nervous system inflammation. Thus, to further investigate this, double immunofluorescence staining was performed to examine CD93 and GIPC colocalization in neurons (NeuN) and microglia (CD11b) during the early stage (9 hours) of central nervous system inflammation. Our results show colocalization of CD93 and GIPC with both CD11b and NeuN (Figure 3A, B). In the cerebral cortex of control and sham surgery rats, CD93 and GIPC were mainly expressed in resting microglia and neurons. In addition, CD93 was mainly expressed in cell membranes while GIPC was distributed throughout the cell membrane and cytoplasm (Figure 3A a3, b3, Figure 3B a3, b3). Nine hours after lipopolysaccharide injection, many microglia had enlarged volumes and round shapes, with activated phenotypes visible. Moreover, CD93-immunoreactive signal appeared to weaken in microglial cell membranes (Figure 3A b3, b6). GIPC localization did not visibly change either before or after lipopolysaccharide stimulation (Figure 3B a3, a6, b3, b6).
Figure 3.

Localization of CD93 and GIPC in the central nervous system of rats with brain inflammation (fluorescence microscope).
(A) a1, a4: NeuN-labeled neurons (green); b1, b4: CD11b-labeled microglia (green); a2, a5, b2, b5: CD93 antibody staining (red); a3, a6, b3, b6: merged images (yellow indicates colocalized green and red signal). a2, a5, b2, b5: CD95 is mainly expressed in cell membranes of microglia and neurons; compared with b3, microglia show bigger volumes, rounder shapes, and activated phenotypes; CD93-positive signal is obviously weakened. (B) a1, a4: NeuN-labeled neurons (green); b1, b4: CD11b-labeled microglia (green); a2, a5, b2, b5: GIPC antibody staining (red); a3, a6, b3, b6: merged images (yellow indicates colocalized green and red signal). a2, a5, b2, b5: GIPC extensively expressed in the cell membrane and cytoplasm of microglia and neurons; compared with controls, GIPC localization was not significantly altered. Arrows show NeuN-, CD11b-, CD93- and GIPC-positive signals, and colocalized signals in overlay chart. Scare bars: 40 μm. LPS: Lipopolysaccharide. GIPC: GAIP-interacting protein, C terminus.
Discussion
Inflammatory reactions are involved in pathogenesis of certain central nervous system diseases (Gresham et al., 1986), and are a major cause for occurrence and development of neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and multiple sclerosis (Tansey and Goldberg, 2010; Alvarez et al., 2011; Xiong et al., 2011). Nerve inflammation mainly participates in glial cell activation and neuronal injury induced by releasing inflammatory mediators. Glial cells are resident phagocytes of the central nervous system, and play a key role in maintaining homeostasis (Kettenmann et al., 2011; Li et al., 2013). Many dissolved factors (e.g., cytokines and bacterial lipopolysaccharides) and extracellular matrix proteins (e.g., laminin and fibronectin) induce certain inflammatory responses, and enhance phagocytosis of monocytes, macrophages, and neutrophils (Griffin and Mullinax, 1990; Onodera et al., 1997). In this study, we injected lipopolysaccharide into the lateral ventricle of rats, generating a stable rat model of brain inflammation. Next, we investigated CD93 and GIPC expression and localization during central nervous system inflammation to determine if CD93 and GIPC participate in pathophysiological processes of inflammation and examine CD93 and GIPC effects in these processes.
A previous study confirmed that CD93 is an adhesion molecule (Petrenko et al., 1999). Nevertheless, some studies have also shown that CD93 is an inflammatory factor and actively involved in immune phagocytosis of various inflammatory reactions (Tsukita et al., 1999; Ivetic et al., 2002). In combination with related proteins, GIPC regulates endocytosis and cell migration (Muders et al., 2006). In the present study, we investigated the expression and localization of two molecules (CD93 and GIPC) during central nervous system inflammation, which differs from the conventional process of studying a single molecule at a time. Our western blot results show that in our rat model of brain inflammation, CD93 expression is dramatically altered after lipopolysaccharide stimulation. CD93 expression increases and then gradually decreases with prolonged inflammatory reaction time, possibly reflecting behavioral feedback to the inflammatory reaction and suggesting that CD93 participates and plays a specific role in pathophysiological processes of central nervous system inflammation. In contrast, expression of GIPC, a CD93-related protein, was not visibly altered by the inflammatory reaction, suggesting that GIPC is not involved in the inflammatory process.
