

Abstract
The efficient folding of membrane and secreted proteins relies on the unfolded protein response (UPR) to buffer fluctuations in the load of misfolded proteins. Although the UPR is thought to operate on a generic manner to maintain ER proteostasis, a recent study revealed the existence of a novel mechanism to eliminate misfolded GPI-anchored proteins via the secretory pathway, termed ‘rapid ER stress-induced export’ (RESET) (Satpute-Krishnan et al, 2014). RESET involves the export of misfolded GPI proteins to the plasma membrane for subsequent degradation by the lysosome.
See also: P Satpute-Krishnan et al (July 2014)
Membrane and secreted proteins are synthesized, modified, and targeted using the ensemble of organelles of the secretory route. Upon overload, a large fraction of these proteins are not folded properly and are removed before reaching their final destination. Several quality control mechanisms monitor the folding status of proteins at the ER. For example, the calnexin and calreticulin cycle operates on misfolded glycosylated proteins. Terminally misfolded glycosylated proteins undergo ER-associated degradation (ERAD), consisting of retrotranslocation to the cytosol and degradation by the proteasome.
Multiple pathological perturbations to the secretory pathway result on ER stress. To cope with ER stress, cells engage an adaptive reaction known as the unfolded protein response (UPR), which controls a series of downstream mechanisms to recover proteostasis (Hetz, 2012). Hours after the stress insult, activation of the UPR triggers the upregulation of a variety of target genes involved in almost every aspect of the secretory pathway. However, rapid reactions are also induced to reduce the load of misfolded protein, for example by the transient inhibition of protein synthesis (Walter & Ron, 2011). All these responses impact the secretory proteome globally. Nevertheless, mechanisms that selectively control the synthesis of defined groups of secretory proteins have begun to emerge. One such pathway has recently been uncovered for GPI-anchored proteins (Satpute-Krishnan et al, 2014) (see below).
Some misfolded variants of the GPI-anchored prion protein (PrP) are not good substrates of ERAD and instead are degraded by lysosomes (Ashok & Hegde, 2008). Interestingly, upon removal of the GPI anchor, PrP becomes an efficient ERAD substrate (Ashok & Hegde, 2008). PrP misfolding is the leading cause of a variety of diseases classified as prion-related disorders, which also involve pathological levels of ER stress. In turn, ER stress can induce a partial misfolding of PrP, suggesting that ER-related factors actively participate in its folding and quality control (Hetz et al, 2007). Satpute-Krishnan et al have now used an artificial version of PrP that is spontaneously misfolded and retained in the ER as a model system to study the pathways regulating its degradation. Unexpectedly, upon experimental induction of ER stress, mutant PrP was exported to the plasma membrane and then rapidly internalized and targeted to lysosomes for degradation (Satpute-Krishnan et al, 2014) (Fig1). The authors noticed this phenomenon had been reported before for the T-cell receptor (Suzuki et al, 1991). Domain analysis to identify the structural determinants of the clearance pathway identified the GPI anchor as a key element. This novel ER protein degradation pathway was designated RESET for ‘rapid ER stress-induced export’. Importantly, RESET was confirmed for other artificial mutants of GPI-anchored proteins, in addition to several natural mutants involved in human disease.
Figure 1.
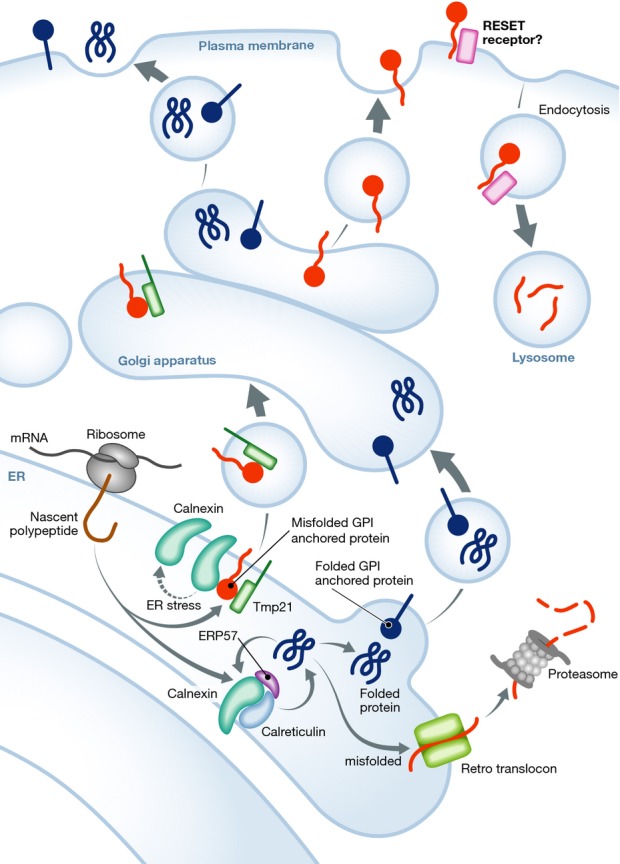
The newly discovered RESET pathway
Membrane and secreted proteins are synthesized in ER membrane-associated ribosomes. Global quality control mechanisms, such as the calnexin–calreticulin cycle, allow folded proteins to exit the ER and enter the secretory route (generic blue proteins and blue GPI-anchored proteins). Alternatively, unfolded proteins are targeted to ERAD for proteasome-mediated degradation (generic red proteins). In the RESET pathway, unfolded GPI-anchored proteins (red GPI-anchored proteins) bind calnexin and Tmp21. ER stress triggers the dissociation of calnexin from GPI proteins, while Tmp21 acts as an export receptor. Unfolded GPI proteins reach the plasma membrane via the Golgi apparatus. Unknown signals and accessory molecules target these proteins for endocytosis and lysosome degradation, thus alleviating ER protein overload.
