

Abstract
The urokinase-type plasminogen activator receptor (uPAR) is a non-integrin vitronectin (VN) cell adhesion receptor linked to the plasma membrane by a glycolipid anchor. Through structure–function analyses of uPAR, VN and integrins, we document that uPAR-mediated cell adhesion to VN triggers a novel type of integrin signalling that is independent of integrin–matrix engagement. The signalling is fully active on VN mutants deficient in integrin binding site and is also efficiently transduced by integrins deficient in ligand binding. Although integrin ligation is dispensable, signalling is crucially dependent upon an active conformation of the integrin and its association with intracellular adaptors such as talin. This non-canonical integrin signalling is not restricted to uPAR as it poses no structural constraints to the receptor mediating cell attachment. In contrast to canonical integrin signalling, where integrins form direct mechanical links between the ECM and the cytoskeleton, the molecular mechanism enabling the crosstalk between non-integrin adhesion receptors and integrins is dependent upon membrane tension. This suggests that for this type of signalling, the membrane represents a critical component of the molecular clutch.
Keywords: integrin, membrane tension, signalling, uPAR, vitronectin
See also: RT Böttcher & R Fässler (November 2014)
Introduction
The physical contact between cells and the extracellular matrix (ECM) is a highly dynamic process crucial for basic cellular functions and tissue homoeostasis under physiological and pathological conditions. The main class of cell surface receptors responsible for the interaction between cells and the ECM is the integrin family of adhesion receptors that are capable of creating a direct physical link, known as the molecular clutch, between the ECM and the intracellular cytoskeleton.
In addition to integrins, other types of cell surface receptors interact with the ECM and act as bona fide cell adhesion receptors. These non-integrin adhesion receptors, including syndecans, discoidin domain receptors and CD44, are thought to mediate signal transduction and cytoskeleton coupling by lateral associations with integrins (Schmidt & Friedl, 2010). One such non-integrin adhesion receptor is the urokinase-type plasminogen activator receptor (uPAR) that promotes cell adhesion through its direct interaction with the provisional ECM protein vitronectin (VN) (Wei et al, 1994; Madsen et al, 2007a). It is well described that overexpression of uPAR results in altered cell adhesion, morphology, migration and proliferation on VN-containing extracellular matrices (Wei et al, 1994; Kjøller & Hall, 2001; Madsen et al, 2007a; Smith et al, 2008; Pirazzoli et al, 2013). Noteworthy the interaction with VN is critical for the tumour growth-promoting activity of the receptor (Pirazzoli et al, 2013). Ligation of uPAR to matrix-bound VN induces integrin-dependent activation of the SRC/FAK/p130Cas/DOC180/Rac1 signalling axis resulting in increased cell spreading, migration and invasion (Kjøller & Hall, 2001; Smith et al, 2008). The lack of transmembrane and cytoplasmic domains makes uPAR incapable of direct signal transduction over the plasma membrane, and as for the other non-integrin adhesion receptors, a popular signalling paradigm for uPAR is that it interacts directly with integrins, altering their substrate specificity and modulating their signalling activity (Wei et al, 1996). Nevertheless, exhaustive mutational analysis of uPAR has shown that VN is the only required direct interaction partner of uPAR in the process (Madsen et al, 2007b).
Using uPAR-mediated cell adhesion to VN as a paradigm of signalling crosstalk between non-integrin and integrin adhesion receptors, we here uncover a novel signalling mechanism whereby integrins transduce adhesion signalling in the absence of direct matrix contact (i.e. ligand-independent integrin signalling). The process does not require direct lateral interactions between the receptor mechanically mediating cell adhesion to the ECM and the integrin responsible for signalling, but relies on plasma membrane tension for their functional coupling. No particular constrains can be identified to the adhesion receptors and their matrix ligand, suggesting that any type of membrane component, with sufficiently high avidity for the ECM, will activate ligand-independent integrin signalling.
Results
uPAR-induced cell adhesion and signalling require a direct uPAR/VN interaction
To investigate the molecular mechanisms of uPAR-mediated cell signalling, we exploited 293 cells engineered to express either wild-type uPAR (uPARWT), a single alanine substitution variant (uPART54A) that displays conditional binding to VN only in the presence of uPA and a variant (uPARW32A) with impaired VN binding even in the presence of uPA (Gardsvoll & Ploug, 2007; Madsen et al, 2007a) as shown in Fig 1A. The adhesive properties of the different uPAR variants were also investigated and confirmed by real-time cell analysis employing impedance-based measurements of cell adhesion (Supplementary Fig S1A). Under standard culture conditions, the expression of uPARWT, but not uPART54A, or uPARW32A results in a ˜2.5-fold increase in cell spreading compared to mock-transfected cells (Fig 1B) and induces p130Cas substrate domain phosphorylation and MAP kinase activation (Supplementary Fig S1B and C). Treatment with uPA rapidly induces cell spreading and signalling of uPART54A cells to levels comparable with those observed in uPARWT cells, but has little effect on cell spreading in mock-, uPARWT- and uPARW32A-transfected cells (Fig 1B). This is better illustrated by the rapid induction of lamellipodia extensions and cell spreading shown in Fig 1C and in the complete time-lapse recording provided as Supplementary Movie S1. These data show that uPAR-mediated cell adhesion to VN induces mitogenic and migratory cell signalling and document the experimental advantages of the conditional uPART54A mutant that displays low baseline VN binding, which may be fully restored by treatment with uPA. The effect of uPA treatment on 293/uPART54A cells is specifically mediated by activation of uPAR binding to VN as the effect is strongly attenuated in cells expressing uPARW32A that is deficient in VN binding, but binds uPA normally.
Figure 1. uPAR-mediated VN adhesion triggers signalling and cell spreading through αvβ5 and β1 integrins.

A VN adhesion of cells expressing uPAR variants in the presence or absence of uPA. Mock-, uPARWT- and uPART54A-transfected 293 cells were plated for 30 min on VN with or without uPA. Adherent cells were fixed and stained with crystal violet. VN adhesion is presented in percentage of cell adhesion to poly-D-lysine (PDL shown as 100%). The data are from three independent experiments and are presented as the mean ± s.e.m.
B Increased cell-matrix contact area upon uPAR-mediated VN adhesion. Cells were grown overnight and stimulated for 30 min with uPA. After fixation, DIC images were taken, and cell areas were quantified using ImageJ software. The figure shows the contact areas of individual cells (n = 56–93) taken from two independent experiments. Whiskers indicate the geometric mean and 95% confidence intervals (95% CI) for each condition.
C Selected frames from a time-lapse phase contrast recording of 293/uPART54A cells after the addition of uPA. Membrane ruffles/processes are indicated by white arrows and rapidly convert into lamellipodia, indicated by black arrows, which progressively expand and lead to the acquisition of a migratory phenotype. The complete time-lapse recording can be found in Supplementary Movie S1.
D, E uPAR/VN signalling and cell spreading require integrins. 293/uPART54A cells were seeded on VN for 30 min with uPA and treated with the indicated antibodies. After lysis or fixation, cell-matrix contact areas (D) and phosphorylation of p130Cas SD (E) were assayed and quantified. Cell-matrix contact area measurements represent at least 50 cells with indicated geometrical means ± 95% CI, in two independent experiments. Some cells (n = 4) with contact areas exceeding the chosen y-axis range are not shown. The grey bars represent the range of cell area of not spread or fully spread uPART54A cells based on 95% CI on poly-D-lysine or on VN with uPA, respectively (Fig 2B). Representative DIC images are shown (scale bar: 10 μm). Western blot data are represented as percentage of phosphorylated p130Cas SD of cells treated with uPA on VN (n ≥ 3, mean ± s.e.m.). Representative blots for phosphorylated p130Cas (pY Cas), total p130Cas (tot cas) and vinculin are shown.
F Cartoon illustrating the signalling downstream of uPAR mediated by αvβ5 and β1 integrins.
uPAR/VN-induced signalling in 293 cells is mediated by αVβ5 and β1 integrins
An attractive explanation for the signalling activity of uPAR on VN-containing matrices is that the increased cell–matrix contact enforced by uPAR enhances the proximity between VN-binding integrins and their ligands in the matrix thereby triggering increased avidity and canonical outside-in signalling (Madsen et al, 2007a). To investigate the role of integrins in the signalling induced by uPAR-mediated cell adhesion to VN, we tested the inhibitory activity of a series of integrin antibodies. The weak integrin-mediated cell adhesion to VN is mediated exclusively by the αVβ5 integrin as evidenced by the inhibitory effect of the function-blocking P1F6 antibody, whereas β1-containing integrins do not contribute to cell adhesion on this ECM component (Supplementary Fig S1D). Similarly, an α5 function-blocking antibody, P1D6, as well two different allosteric inhibitory β1-antibodies, 4B4 and mAb13, specifically impaired cell adhesion to FN. None of the tested antibodies had any effect on uPAR-mediated cell adhesion to VN in accordance with cell binding to this substrate being driven by the direct uPAR/VN interaction. The activation of cell spreading and signalling, induced by treatment of uPART54A cells with uPA, did not modulate the cell surface expression of α5, β1 and αvβ5 and also did not induce notable expression of the VN-binding β3 integrin, as evidenced by flow cytometry analysis (Supplementary Fig S1E).