We used immunohistochemical staining to investigate CD93 immunoreactivity and localization in the cerebral cortex of rats with lipopolysaccharide-induced central nervous system inflammation. Our immunohistochemical results are not consistent with those by western blot assay. We found that CD93 is widely distributed in the cortex, mainly within cell membranes. CD93 immunoreactivity was low in control and sham surgery groups, but increased during inflammatory reaction in the cerebral cortex, reaching a peak at 9 hours after stimulation, and showing distinct cytoplasmic and nuclear staining. These findings suggest that the CD93 cytoplasmic tail is mainly expressed in cell membranes yet may migrate towards the cytoplasm or nucleus with increasing inflammatory reaction. This complicated process warrants further investigation. Double immunofluorescence staining showed that CD93 and GIPC colocalize in neurons and microglia in the early stage (9 hours) of central nervous system inflammation. In the cerebral cortex of normal rats, CD93 and GIPC are mainly expressed in resting microglia and neurons. Moreover, CD93 localizes to cell membranes while GIPC distributes to both the cell membrane and cytoplasm, inconsistent with our protein expression results. There was no significant change in localization before and after lipopolysaccharide stimulation. Thus, alterations in CD93 localization are of interest. At 9 hours after lipopolysaccharide injection, many microglia had increased volumes and rounded shapes with activated phenotypes apparent. Weakened cell membrane-related CD93-immunoreactive signal was also detected in microglia. Normal microglia are in the resting state, becoming activated once a stimulus signal is detected (Akira et al., 2006; Takeuchi and Akira, 2010). Activated microglia can phagocytose and scavenge apoptotic cells, invading pathogens, exogenous substances, and debris (Ulvestad et al., 1994; Van der Laan et al., 1996), and thereby maintain homeostasis. During central nervous system inflammation, glial cell activation is commonly accompanied by inflammatory and proinflammatory cytokine release. However, release of abundant inflammatory cytokines often induces injury to surrounding nerves. After 2 hours of inflammation, CD93 expression increased and then reduced, while at 9 hours, there was weakened CD93-immunoreactive signal in microglia cell membranes. A reasonable explanation for these dramatic changes may be that glial cells promote the early stage of an inflammatory reaction and phagocytize apoptotic cells by synthesizing abundant CD93. Thus, inflammatory cytokine release must damage surrounding nerves. Reducing expression and release of inflammation-related molecules such as CD93 may diminish neuronal injury, and suggests that CD93 may play a role in neuronal protecting during central nervous system inflammation. Consequently, we propose that CD93 participates in early pathophysiological processes of central nervous system inflammation, and may contribute to glial cell inflammation and neuronal protection. The exact function of CD93 and its mechanism of action deserve further investigation.
This study is the first to report changes in CD93 and GIPC expression and localization during central nervous system inflammation. Despite comprehensive analysis of our results we are unable to determine if GIPC is involved in inflammatory pathophysiological processes. However, our results confirm alterations in expression, localization, and involvement of CD93 in central nervous system inflammation. We believe that CD93 has dual effects, promoting glial cell inflammation and protecting neurons. In the early stage of an inflammatory reaction, increased CD93 expression may play a proinflammatory role, strengthening the effect of glial cells in phagocytizing apoptotic cells and debris, and clearing exogenous foreign bodies. In later stages, decreased CD93 expression and release may diminish injury to surrounding neurons. Specific biological effects and molecular mechanisms of CD93 in central nervous system inflammation require further study.
Footnotes
Funding: This study was supported by the National Natural Science Foundation of China, No. 31170766; the Nantong Municipal Social Undertakings Technological Innovation and Demonstration Project Foundation, No. HS2012032; the Natural Science Pre-research Project Foundation of Nantong University in 2012, No. 12ZY020.
Conflicts of interest: None declared.