To uncover the mechanistic basis of RESET, the authors screened for factors mediating the retention of misfolded GPI-anchored proteins at the ER and components that operate as export or release receptors. An interactome study identified the association of ER-retained mutant PrP with the chaperone calnexin, an interaction that was, noteworthy, disrupted by ER stress. Known components involved in the trafficking of GPI-anchored proteins were then tested, revealing that ER export factor Tmp21 physically interacted with misfolded PrP. Knocking down Tmp21 triggered a drastic aggregation of PrP in the ER, suggesting that ER exit may contribute to prevent abnormal aggregation of this mutant GPI-anchored protein.
Overall, this study reveals a novel pathway that uses a post-ER conventional secretory route to selectively eliminate misfolded GPI-anchored proteins that did not pass classical quality control mechanisms. The authors speculate that RESET may help attenuate ER proteostasis impairment before a strong UPR reaction is established. However, it is important to highlight the fact that UPR stress sensors are activated minutes after the exposure to stress, regulating early non-transcriptional responses (DuRose et al, 2006; Hetz, 2012).
Based on the fact that the physicochemical properties of the GPI anchor reduces the efficiency of ERAD-mediated clearance of misfolded proteins, RESET offers a rapid escape response to avoid further alterations in ER function and to decrease the saturation of ER chaperones. To reinforce the biological relevance of this newly identified mechanism, several essential questions remain to be answered. For example, since GPI-anchored proteins lack a cytosolic domain, what constitutes the tag that targets them to lysosomal degradation when misfolded? Or what is the relationship of RESET to post-ER export quality control mechanisms (Hicks & Machamer, 2005). In addition, further studies are necessary to explore the universality of the RESET ‘clientele’ and the relevance of RESET after long-term ER stress. Additionally, other clearance pathways, such as autophagy, trigger the secretion of cellular components (Manjithaya & Subramani, 2011). Whether RESET also results in the secretion of misfolded proteins remains to be determined. Assessing the relevance of RESET in the quality control of endogenous GPI-anchored proteins and in vivo will help expand the biological significance of RESET. Finally, due to the fact that extracellular misfolded PrP is essential for the propagation of prion diseases, and because cell-to-cell transfer of misfolded proteins is becoming a common theme in most neurodegenerative diseases (Soto, 2012), it is tempting to speculate that RESET may even contribute to disease propagation. The discovery of RESET underscores the functional precision of the molecular machineries operating within conventional and well-studied secretory routes.
Acknowledgments
FONDECYT 1140549, CONICYT-EEUU USA2013-0003, and ACT1109 grants (CH), FONDECYT 1140617 (AC) and P09-015-F (AC and CH).
References
- Ashok A, Hegde RS. Retrotranslocation of prion proteins from the endoplasmic reticulum by preventing GPI signal transamidation. Mol Biol Cell. 2008;19:3463–3476. doi: 10.1091/mbc.E08-01-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuRose JB, Tam AB, Niwa M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol Biol Cell. 2006;17:3095–3107. doi: 10.1091/mbc.E06-01-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Castilla J, Soto C. Perturbation of endoplasmic reticulum homeostasis facilitates prion replication. J Biol Chem. 2007;282:12725–12733. doi: 10.1074/jbc.M611909200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- Hicks SW, Machamer CE. Golgi structure in stress sensing and apoptosis. Biochim Biophys Acta. 2005;1744:406–414. doi: 10.1016/j.bbamcr.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Manjithaya R, Subramani S. Autophagy: a broad role in unconventional protein secretion? Trends Cell Biol. 2011;21:67–73. doi: 10.1016/j.tcb.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpute-Krishnan P, Ajinkya M, Bhat S, Itakura E, Hegde RS, Lippincott-Schwartz J. ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway. Cell. 2014;158:522–533. doi: 10.1016/j.cell.2014.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C. Transmissible proteins: expanding the prion heresy. Cell. 2012;149:968–977. doi: 10.1016/j.cell.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki CK, Bonifacino JS, Lin AY, Davis MM, Klausner RD. Regulating the retention of T-cell receptor alpha chain variants within the endoplasmic reticulum: Ca(2+)-dependent association with BiP. J Cell Biol. 1991;114:189–205. doi: 10.1083/jcb.114.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]