When the same antibodies were tested individually for their inhibitory effect on uPAR-induced cell spreading and p130Cas signalling on VN, only modest effects were observed (Fig 1D and E). However, a virtually complete inhibition was obtained by the combined inhibition of αVβ5 and β1. The data thus confirm that integrins are indeed crucial signalling transducers downstream of uPAR-mediated cell adhesion to VN and document a functional redundancy between αVβ5 and β1 integrins in the process.
The fact that inhibition of β1 reduces uPAR signalling, even if this integrin is not a functional VN adhesion receptor in the 293 cells, suggests that the β1-dependent branch of uPAR/VN signalling may occur independently of integrin binding to the matrix, as illustrated in the cartoon shown in Fig 1F.
uPAR-induced cell spreading and signalling on an integrin refractory VN substrate is mediated by β1 integrins
To address the possibility that uPAR induces integrin signalling independently of integrin ligation, we employed recombinant VN variants where the integrin binding site had been disrupted by alanine substitution of the glycine residue in the RGD motif (VNRAD). It is well established that the RGD sequence represents the only important integrin binding site in VN and that this motif is both functionally and physically separated from the uPAR binding site located in the proximal somatomedin-B (SMB) domain (Madsen et al, 2007a). The predicted properties of the recombinant VN molecules were confirmed in adhesion assays using uPART54A cells (Fig 2A). When uPART54A is active in VN binding (i.e. in the presence of uPA), cell adhesion to VN was strong and RGD-independent. When uPART54A is inactive in VN binding (i.e. in the absence of uPA), cell adhesion was ˜ threefold weaker and entirely RGD-dependent as no residual adhesion was observed on VNRAD. Treatment with uPA did not affect cell adhesion on FN, poly-D-lysine (PDL) or a VN variant in which the SMB domain was deleted (VNΔSMB), demonstrating the specificity of uPA in modulating uPAR-mediated cell adhesion to VN. 293/uPART54A cells used in combination with the VN variants thus allowed us to functionally dissect the contribution of uPAR and integrins to the process of cell adhesion, spreading and signalling on VN.
Figure 2. The RGD motif VN is required for integrin-mediated cell adhesion but dispensable for uPAR-induced β1 integrin signalling and cell spreading.

A Effect of VN variants on uPAR and integrin-dependent cell adhesion. 293 uPART54A cells were seeded, in the presence or absence of uPA, on wells coated with WT VN or recombinant VN variants in which the integrin binding site is inactivated (VNRAD), the SMB domain is deleted (VNΔSMB) or both (VNRADΔSMB) for 30 min. Cell adhesion was assayed and expressed as percentage of PDL. For comparison, the adhesion to fibronectin (FN) and PDL is shown. The data are means ± s.e.m. (n = 3).
B, C uPAR-induced cell spreading and signalling do not require vitronectin integrins engagement. 293/uPART54A cells were seeded on the indicated substrate with or without uPA for 30 min. Cell-matrix contact area (B) and p130Cas SD phosphorylation (C) were analysed and quantified. Cell-matrix contact area measurements represent at least 50 cells with indicated geometrical means ± 95% CI, in two independent experiments. Cells (n = 10) having contact areas exceeding the scale of the y-axis are not shown. Cells do not adhere to VNRAD in the absence of uPA, and the quantification of cells spreading is therefore not applicable (n.a.). The grey bars represent the range of cell area of not spread or fully spread uPART54A cells based on 95% CI on poly-D-lysine or on VN with uPA, respectively. Western blot data are represented as percentage of phosphorylated p130Cas SD of cells treated with uPA on VN (n ≥ 6, mean ± s.e.m.). Representative blots are shown.
D, E β1 integrin mediates uPAR-induced signalling and spreading on an integrin refractory VN variant. 293/uPART54A cells were treated with the indicated antibody and seeded on VNRAD with uPA for 30 min. p130Cas SD phosphorylation (D) and cell-matrix contact area (E) were analysed and quantified. Cell-matrix contact area measurements represent at least 50 cells with indicated geometrical means ± 95% CI, in two independent experiments. The grey bars represent the range of cell area of not spread or fully spread uPART54A cells based on 95% CI on poly-D-lysine or on VN with uPA, respectively (B). Representative DIC images are shown (scale bar: 10 μm). The Western blot data are represented as percentage of phosphorylated p130Cas SD of cells seeded on VNRAD with uPA in absence of antibody (n ≥ 3, mean ± s.e.m.). Representative blots are shown.
F Cartoon illustrating the β1 integrin signalling triggered by uPAR on VNRAD.
To probe the importance of direct integrin–matrix interactions in uPAR-induced cell spreading and signalling, we seeded 293/uPART54A cells in the absence or presence of uPA on VN, VNRAD, FN and PDL and assayed cell spreading and p130Cas phosphorylation (Fig 2B and C). Remarkably, cell spreading and p130Cas phosphorylation were comparable on VN and VNRAD, suggesting that integrin binding to VN is indeed dispensable in the process. In the absence of uPAR binding to VN, cell spreading and p130Cas phosphorylation were reduced and comparable to the one observed on PDL and in mock-transfected cells on serum-coated surfaces (see Fig 1B). Again, the presence or absence of uPA had no effect on cell spreading and p130Cas phosphorylation on FN and PDL. It has been shown that the kringle domain of uPA interacts with integrins (Tarui et al, 2006), and it is therefore possible that uPA may physically connect uPAR and integrins via this domain. However, this potential bridging does not seem to be of relevance here as the small N-terminal growth factor-like domain of uPA (GFD), lacking the kringle domain, is equally efficient in inducing p130Cas phosphorylation and cell spreading as intact uPA (Supplementary Fig S2A and B). These data show that the uPAR-induced signalling requires the direct interaction between uPAR and VN, but not the integrity of the RGD motif, and that it is not mediated by contemporary binding of uPA to both uPAR and integrins. The RDG-independent signalling downstream of uPAR is not restricted to 293 cells as similar results were observed in Chinese hamster ovary (CHO) cells overexpressing uPARWT and uPART54A (Supplementary Fig S2C). Also in these cells, the expression of uPART54A, in the presence of uPA, induced cell spreading to a similar extent on VN and VNRAD.
As integrins are responsible for the transduction of uPAR signalling on VN surfaces, we next tested if they are also responsible for signalling on VNRAD to which they do not bind. As for wild-type VN, both p130Cas phosphorylation and cell spreading (Fig 2D and E) were fully suppressed by inhibitory integrin antibodies, demonstrating that also on this substrate, signalling is transduced by integrins. Consistently with signalling being mediated by integrins, both p130Cas phosphorylation and cell spreading were sensitive to SRC kinase inhibition. Furthermore, uPAR-mediated cell spreading was insensitive to MEK or myosin inhibition (Supplementary Fig S2B, D and E). In contrast to wild-type VN, where both αvβ5 and β1 contribute to the signalling, inhibition of β1 alone was sufficient to impair signalling on VNRAD, showing that the αvβ5 integrin only contributes to uPAR signalling when its binding site in VN is intact, as illustrated in the cartoon shown in Fig 2F. To address the possible relevance of ligand-independent β1 signalling in cells expressing (patho) physiological levels of uPAR, we also analysed the effect of β1 inhibition in the triple negative MDA-MB-231 breast carcinoma cell line seeded on different substrates (Supplementary Fig S3A). These cells express high levels of uPAR, and their growth in xenograft models has been documented to be uPAR- and VN-dependent (Kunigal et al, 2007; Lebeau et al, 2013; Pirazzoli et al, 2013). These cells do not adhere to VNRAD independently of the addition of exogenous uPA (data not show), but do adhere and spread on an immobilised anti-uPAR antibody allowing us to isolate and analyse uPAR dependent cell adhesion and spreading also in this cell line. Consistently with our findings in the 293 cells, spreading of MDA-MB-231 cells on the uPAR antibody was ablated by the inhibitory β1 antibody 4B4. Finally, we analysed the localisation of uPAR and β1 integrin in MDA-MB-231 cells seeded on FN and VN (Supplementary Fig S3B). On FN, uPAR was distributed diffusely under the cell body, whereas β1 integrin was located primarily in the periphery of the cells. On VN, uPAR formed small clusters especially in cell edges, where also β1 integrin was localised. Although both uPAR and β1 integrin were found close to the borders of protrusions, only limited co-localisation was observed.