Copyedited by James R, Wysong S, Wang J, Qiu Y, Li CH, Song LP, Zhao M
References
- 1.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Almolda B, Villacampa N, Manders P, Hidalgo J, Campbell IL, González B, Castellano B. Effects of astrocyte-targeted production of interleukin-6 in the mouse on the host response to nerve injury. Glia. 2014;62:1142–1161. doi: 10.1002/glia.22668. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez JI, Cayrol R, Prat A. Disruption of central nervous system barriers in multiple sclerosis. Biochim Biophys Acta. 2011;1812:252–264. doi: 10.1016/j.bbadis.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 4.Anthony DC, Couch Y. The systemic response to CNS injury. Exp Neurol. 2014;258:105–111. doi: 10.1016/j.expneurol.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 5.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 6.Bohlson SS, Zhang M, Ortiz CE, Tenner AJ. CD93 interacts with the PDZ domain-containing adaptor protein GIPC: implications in the modulation of phagocytosis. J Leukoc Biol. 2005;77:80–89. doi: 10.1189/jlb.0504305. [DOI] [PubMed] [Google Scholar]
- 7.Booth RA, Cummings C, Tiberi M, Liu XJ. GIPC participates in G protein signaling downstream of insulin-like growth factor 1 receptor. J Biol Chem. 2002;277:6719–6725. doi: 10.1074/jbc.M108033200. [DOI] [PubMed] [Google Scholar]
- 8.Brown GC, Bal-Price A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate and mitochondria. Mol Neurobiol. 2003;27:325–355. doi: 10.1385/MN:27:3:325. [DOI] [PubMed] [Google Scholar]
- 9.Cekanaviciute E, Dietrich HK, Axtell RC, Williams AM, Egusquiza R, Wai KM, Koshy AA, Buckwalter MS. Astrocytic TGF-β signaling limits inflammation and reduces neuronal damage during central nervous system toxoplasma infection. J Immunol. 2014;193:139–149. doi: 10.4049/jimmunol.1303284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong F, Li JH, Wu ZY. Expression of tumor necrosis factor-alpha and interleukin 1 beta in spinal cord injury rats undergoing intravenous transplantation of bone marrow mesenchymal stem cells. Zhongguo Zuzhi Gongcheng Yanjiu. 2012;16:6730–6735. [Google Scholar]
- 11.Fenn AM, Hall JC, Gensel JC, Popovich PG, Godbout JP. IL-4 signaling drives a unique Arginase+/IL-1β+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in il-4rα after traumatic spinal cord injury. J Neurosci. 2014;34:8904–8917. doi: 10.1523/JNEUROSCI.1146-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao Y, Li M, Chen W, Simons M. Synectin syndecan 4 cytoplasmic domain binding PDZ protein, inhibits cell migration. J Cellular Physiol. 2000;184:373–379. doi: 10.1002/1097-4652(200009)184:3<373::AID-JCP12>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 13.Greenlee MC, Sullivan SA, Bohlson SS. Detection and characterization of soluble CD93 released during inflammation. Inflammation Res. 2009;58:909–919. doi: 10.1007/s00011-009-0064-0. [DOI] [PubMed] [Google Scholar]
- 14.Greenlee-Wacker MC, Briseño C, Galvan M, Moriel G, Velázquez P, Bohlson SS. Membrane-associated CD93 regulates leukocyte migration and C1q-hemolytic activity during murine peritonitis. J Immunol. 2011;187:3353–3361. doi: 10.4049/jimmunol.1100803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffin FM, Mullinax PJ. High concentrations of bacterial lipopolysaccharide but not microbial infection-induced inflammation, activate macrophage C3 receptors for phagocytosis. J Immunol. 1990;145:697–701. [PubMed] [Google Scholar]
- 16.Harhausen D, Prinz V, Ziegler G, Gertz K, Endres M, Lehrach H, Trendelenburg G. CD93/AA4. 1: a novel regulator of inflammation in murine focal cerebral ischemia. J Immunol. 2010;184:6407–6417. doi: 10.4049/jimmunol.0902342. [DOI] [PubMed] [Google Scholar]
- 17.Harry GJ, Kraft AD. Neuroinflammation and microglia: considerations and approaches for neurotoxicity assessment. Expert Opin Drug Metab Toxicol. 2008;4 doi: 10.1517/17425255.4.10.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henson P. The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays Biochem. 2003;39:105–117. doi: 10.1042/bse0390105. [DOI] [PubMed] [Google Scholar]
- 19.Ivetic A, Deka J, Ridley A, Ager A. The cytoplasmic tail of l-selectin interacts with members of the ezrin-radixin-moesin (ERM) family of proteins cell activation-dependent binding of moesin but not ezrin. J Biol Chem. 2002;277:2321–2329. doi: 10.1074/jbc.M109460200. [DOI] [PubMed] [Google Scholar]
- 20.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 21.Li M, Liu J, Zhong YH, Peng FH. Changes of gliacytes after spinal cord transection in Ambystoma mexicanum. Zhongguo Zuzhi Gongcheng Yanjiu. 2013;17:1993–2000. [Google Scholar]