Integrins and focal adhesion components localise differently in canonical integrin and uPAR-mediated cell adhesion
To investigate if the localisation of integrins and typical focal adhesion components differs between uPAR/VN-induced cell adhesion and canonical integrin-mediated cell adhesion, we performed immunofluorescence analysis on 293/uPART54A cells seeded on FN and VNRAD (Fig 3). On FN, both total and activated β1 integrin localised in focal adhesion-like structures in the cell periphery (Fig 3A). On VNRAD, similar structures were not observed, and β1 displayed a more diffuse staining in zones close to the leading edge. Only poor staining for active β1 was observed. Furthermore, the localisation of β1 integrin was not remarkably different between cells seeded on VN and VNRAD (Supplementary Fig S4A).
Figure 3. Localisation of β1 integrin and focal adhesion components during uPAR-mediated cell spreading.
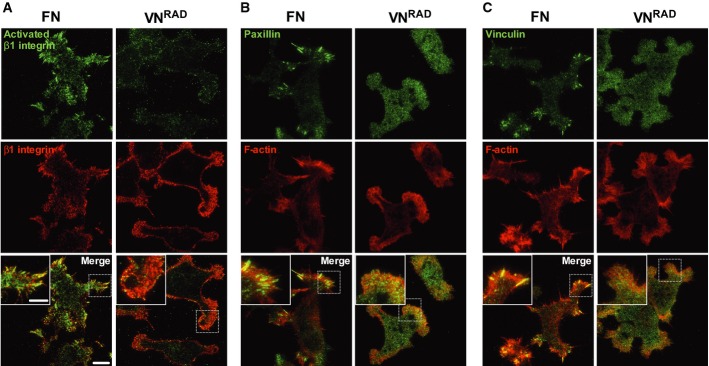
A–C 293 uPART54A were seeded on FN or VNRAD in the presence of uPA for 30 min and stained with the indicated antibodies. Representative TIRF-FM images are shown. (A) Activated β1 integrin (9EG7): green, total β1 integrin (K20): red. (B) Paxillin: green, actin: red. (C) Vinculin: Green, actin: red. Scale bar: 10 μm (whole image), 5 μm (insert).
When seeded on FN, immunofluorescence analysis for two distinct focal adhesion components (vinculin and paxillin) evidenced localisation to focal adhesion-like structures (Fig 3B and C) similar to those observed for β1 (Fig 3A). In contrast, both paxillin and vinculin displayed a diffuse dotted staining in cells seeded on VNRAD.
A previous study has identified activated, but unligated, α5β1 integrins along the leading edge of lamellipodia (Galbraith et al, 2007). To investigate the localisation of this receptor in the process of uPAR-induced cell spreading on VN, we conducted time-lapse recordings of 293/uPART54A cells transiently transfected with a GFP-tagged α5 (Supplementary Fig S4B and Movie S2). In accordance with the localisation of β1, also α5 was located at the leading edge of protruding lamellipodia during cell spreading, suggesting that the α5β1 heterodimer might indeed be responsible for the signalling downstream of uPAR. Consistently, siRNA-mediated depletion of α5 integrin impaired uPAR-mediated cell spreading (Supplementary Fig S4C).
β1 signalling on VNRAD requires the binding of cytoplasmic factors, an active conformation of the receptor, but no ligand engagement
The uPAR-induced signalling via αVβ5 on VN is coherent with the current paradigm for ligand-induced outside-in signalling. We therefore focused our attention on the dominant and unusual ligand-independent β1 signalling observed on VNRAD. To determine the structural requirements of the integrin in this type of signalling, we conducted a structure–function analysis of β1 in this process (Fig 4). For this purpose, we generated a stable clone of 293 Flp-In T-REx cells constitutively expressing uPART54A and subsequently used this cell line to express the different β1 variants using the Flp-In cassette ensuring comparable expression of the different variants. To functionally discriminate the biological activity of the transgenic β1 chains from the activity of residual endogenous β1, we rendered the transfected β1 chains refractory to inhibition by the 4B4 antibody using the K218A substitution reported to impair antibody binding (Luo et al, 2004). The cell system was validated in FN adhesion assays in the presence or absence of mAb13 and 4B4, using cells transfected with either empty vector or with vectors encoding β1WT and β1K218A (Supplementary Fig S5A). As predicted, both antibodies strongly impaired adhesion of mock- and β1WT-transfected cells to FN, while only mAb13 inhibited adhesion of cells expressing β1K218A, confirming that this β1 chain is resistant to inhibition by 4B4 but otherwise functionally intact.
Figure 4. Structure–function analysis of the β1 integrin in uPAR/VN-induced cell spreading and signalling.
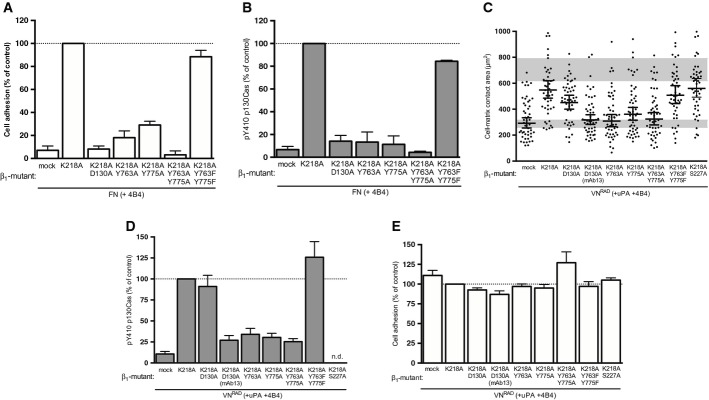
A, B Effect of β1 integrin mutants on canonical integrin-mediated FN adhesion and signalling. 293 cells co-expressing uPART54A and the indicated β1 variants were seeded on FN for 30 min. Cell adhesion (A) and p130Cas SD phosphorylation (B) were assayed and quantified. Adhesion is expressed in percentage of adhesion to PDL (n = 3, mean ± s.e.m.). The Western blot data are represented as percentage of phosphorylated p130Cas SD of β1K218A-expressing cells (n = 3, mean ± s.e.m.).
C–E Structural requirements for ligand-independent β1 integrin signalling and cell spreading. 293 cells co-expressing uPART54A and the indicated β1 variants were seeded on VNRAD in the presence of uPA and 4B4 for 30 min. Cell area (C), p130Cas SD phosphorylation (D) and cell adhesion (E) were assayed and quantified. Adhesion is shown in percentage of PDL (n = 3, means ± s.e.m.). The Western blot data are represented as percentage of phosphorylated p130Cas SD of β1K218A-expressing cells (n ≥ 3, mean ± s.e.m.). Cell-matrix contact area measurements represent at least 50 cells with indicated geometrical means ± 95% CI, in two independent experiments. The grey bars represent the range of cell area of not spread or fully spread uPART54A cells based on 95% CI on poly-D-lysine or on VN with uPA, respectively (Fig 2B).
Using this β1 replacement system, we first sought complementary evidence for the ligand independence of the β1 signalling induced by the uPAR/VNRAD interaction. To this end, we utilised the D130A alanine substitution that disrupts the metal ion-dependent adhesion binding site in β1 resulting in abolished ligand binding (Takada et al, 1992). As predicted, the expression of β1K218/D130A failed to rescue the 4B4 block on cell adhesion to FN (Fig 4A) and downstream p130Cas phosphorylation (Fig 4B). In strong contrast, the β1K218/D130A chain almost fully rescued the transmission of uPAR-induced cell spreading on VNRAD (Fig 4C) and p130Cas phosphorylation (Fig 4D), conclusively demonstrating that matrix engagement by the β1 integrin is dispensable for transduction of uPAR-induced adhesion signalling. Importantly, we found that the uPAR-mediated cell spreading and p130Cas activation on VNRAD transduced by the ligand-binding deficient β1K218/D130A was fully suppressed by mAb13 (Fig 4C and D), documenting that even if ligand binding is dispensable, an active conformation of the integrin is still required.
The conformation and activation state of integrins is largely determined from the intracellular side of the plasma membrane through the binding of scaffolding proteins to their cytoplasmic tails (Moser et al, 2009). We therefore also analysed the structural requirements to this region of β1. For this purpose, we generated β1 mutants with inactivated membrane-proximal and membrane-distal NPxY motifs alone or in combination (β1K218/Y763/775A), as well as a control mutant in which both tyrosine residues were replaced with functionally permissive phenylalanine residues (β1K218/Y763/775F) (Czuchra et al, 2006). Control experiments with cells seeded on FN confirmed that alanine substitution of these tyrosine residues, individually or together, efficiently abrogated cell adhesion (Fig 4A) and subsequent p130Cas phosphorylation (Fig 4B), while the double phenylalanine substitution retained full functionality. Disruption of either one or both the NPxY motifs also impaired uPAR-induced cell spreading (Fig 4C) and p130Cas phosphorylation (Fig 4D) on VNRAD, even if cell adhesion on this substrate was not affected by these mutations (Fig 4E), demonstrating that binding of cytoplasmic effectors is also required for ligand-independent integrin signalling. Consistent with these observations, siRNA-mediated depletion of talin was found to impair uPAR-mediated cell spreading on VNRAD (Supplementary Fig S5D). uPAR has been reported to interact directly with β1 integrin in 293 cells with β1 S227 being a critical residue for this interaction(Wei et al, 2005). However, an alanine substitution at this position failed to compromise uPAR-mediated cell spreading on VNRAD (Fig 4C), consistent with previous findings that mutational disruption of all published integrin interaction sites in uPAR leaves the receptor fully competent in promoting cell spreading on VN (Madsen et al, 2007a).
Control experiments by flow cytometry demonstrated that representative β1 variants competed comparably with endogenous receptor. This results in a similar reduction in surface expression of endogenous β1 recognised specifically by the 4B4 antibody (Supplementary Fig S5B). Real-time cell adhesion analysis (Supplementary Fig S5C) furthermore evidenced a similar kinetics of cell adhesion to VNRAD and FN among the different β1 variants, suggesting that the observed functional differences are also not related to different kinetics of cell adhesion.
Ligand-independent signalling downstream of β3 integrin
To determine if ligand-independent signalling is specific for β1, or a more general property of integrins, we assayed the activity of β3 in rescuing signalling in conditions where the αvβ5 pathway is non-operative (i.e. on VNRAD) and where the β1 integrin pathway is silenced by the 4B4 antibody. The 293 cells used in this study do not express notable endogenous β3 integrin, as judged by flow cytometry (Supplementary Fig S1E) or by function inhibition (Madsen et al, 2007a), and the ectopic expression of β3 caused enhanced cell adhesion to VN (Fig 5A). In contrast, the expression of ligand-binding deficient (β3D119Y; Loftus et al, 1990) or talin/kindlin-deficient (β3Y747/759A; Moser et al, 2008; Tadokoro et al, 2003) β3 variants reduced cell adhesion to VN, presumably by sequestering endogenous αV into inactive αVβ3D119Y and αVβ3Y747/759A heterodimers. The expression of the different β3 variants did not affect uPAR-mediated cell adhesion to VNRAD or integrin-mediated adhesion to FN. Importantly, however, the expression of both β3WT and β3D119Y, but not β3Y747/759A, rescued the 4B4 inhibition of p130Cas phosphorylation and cell spreading (Fig 5B). These data thus show that also β3 is capable of signalling independently of ligand binding. Analysis of cell surface expression by flow cytometry (Supplementary Fig S6A) confirmed that all the β3 variants were efficiently expressed and sorted to the cell surface, even if the β3Y747/759A variant was expressed at mildly reduced levels.
Figure 5. Ligand-independent integrin signalling by β3 integrin.
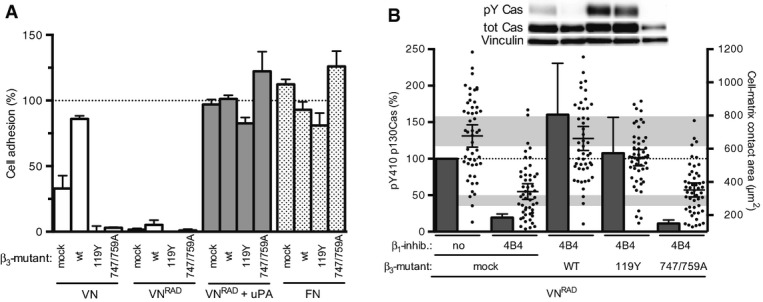
- Adhesive properties of 293 uPART54A cells expressing β3 integrin mutants. Cells were plated on the indicated substrate with uPA (where indicated) for 30 min. Cell adhesion was assayed and quantified. Data are shown in percentage of PDL (n = 3, mean ± s.e.m.).
- β3 integrin transmits uPAR/VN signalling and cell spreading independently of ligand binding. 293 cells co-expressing uPART54A and different β3 variants were seeded on VNRAD for 30 min in presence of 4B4. Cell area (dots) and p130Cas phosphorylation (columns) were assayed and quantified. Western blot data were normalised setting phosphorylation level of mock-transfected uPART54A cells without inhibitory antibody as 100% and expressing them as means ± s.e.m., n ≥ 3. Representative blots are shown. Cell-matrix contact area measurements represent at least 50 cells with indicated geometrical means ± 95% CI, in two independent experiments. The grey bars represent the range of cell area of not spread or fully spread uPART54A cells based on 95% CI on poly-D-lysine or on VN with uPA, respectively (Fig 2B).
Structural requirements to the anchoring receptor in ligand-independent integrin signalling
To determine if ligand-independent integrin signalling is specific for uPAR or if it may be induced by other ECM-binding membrane receptors, we analysed the activity of an artificial VN receptor generated by attaching a GPI-anchoring signal on the plasminogen activator inhibitor-1 (PAI-1GPI (Madsen et al, 2007a), illustrated in Fig 6D). This receptor shares no sequence or structure homology with uPAR, but still induced strong cell adhesion to VN (Supplementary Fig S6B and Madsen et al, 2007a). Like uPAR, the expression of PAI-1GPI induced robust cell spreading and p130Cas phosphorylation on VNRAD (Fig 6A). The ability of PAI-1GPI to induce adhesion and signalling required its binding to the matrix, as a VN-binding deficient variant of this receptor (PAI-1GPIVN-/-) was inactive in promoting cell adhesion (Supplementary Fig S6B), cell spreading and p130Cas phosphorylation (Fig 6A). Importantly, the signalling pathway activated upon PAI-1GPI-mediated cell adhesion to VNRAD was inhibited by 4B4 and therefore functionally similar, if not identical, to the pathway triggered by uPAR (Fig 6A). Both uPAR and the artificial PAI-1GPI receptor are tethered to the cell membrane by a GPI anchor. To determine the possible importance of this type of membrane anchorage, we analysed uPART54A variants engineered to carry different membrane anchorage sequences. Two of these were engineered to carry GPI-anchoring signals from other known GPI-anchored membrane proteins (CEA S4 and N-CAM GPI-anchored isoform), and two were tethered to the membrane using transmembrane segments copied from typical type-1 membrane receptors (EGFR and N-CAM transmembrane isoform). All four uPAR variants were comparably expressed on the cell surface (data not shown) and induced cell adhesion (Supplementary Fig S6C), spreading and p130Cas phosphorylation (Supplementary Fig S6D), suggesting that the mode of membrane anchorage is of little importance. The fact that both uPAR and PAI-1GPI share the same extracellular matrix ligand (VN) and have overlapping binding sites in this molecule could suggest that the observed signalling may be specific for VN. To address this, we seeded uPAR-expressing cells on surfaces coated with an anti-uPAR antibody and measured cell spreading and p130Cas phosphorylation in the absence or presence of 4B4 (Fig 6B). Consistent with the absence of specific structural requirement to the matrix ligand, cells seeded on the antibody displayed prominent cell spreading and p130Cas phosphorylation that was inhibited by the 4B4 antibody.
Figure 6. Structural requirements to the anchoring receptor, its ligand and type of membrane anchorage.

- Matrix-binding activity is required for the induction of intracellular signalling and spreading. 293 cells expressing an engineered cell surface VN receptor (PAI-1WT/GPI) or a VN-binding incompetent variant of the same receptor (PAI-1VN-/GPI) were seeded on VNRAD in presence of 4B4 for 30 min. Cell area (dots) and p130Cas phosphorylation (columns) were assayed and quantified. Phosphorylation of p130Cas is shown in percentage of the levels observed in uPART54A cells seeded in the presence of uPA on the same substrate (n ≥ 3, mean ± s.e.m.). Representative blots are shown. Cell-matrix contact area measurements represent at least 50 cells with indicated geometrical means ± 95% CI, in two independent experiments. The grey bars represent the range of cell area of not spread or fully spread uPART54A cells based on 95% CI on poly-D-lysine or on VN with uPA, respectively (Fig 2B).
- Cell signalling and spreading occur independently of the nature of the matrix component. Cells expressing uPART54A were seeded on an anti-uPAR monoclonal antibody (α-uPAR) and, where indicated, treated with 4B4 for 30 min. Signalling to p130Cas (columns) and cell spreading (dots) were quantified. Phosphorylation of p130Cas SD is shown in percentage of the levels observed in cells seeded on VNRAD in the presence of uPA (n ≥ 3, mean ± s.e.m.). Representative blots are shown. Cell-matrix contact area measurements represent at least 50 cells with indicated geometrical means ± 95% CI, in two independent experiments. The grey bars represent the range of cell area of not spread or fully spread uPART54A cells based on 95% CI on poly-D-lysine or on VN with uPA, respectively (Fig 2B).
- Cross signalling between β1 and β3 integrins. The indicated cells treated with 4B4 (where indicated) were plated for 30 min on LM609-coated plates. Phosphorylated p130Cas SD (columns) and cell-matrix contact area (dots) were quantified. Cell-matrix contact area measurements represent at least 50 cells with indicated geometrical means ± 95% CI, in two independent experiments. p130Cas SD phosphorylation of 293 uPART54A cells on VNRAD with uPA was set as 100%. Data are means ± s.e.m., n = 3.