- 22.Ligensa T, Krauss S, Demuth D, Schumacher R, Camonis J, Jaques G, Weidner K M. A PDZ domain protein interacts with the C-terminal tail of the insulin-like growth factor-1 receptor but not with the insulin receptor. J Biol Chem. 2001;276:33419–33427. doi: 10.1074/jbc.M104509200. [DOI] [PubMed] [Google Scholar]
- 23.Lou X, Yano H, Lee F, Chao MV, Farquhar MG. GIPC and GAIP form a complex with TrkA: a putative link between G protein and receptor tyrosine kinase pathways. Mol Biol Cell. 2001;12:615–627. doi: 10.1091/mbc.12.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu K, Wang J, Hu B, Shi X, Zhou J, Tang Y, Peng Y. Brilliant blue G attenuates lipopolysaccharide-mediated microglial activation and inflammation. Neural Regen Res. 2013a;8:599–608. doi: 10.3969/j.issn.1673-5374.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu MJ, Wang SS, Zhu Y. Microglia-mediated oxidative stress injury in a mouse model of Parkinson's disease. Zhongguo Zuzhi Gongcheng Yanjiu. 2013b;17:2001–2006. [Google Scholar]
- 26.Muders MH, Dutta SK, Wang L, Lau JS, Bhattacharya R, Smyrk TC, Mukhopadhyay D. Expression and regulatory role of GAIP-interacting protein GIPC in pancreatic adenocarcinoma. Cancer Res. 2006;66:10264–10268. doi: 10.1158/0008-5472.CAN-06-2321. [DOI] [PubMed] [Google Scholar]
- 27.Norsworthy PJ, Fossati-Jimack L, Cortes-Hernandez J, Taylor PR, Bygrave AE, Thompson RD, Botto M. Murine CD93 (C1qRp) contributes to the removal of apoptotic cells in vivo but is not required for C1q-mediated enhancement of phagocytosis. J Immunol. 2004;172:3406–3414. doi: 10.4049/jimmunol.172.6.3406. [DOI] [PubMed] [Google Scholar]
- 28.Onodera S, Suzuki K, Matsuno T, Kaneda K, Takagi M, Nishihira J. Macrophage migration inhibitory factor induces phagocytosis of foreign particles by macrophages in autocrine and paracrine fashion. Immunology. 1997;92:131–137. doi: 10.1046/j.1365-2567.1997.00311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paxinos G, Watson C. Academic Press; 2006. The rat brain in stereotaxic coordinates:hard cover edition. [Google Scholar]
- 30.Petrenko O, Beavis A, Klaine M, Kittappa R, Godin I, Lemischka IR. The molecular characterization of the fetal stem cell marker AA4. Immunity. 1999;10:691–700. doi: 10.1016/s1074-7613(00)80068-0. [DOI] [PubMed] [Google Scholar]
- 31.Sheng JG, Mrak RE, Griffin WST. Enlarged and phagocytic, but not primed, interleukin-1α-immunoreactive microglia increase with age in normal human brain. Acta Neuropathol. 1998;95:229–234. doi: 10.1007/s004010050792. [DOI] [PubMed] [Google Scholar]
- 32.Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsukita S, Yonemura S. Cortical actin organization: lessons from ERM (ezrin/radixin/moesin) proteins. J Biol Chem. 1999;274:34507–34510. doi: 10.1074/jbc.274.49.34507. [DOI] [PubMed] [Google Scholar]
- 34.Ulvestad E, Williams K, Bjerkvig R, Tiekotter K, Antel J, Matre R. Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J Leukoc Biol. 1994;56:732–740. doi: 10.1002/jlb.56.6.732. [DOI] [PubMed] [Google Scholar]
- 35.van der Laan LJ, Ruuls SR, Weber KS, Lodder IJ, Döpp EA, Dijkstra CD. Macrophage phagocytosis of myelin in vitro determined by flow cytometry: phagocytosis is mediated by CR3 and induces production of tumor necrosis factor-alpha and nitric oxide. J Neuroimmunol. 1996;70:145–152. doi: 10.1016/s0165-5728(96)00110-5. [DOI] [PubMed] [Google Scholar]
- 36.Vidal PM, Lemmens E, Geboes L, Vangansewinkel T, Nelissen S, Hendrix S. Late blocking of peripheral TNF-α is ineffective after spinal cord injury in mice. Immunobiology. 2013;218:281–284. doi: 10.1016/j.imbio.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 37.Wyss-Coray T. Inflammation in Alzheimer disease: driving force bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 38.Xiong H, Callaghan D, Wodzinska J, Xu J, Premyslova M, Liu QY, Zhang W. Biochemical and behavioral characterization of the double transgenic mouse model (APPswe/PS1dE9) of Alzheimer's disease. Neurosci Bull. 2011;27:221–232. doi: 10.1007/s12264-011-1015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Youn JC, Yu HT, Jeon JW, Lee HS, Jang Y, Park YW, Park YB, Shin EC, Ha JW. Soluble CD93 levels in patients with acute myocardial infarction and its implication on clinical outcome. PLoS One. 2014;9:e96538. doi: 10.1371/journal.pone.0096538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao YY, Yan DJ, Chen ZW. Role of AIF-1 in the regulation of inflammatory activation and diverse disease processes. Cell Immunol. 2013;284:75–83. doi: 10.1016/j.cellimm.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 41.Zindler E, Zipp F. Neuronal injury in chronic CNS inflammation. Best Pract Res Clin Anaesthesiol. 2010;24:551–562. doi: 10.1016/j.bpa.2010.11.001. [DOI] [PubMed] [Google Scholar]