- Cartoon illustrating the different receptors and ligands used to trigger non-integrin cell adhesion.
Ligand-independent cross signalling between integrins
An intriguing prediction of ligand-independent integrin signalling is that it may occur between different integrin family members co-expressed by the same cell. If this prediction is true, a signalling-deficient integrin with affinity for the ECM should still be able to promote cell spreading by ‘hijacking’ the signalling activity of other integrins co-expressed by the same cell. To test this hypothesis, we assayed signalling in cells expressing signalling-proficient β3WT or signalling-deficient β3Y747/759A seeded on the anti-αvβ3 antibody LM609 to uncouple the matrix-binding capacity of the integrin from its activation state (Fig 6C). Seeding β3WT cells on LM609 resulted in robust p130Cas phosphorylation and cell spreading that was largely refractory to inhibition by 4B4, consistent with the β3WT chain being responsible for both adhesion and signalling on this substrate. Under the same conditions, cells expressing β3Y747/759A displayed mildly reduced cell adhesion to LM609 (Supplementary Fig S6E) and partially impaired p130Cas phosphorylation and cell spreading (Fig 6C), consistent with β3Y747/759A being signalling incompetent. Importantly, however, the residual p130Cas phosphorylation and cell spreading observed for β3Y747/759A cells were fully inhibited by the 4B4 antibody, demonstrating that in these cells, the β1 integrin is responsible for signalling even if cell adhesion is mechanically sustained by the β3 integrin.
Ligand-independent integrin signalling involves mechanotransduction
The apparent lack of structural requirements to the receptor and its matrix ligand suggests that the sole requirement to these structures is that they physically connect the cells to the matrix (i.e. promote cell adhesion). Consistently, both VNRAD and the anti-uPAR antibody failed to induce p130Cas activation when presented to cells in a soluble form (Fig 7A). The finding that the ligands need to be immobilised on a rigid substrate suggests that mechanotransduction is involved in the process. To address this point, we produced hydrogels with different rigidity (Tse & Engler, 2010), coated these with VNRAD and examined the cell morphology by phase contrast microscopy (Fig 7B). The different stiffness of the hydrogels did not notably perturb uPAR-mediated adhesion to VNRAD (data not shown); however, cells displayed prominent cell spreading only on the rigid (40 kPa) substrate, whereas they remained mostly rounded on the soft (0.7 kPa) substrate. These data show that even in the absence of direct contact between the integrin and the ECM, signalling and cell spreading require force generation between the cells and the ECM as it has already been extensively documented for canonical integrin signalling.
Figure 7. uPAR/VN signalling requires cell adhesion, ECM rigidity and membrane tension.

- uPAR-induced signalling requires cell attachment. Cells expressing uPART54A were seeded on an anti-uPAR monoclonal antibody (α-uPAR) or on VNRAD (with uPA) for 30 min. For ‘off-plate’ experiments, cells were incubated in suspension with these components. Phosphorylation of p130Cas SD is shown in percentage of the positive control, that is the same cells seeded on VNRAD in the presence of uPA (n ≥ 3, mean ± SEM). Representative blots are shown.
- uPAR-induced cell spreading requires adhesion to a rigid matrix. Representative images of 293 cells expressing uPART54A plated in presence of uPA on hydrogels coated with VNRAD at different rigidity (soft: 0.7 kPa, Rigid: 40 kPa) for 30 min. Scale bar: 10 μm.
- Effect of plasma membrane relaxation on uPAR or integrin-induced cell spreading. 293 cells expressing uPART54A were seeded on FN or VNRAD (with uPA) in presence of sucrose (150 mM) or deoxycholate (0.4 mM) for 30 min. Cell-matrix contact area was quantified. Measurements represent at least 50 cells with indicated geometrical means ± 95% CI, in at least two independent experiments. The grey bars represent the range of cell area of not spread or fully spread uPART54A cells based on 95% CI on poly-D-lysine or on VN with uPA, respectively (Fig 2B).
- Increased membrane tension induces integrin-dependent cell spreading. Cells were seeded on PDL for 30 min in presence or absence of 4B4. Where indicated, an equal volume of hypotonic buffer (H2O with 1 mM MgCl2 and 1.2 mM CaCl2) or isotonic (medium) buffer was added to increase plasma membrane tension. Cell-matrix contact area was assayed and quantified. Measurements represent at least 50 cells with indicated geometrical means ± 95% CI, in at least two independent experiments. Representative DIC images of cells on PDL treated with isotonic or hypotonic medium are shown. Scale bar: 10 μm.
Ligand-independent integrin signalling requires membrane tension
The data suggest the existence of an unconventional mechanism of mechanotransduction where uPAR-induced cell adhesion to VN mechanically acts on a cellular component triggering ligand-independent integrin signalling. Having excluded functionally relevant lateral interactions between uPAR and integrins as the driving force, the only cellular component that mechanically connects the adhesion and signalling receptors is the plasma membrane itself. Analysis of membrane tension by atomic force microscopy (Supplementary Fig S7A and B) revealed that the lamellipodia of 293/uPART54A cells seeded on VN in the presence of uPA displayed increased membrane tension (young modulus ˜1,375 Pa) as compared to the cell body (young modulus ˜450 Pa). These data show that cell protrusions, induced by the uPAR/VN interaction like those formed during canonical integrin-mediated cell spreading (Lieber et al, 2013), are associated with an increased membrane tension.
To test if membrane tension is functionally involved in the coupling between uPAR and integrins, we analysed the effect of decreasing membrane tension using either a hypertonic medium that deflates cells (Houk et al, 2012) or low concentration of deoxycholate that relaxes the plasma membrane without affecting the cell volume (Raucher & Sheetz, 2000). Remarkably, the treatment with either sucrose or deoxycholate strongly impaired cell spreading on VNRAD, while the same treatments had no or little effect on integrin-mediated cell spreading on FN (Fig 7C). The molecular mechanisms of canonical and ligand-independent integrin signalling are thus clearly differentiated by the requirement for membrane tension.
The finding that membrane tension is required for ligand-independent integrin signalling suggests that an increase in plasma membrane tension might promote cell spreading under conditions of weak cell adhesion to non-integrin substrates. One such substrate, poly-D-lysine (PDL), favours cell adhesion by electrostatic interactions with charged molecules on the cell surface but typically does not induce efficient cell spreading. We therefore seeded cells on PDL-coated plates and analysed cell spreading after induction of plasma membrane tension by hyposmotic swelling (Raucher & Sheetz, 2000). Consistently with membrane tension being required for cell spreading on non-integrin substrates, the hyposmotic treatment triggered major changes in cell shape with the appearance of large lamellipodia-like protrusions, while cells in isotonic medium remain rounded (Fig 7D). Interestingly, cell spreading on PDL induced by hyposmotic treatment was mildly inhibited by the 4B4 antibody consistent with integrin signalling being, at least partially, involved also on PDL.
Discussion
Signalling by uPAR has been extensively studied and it is widely accepted that members of the integrin family are required and responsible for the transmission of the ‘uPAR signal’ across the plasma membrane (Smith & Marshall, 2010). The exact mechanism of the functional coupling between uPAR and integrins has, however, been elusive and a matter of debate. The coupling was initially believed to be direct with uPAR physically interacting with the integrin thereby changing its substrate specificity and signalling activity (Wei et al, 1996). Later studies have indicated that the direct interaction between uPAR and the ECM component VN is the real driver of the signalling and that direct specific protein–protein interactions between uPAR and integrins are functionally dispensable in the process (Kjøller & Hall, 2001; Madsen et al, 2007a; Smith et al, 2008). We here present evidence supporting a versatile mechanism in which the plasma membrane is responsible for the mechanical coupling between uPAR and integrins, as illustrated in Fig 8. In contrast to other paradigms for the uPAR/integrin coupling, based on specific direct receptor–receptor interactions, this novel paradigm for the first time explains how uPAR is able to functionally couple with a variety of structurally diverse integrins (Smith & Marshall, 2010).
Figure 8. Comparison between canonical and ligand-independent integrin signalling.
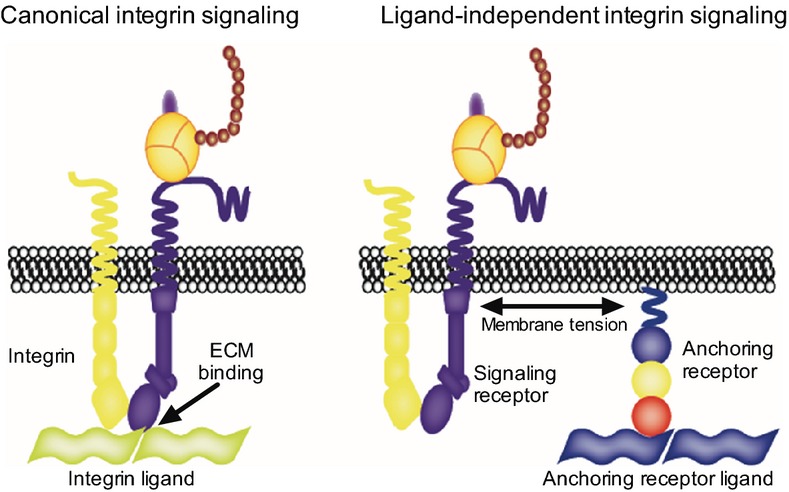
In canonical signalling, integrins engage the specific ligands in the ECM and signal by recruiting proteins on the cytoplasmic tails. In ligand-independent integrin signalling, the anchoring receptor-mediated cell adhesion, through plasma membrane, transmits a mechanical stimulus to the integrin that signals independently of ECM binding.
The biological relevance of ligand-independent integrin signalling is likely to go well beyond uPAR. In fact, it seems to be induced by any type of receptor–ECM interactions as long as cell adhesion is generated. Indeed, we have not been able to identify any specific structural constrains to the type of adhesion receptor and ECM component involved in the process. Consequently, the same signalling pathway is triggered by highly specific receptor–ECM interactions (i.e. uPAR/VN) as well as by non-selective membrane–ECM interactions (i.e. on poly-D-lysine). Ligand-independent integrin signalling does not seem to require a specific type of integrin, as both β1 and β3 integrins are proficient to support it.
The competence of integrins in signalling without engaging their specific ECM ligands is supported by independent studies. The disruption of the salt bridge in the membrane-proximal region (Hughes et al, 1996) or the introduction of polar residues in the integrin transmembrane segment (Li et al, 2003) result in enhanced integrin activation and signalling in cells in suspension in absence of ligands. These findings are, however, clearly differentiated from those presented here as those integrin mutations cause constitutive and adhesion-independent signalling. The ligand-independent signalling described here is not constitutive, it does not require activating mutations and it is both adhesion-induced and adhesion-dependent. The relevance of ligand-independent integrin signalling in vivo is supported by observations that the expression of critical genes, required for Drosophila embryo development, is supported by integrin chimeras lacking the ligand-binding domain (Martin-Bermudo & Brown, 1999). Furthermore, ligand-binding deficient mutants of αvβ3 are competent in supporting tumour growth through the formation of an oncogenic complex with SRC kinase (Desgrosellier et al, 2009).
Ligand-independent integrin signalling shares many common features with canonical integrin signalling including the requirement for an active conformation of the integrin, the binding of intracellular scaffolding proteins, as well as force generation on a rigid ECM. What clearly distinguishes the two types of integrin signalling, aside from the requirement for ligand binding, is the role of membrane tension. In canonical integrin signalling, the relaxation of membrane tension does not impair cell spreading but rather increases it (Raucher & Sheetz, 2000). Membrane tension is in fact known to antagonise cell protrusions and to rise during cell spreading and polarisation (Raucher & Sheetz, 2000; Houk et al, 2012). In the ligand-independent integrin signalling, described here, the relaxation of membrane tension abrogates cell spreading, while increasing membrane tension enhances cell spreading. This is possibly explained by the finding that in ligand-independent integrin signalling, the (tense) membrane is a critical component of the molecular clutch responsible for force transmission between the extracellular matrix and the cytoskeleton. In canonical ligand-dependent integrin signalling, the membrane is not an integral component of the clutch as integrins directly connect the ECM and the cytoskeleton (see cartoon in Fig 8).
Consistent with our finding that membrane tension is critical for cell spreading on non-integrin substrates, it has previously been reported that non-ligated β1 integrins are localised at the leading edge during cell protrusion (Galbraith et al, 2007), coinciding with zones of high membrane tension (Houk et al, 2012). The biological importance of membrane tension is furthermore substantiated by studies showing that membrane tension is required for the polarisation of neutrophils (Houk et al, 2012) and for efficient cell migration and lamellipodia organisation (Batchelder et al, 2011).
Material and Methods
Materials
HEK 293 Flp-In T-REx cells, expression vectors pcDNA5/FRT/TO and pOG44, zeocin, blasticidin S HCl and F-12 (Ham) medium were from Invitrogen. Dulbecco's modified Eagle's medium (DMEM) was from Lonza. PBS, trypsin, glutamine, penicillin and streptomycin were obtained from EuroClone, while foetal bovine serum (FBS) was from HyClone. Non-tissue culture plates were from Falcon Becton Dickinson. Tetracycline, poly-L-lysine, anti-vinculin antibody (hVIN-1) and CHO protein-free culture medium were from Sigma. FuGENE 6, fibronectin and hygromycin B were from Roche. Pro-uPA was kindly provided by Dr. Jack Henkin (Abbott Laboratories). Antibodies against total (cat no. 13383) and phosphorylated p130Cas (cat no. 4011), total ERK1/2 (cat. no. 9102) and phosphorylated ERK1/2 (cat. no. 9101) were from Cell Signalling Technology. The talin monoclonal antibody (cat. no. T3287) was from Sigma. Blocking antibodies against αvβ3 (LM609), α5β1 (P1D6) and αvβ5 (P1F6) integrins were obtained from Immunological Sciences or Millipore. Monoclonal antibodies against the β1 integrin were from BD Pharmingen (mAb13 and 9EG7), Beckman Coulter (4B4) and Santa Cruz (K-20). The monoclonal anti-uPAR R4 and R2 antibodies were kindly provided by Dr. Gunilla Høyer-Hansen (Finsen Laboratory, Copenhagen, Denmark). Paxillin antibody was from BD Pharmingen. Vinculin antibody was from Sigma. Glass-bottom 12-well plates used for DIC and time-lapse microscopy were from MatTek Corporation. SRC inhibitors, PP1 and PP2, were from Calbiochem, and the MEK inhibitor UO126 from Cell Signalling Technology. SiRNA oligos for luciferase and talin were from Riboxx. SiRNA oligos for α5 integrin are from Ambion.
Expression vector construction
β1 integrin oligos and cloning: To generate Flp-In expression vectors for the β1 integrin (i.e.), a β1A cDNA (OpenBiosystems, Cat# MHS1010-58245) was amplified with oligos B1f/B1ageR (product digested KpnI/AgeI) and B1tmF/B1r (product digested AgeI/NotI) and assembled in KpnI/NotI digested pcDNA5/FRT/TO generating pFRT/TO-β1WT. This procedure introduces a unique AgeI restriction site by silent mutagenesis allowing for the easy swapping of the extracellular and membrane/intracellular coding regions between different constructs. The single substitution mutants β1763A, β1763F, β1775A and β1775F were generated by amplification of pFRT/TO-β1WT with oligos B1tmF and B1Y763Ar, B1Y763Fr, B1Y775Ar or B1Y775Fr followed by re-cloning of the products AgeI/NotI in the parental vector. The single amino acid substitution mutants, β1218A, β1130D and β1227A, were generated by site-directed mutagenesis using oligos B1K218f/r, B1D130f/r and B1S227f/r, respectively. Constructs containing multiple substitutions were generated by multiple rounds of mutagenesis.
The expression vector for β3 integrin (pFRT/TO-B3) was generated by amplifying a human β3 integrin cDNA (OpenBiosystems, Cat# MHS4426-99626129) with oligos B3fn4/B3tmcR (product digested HinDIII/AgeI) and B3tmcF/B3rxx (product digested AgeI/XhoI) and assembling the fragments in HinDIII/XhoI-digested pcDNA5/FRT/TO. This procedure introduces a unique AgeI restriction site allowing for the easy swapping of the extracellular and membrane/intracellular coding regions between different constructs. The introduction of this restriction site causes a single amino acid substitution (K689T) that apparently does not compromise receptor function. The D119Y mutation in pFRT/TO-B3119Y was generated by site-directed mutagenesis using oligos B3119Yf/B3119Yr. Mutations preventing the interaction with intracellular adaptor proteins (Y747A, S752P and Y759A) were introduced by amplification of pFRT/TO-B3 with oligos B3tmcF/B3YSYrx and re-cloning the product AgeI/XhoI in the parental vector. VN cloning: a human VN cDNA (RZPD Clone ID: IRAUp969G1135D6)) was amplified with oligos hVNu (Bam)/hVNd (Xba) and the PCR product cloned BamHI/XbaI in pBluescript. A 6xHis tag was introduced at the C-termini by digestion with XbaI/NotI and insertion of a linker made by annealing oligos XbNhisf and XbNhisR. The 6xHis-tagged VN coding region was transferred BamHI/NotI to pcDNA5/FRT-TO generating the expression vector pFRT/TO-VN. The expression vector encoding VN lacking the SMB domain (pFRT/TO-VNΔSMB) was generated by amplification of pFRT/TO-VN with oligos SigUd40 and re-cloning the product BamHI/NotI. This strategy replaces amino acids 2–40 of VN with a single leucine residue.
Oligonucleotide sequences
B1f: 5′-cggggtacccgccgcgcggaaaagatgaatttacaaccaattttctgg-3′
B1ageR: 5′-ggaccggtgggacactctggattctc-3′
B1tmF: 5′-ccaccggtccagacatcattccaattgta-3′
B1r: 5′-tgcgcggccgctcattttccctcatacttcggattgacca-3′
B1K218f: 5′-aatgaacttgttggagcacagcgcatatctgga-3′
B1K218r: 5′-tccagatatgcgctgtgctccaacaagttcatt-3′
B1D130f: 5′-ctctactaccttatggccctgtcttactcaatg-3′
B1D130r: 5′-cattgagtaagacagggccataaggtagtagag-3′
B1S227f: 5′-tctggaaatttggatgctccagaaggtggtttc-3′
B1S227r: 5′-gaaaccaccttctggagcatccaaatttccaga-3′
B1Y763Ar: 5′-tgcgcggccgctcattttccctcatacttcggattgaccacagttgttacggcactcttagcaataggattttcacc-3′
B1Y763Fr: 5′-tgcgcggccgctcattttccctcatacttcggattgaccacagttgttacggcactcttaaaaataggattttcacc-3′
B1Y775Ar: 5′-tgcgcggccgctcattttccctcagccttcggattgacca-3′
B1Y775Fr: 5′-tgcgcggccgctcattttccctcaaacttcggattgacca-3′
B3fn4: 5′-aaaaagcttccaccatgcgagcacggccgcggcccc-3′
B3tmcF: 5′-ccaccggtcctgacatcctggtggtcctgctc-3′
B3tmcR: 5′-tgcgcggccgcttaagtgccccggtagctgat-3′
B3rxx: 5′-tgcctcgagttaagtgccccggtagctgat-3′
B3119Yf: 5′-atctactacttgatgtacctgtcttactccatg-3′
B3119Yr: 5′-catggagtaagacaggtacatcaagtagtagat-3′
B3YSYrx: 5′- gcctcgagttaagtgccccgggccgtgatattggtgaaggtgggcgtggcctctttagccagtgggttgtt-3′
HVnu: 5′-gcggatccagccctgccatggcacccctgag-3′
XbNhisF: 5′-ctagagggcatcatcaccatcaccattgagc-3′
XbNhisR: 5′-ggccgctcaatggtgatggtgatgatgccct-3′
SigUd40: 5′-cggggtaccatggcacccctgagaccccttctcatactg, gccctgctggcatgggttgctctggctgacctcccccaagtgactcgcggg-3′
RADf: 5′-ccccaagtgactcgcgcggatgtgttcactatg-3′
RADr: 5′-catagtgaacacatccgcgcgagtcacttgggg-3′
Cell culture and transfection
293 Flp-In T-Rex cells were grown in DMEM supplemented with 10% FBS, penicillin 100 U/ml, streptomycin 100 U/ml, l-glutamine 5 mM, 15 μg/ml blasticidin and 100 μg/ml zeocin at 37° in 5% CO2. CHO Flp-In cells were cultured in Ham's F12 medium supplemented with 10% FBS, penicillin 100 U/ml, streptomycin 100 U/ml, l-glutamine 5 mM and 100 μg/ml zeocin at 37° in 5% CO2. MDA-MB-231 cells were grown in Leibovitz medium supplemented with 10% FBS, penicillin 100 U/ml, streptomycin 100 U/ml and l-glutamine 5 mM at 37° in 0% CO2. The Flp-In system generates pools of isogenic transfectants carrying a single copy of the expression cassette in exactly the same chromosomal position, thus ensuring comparable expression levels of different receptor variants and, in addition, eliminating potential artefacts caused by clonal differences or heterogeneous expression level. The T-REx system permits inducible expression by the addition of tetracycline to the growth medium.
Transfections were performed using FuGENE keeping a 1:10 ratio between pOG44 (Invitrogen) and pcDNA5/FRT/TO-based vectors. HEK 293 and CHO-transfected cells were selected by substituting zeocin with 150 μg/ml or 300 μg/ml hygromycin B, respectively. Cells used in integrin structure–function analysis were first transfected to stably express the inducible uPART54A variant. Briefly, 293 Flp-In T-Rex cells were transfected with a vector encoding for uPART54A and selected with 1 mg/ml G418. The G418-resistant cells were cloned by limiting dilution, and a single clone expressing high levels of uPART54A (clone 22) was further transfected and selected, using the Flp-In system, as described above. Expression in HEK 293 Flp-In T-REx was induced by adding 1 μg/ml tetracycline to the medium overnight or for 2 days, in case of integrin constructs.
Expression and purification of recombinant proteins
Semi-confluent CHO Flp-In cells, stably transfected with the specific vectors encoding for VN variants, were washed with PBS and incubated for 1 or 2 weeks in CHO protein-free medium (SIGMA Aldrich). The supernatants were collected, and the VN variants were purified by immobilised metal affinity chromatography.
Adhesion assay
96-well plates were coated with purified substrates overnight at 4°C substrates (poly-D-lysine 100 μg/ml, FN 10 μg/ml, anti-αvβ3 integrin antibody LM609 20 μg/ml, VN, VNRAD, VNΔSMB, VNRADΔSMB were all coated at 5 μg/ml) and blocked for 2 h at 37°C with 2% heat-inactivated BSA in PBS. Cells were washed, harvested and counted. After three washes with binding buffer (DMEM supplemented with penicillin 100 U/ml, streptomycin 100 U/ml, l-glutamine 5 mM, HEPES 25 mM and 0.1% BSA), equal number of cells were seeded (3 × 104 cells/well) and allowed to adhere for 30 min in presence or absence of uPA (10 nM) and/or integrin-blocking antibodies (anti-αvβ5P1F6, anti-α β P1D6, anti-β 4B4 and mAb13 all used at 10 μg/ml). After washing extensively, adherent cells were fixed with 4% PFA and stained with crystal violet. Cell adhesion was quantified measuring absorbance at 540 nm.
Immunoblot experiments
24-well plates were coated overnight at 4°C with different substrates (poly-D-lysine 100 μg/ml, FN 10 μg/ml, α-uPAR antibody R4 20 μg/ml, anti-αvβ3integrin antibody LM609 20 μg/ml, VN and VNRAD were all coated at 5 μg/ml) and blocked for 2 h at 37°C with 5% heat-inactivated BSA in PBS. Detached cells were washed three times in binding buffer and counted. 2.5 × 105 cells/well were seeded and allowed to adhere for 30 min in presence or absence of uPA or GFD (10 nM). In case of antibody or inhibitors treatment, cells were pre-incubated with integrin-blocking antibodies (anti-αvβ5P1F6, anti-α5β P1D6, anti-β 4B4 and mab13 all used at 10 μg/ml) or inhibitors (PP1 and PP2 10 μM, UO126 20 μM) in suspension for 15 or 30 min, respectively, before plating. For off-plate experiments, cells were seeded on BSA-blocked plates for 30 min in presence of R4 (20 μg/ml) or VNRAD and uPA (respectively 5 μg/ml and 10 nM). For experiments with cells expressing β3integrin constructs, the concentration of 4B4 was increased to 20 μg/ml. Cells were lysed in 95°C reducing Laemmli buffer (60 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 0.01% bromophenol blue, 100 mM DTT) or in ice-cold RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, protease inhibitor cocktail (complete-EDTA-free), 1 mM PMSF, 1 mM EDTA, 1 mM NaF, and 1 mM Na3VO4). Equal amounts of protein were separated by SDS-PAGE, transferred to nitrocellulose membranes and probed as indicated in the figures. Different biological replicates were quantified by densitometry using ImageJ64 (for data recorded on photographic film) or Image Lab for data recorded by CCD (Chemidoc XRS, Biorad). In a first series of experiments, vinculin was used for normalisation of phosphorylated p130Cas, while total p130Cas was used in later experiments.
DIC and phase contrast microscopy
Glass-bottom plates were coated overnight at 4°C with different substrates (poly-D-lysine 100 μg/ml, FN 10 μg/ml, α-uPAR antibody R4 20 μg/ml, anti-αvβ3integrin antibody LM609 20 μg/ml, VN and VNRAD were all coated at 10 μg/ml) and blocked for 2 h at 37°C with 5% heat-inactivated BSA in PBS. Detached 293, CHO and MDA-MB-231 cells, expressing the different constructs, were washed three times in binding buffer and counted. 105 cells/well were seeded and allowed to adhere for 30 min in presence or absence of uPA (10 nM) where indicated. In case of antibody or inhibitors treatment, cells were pre-incubated with integrin-blocking antibodies (anti-αvβ5P1F6, anti-α5β P1D6, anti-β 4B4 and mAb13 all used at 10 μg/ml) or inhibitors (PP1 and PP2 10 μM, UO126 20 μM) in suspension for 15 or 30 min, respectively, before plating. For experiments involving membrane tension modulation, after washing, cells were resuspended in hyperosmotic buffer (binding buffer with 150 mM sucrose) and deoxycholate buffer (binding buffer containing 0.4 mM deoxycholate). For hyposmotic swelling, a volume of hyposmotic buffer (water containing 1.2 mM CaCl2 and 1 mM MgCl2) or isotonic buffer (medium) was added to the cells immediately before plating. For experiments involving membrane tension, the concentration of 4B4 was increased to 20 μg/ml. Adherent cells were fixed with 4% PFA (in PBS) for 10 min at room temperature. Fixed cells were washed with PBS and DIC or phase contrast imaging of cells was performed using an inverted microscope Olympus IX81. Cells were viewed through a high-aperture 60× or 20× objective lens (UIS2 60× TIRFM PlanApo N, NA 1.45; Olympus). Images were acquired using Hamamatsu Orca-ER digital camera with the software Metamorph 7.5.6.0. Cell area was quantified using ImageJ.
Hydrogel production
Hydrogels were produced as described (Tse & Engler, 2010). Acrylamide and bis-acrylamide were mixed to obtain hydrogel at 40 (rigid) and 0.7 (soft) kPa.
SiRNA
For talin siRNA, cells were seeded and directly transfected with Riboxx oligos at 20 nM as final concentration (Luciferase: UCGAAGUAUUCCGCGUACGCCCC, Talin oligo 1: UUUCUGCUUUAGUUUCUCCCCC, Talin oligo 4: UUCUCUUCCAUCAGCUCUCCCCC). For α5 siRNA, cells were transfected with oligos from Ambion (α5 oligo 1: GUUUCACAGUGGAACUUCAtt, (α5 oligo 3: GCAGAGAGAUGAAGAUCUAtt) or with oligos from Darmacon (Luciferase: CAUUCUAUCCUCUAGAGGAUGdTdT) at 50 nM as final concentration.
SiRNA transfection was performed using RNAiMAX according to manufacturer's protocol. After 72 h, cells were detached, plated on VNRAD and processed for the quantification of cell spreading as described before.
Label-Free Real-Time Cell-based Assay (RTCA) experiments
96-well E-plates (Roche) were coated with FN (10 μg/ml), VN (5 μg/ml), VNRAD (5 μg/ml) or PDL (100 μg/ml) overnight at 4°C and then blocked with 5% BSA in PBS for 1 h at 37°C. 15,000 cells were seeded in 100 μl serum-free OptiMEM medium (Life Technologies) supplemented with 100 U/ml penicillin and 100 U/ml streptomycin and allowed to adhere. After 15 min of pre-incubation at RT to settle the cells, the plate was transferred to a real-time cell-based assay instrument (RTCA, xCELLigence SP, Roche) located in a humidified cell culture incubator (37°C and 5% CO2). At regular intervals, the impedance (termed cell index, CI) was recorded. Cells were subjected to treatments as reported in the figures, and the times at which the treatments were performed are indicated in the graphs by vertical lines. Treatments were conducted by removing the E-plate from the instrument and adding 20 μl of sixfold concentrated compounds diluted in OptiMEM. Each condition was analysed in duplicate or triplicate, and the measured cell indexes normalised to the signal measured on PDL at the time of uPA addition.
FACS analysis
Cell surface expression of integrins was analysed by flow cytometry. For detection of integrins surface expression levels upon uPAR/VN interaction, 293 uPART54A cells were grown overnight and stimulated with uPA for 30 min prior to detachment. Integrins were detected using primary antibodies (P1D6 for α5β1, LM609 for αvβ3, P1F6 for αvβ5 and K20 β1 all used at 10 μg/ml). For the analysis of the expression levels of integrin mutants, tetracycline-induced cells were detached and stained with 4B4 antibody (β1 integrin 5 μg/ml) or LM609 antibody (10 μg/ml). Cells were stained with appropriate secondary FITC-labelled antibody (diluted 1:50) and analysed by flow cytometry (FACSCalibur; BD Biosciences).
Total internal reflection fluorescence microscopy (TIR-FM)
293 cells expressing uPART54A were seeded on FN, VN or VNRAD (in presence of uPA) as described above. Cells were then fixed with 4% PFA, eventually permeabilised with 0.1% Triton X-100 (for paxillin, vinculin and phalloidin staining) and blocked with 2% BSA. Cells were then incubated with the indicated primary antibody (K20 10 μg/ml, paxillin 1:50, vinculin 1:400, 1 h at room temperature) followed by secondary antibody (anti-mouse or anti-rat Alexa 488 or Alexa 647 conjugated, phalloidin TRIC conjugated 1 h at room temperature). For K20 phalloidin co-staining, cells were stained with K20 antibody prior to permeabilisation and phalloidin staining. For 9EG7 staining, cells were incubated with 9EG7 antibody (10 μg/ml, 15 min on ice) prior to fixation. For MDA-MB-231 staining, cells were plated on FN or VN, fixed and stained with K20 antibody as described above. Cells were then incubated 1 h at room temperature with anti-uPAR R2 antibody Alexa488 conjugated. TIR-FM imaging of cells was performed using an Olympus Biosystem TIR-FM workstation based on the Cell R Imaging System (Olympus Biosystems, Munich, Germany). A 488-nm Ar laser was coupled in an inverted epifluorescence motorized microscope (Olympus IX81) and focused at an off-axis position of the objective back focal plane. Cells plated on glass coverslips were viewed through a high-aperture 60× objective lens (UIS2 60× TIRFM PlanApo N, NA 1.45, Olympus, Tokyo, Japan). Images (16 bit depth) were acquired using an Orca-ER (C4742-80) Cooled CCD digital camera (Hamamatsu Italy, Milan, Italy). Time-lapse TIR-FM imaging of 293 cells expressing uPART54A and transiently transfected with α5 integrin GFP (kindly provided by Dr. Rick Horwitz) was performed as above with the exception that fixation/permeabilisation/staining was omitted and that the cells were maintained at 37°C in normal growth medium throughout the recordings (every 20 s for a total of 15 min). Adjustment of brightness/contrast and smoothening of images were done using ImageJ and always applied to the entire image.
Time-lapse imaging
Time-lapse live-cell imaging was performed at 37°C, 5% CO2 with an inverted IX70 microscope (Olympus) equipped with an incubation chamber (Solent Scientific). Cells were plated in serum-containing growth medium and viewed through 20× (LCPlanFl, NA 0.4 Ph1, Olympus, Tokyo, Japan) with an additional 1.5× magnification lens. The acquisition system includes a digital camera (Sensys, Roper Scientific) and System Control Software Metamorph 7.0r4 (Universal Imaging). Adjustment of brightness/contrast and smoothening of images were done using ImageJ and always applied to the entire image.
Live-cell atomic force microscopy measurements
The preparation of the cells was done as described in cell culture section, but eventually plating the cells onto VNRGD (10 μg/ml)-coated glass-bottomed dishes (Willco Wells) for AFM measurements. The cells were allowed to adhere in the absence or presence of uPA (10 nM). During the AFM measurements, the temperature of the imaging buffer (25 mM HEPES buffer at physiological pH) was maintained at 37°C by a homemade heating system. For combined topographical/mechanical measurements, a Bioscope Catalyst AFM (Bruker) was operated in force volume mode (Butt et al, 2005) using spherical probes attached to silicon cantilevers (elastic constant 0.22 N/m, measured radius of curvature of the tip 4,850 nm). AFM probes were manufactured and characterised in the laboratory using an established method (Indrieri et al, 2011). Each force volume consists in an array of 64 × 64 force versus distance curves from which the local height of the sample and the local elastic modulus can be extracted; topographic and elastic maps of the sample can therefore be acquired and are in one-to-one correspondence. All the measurements were performed with the following parameters: ramp length for each force curve: 5 μm; approaching speed 43.4 μm/s; scan rate 7.1 Hz; 2,048 points per curve. The lateral scan size varied between 60 × 60 μm and 100 × 100 μm, depending on the cell dimension. Data processing of force volumes was carried out in Matlab™ environment using custom routines. Using the Hertz model enhanced by finite thickness correction (Dimitriadis et al, 2002), each force curve was fitted on the small indentation regime (up to 15% of the total local indentation) to highlight the elasticity of the membrane and actin cytoskeleton.
Acknowledgments
This work was supported by research grants from the Italian Association for Cancer Research (AIRC) and the Cariplo Foundation hold by NS and Research fellowships from the Italian Federation for Cancer Research (FIRC) to GMSF and VDL. Cristina Lenardi e Paolo Milani are acknowledged for providing access to the cell culture laboratory of the CIMaINa centre and Luca Puricelli for support in AFM data analysis.
Author contributions
GMSF, CDM and NS conceived the study, analysed the data and wrote the manuscript. GMSF, CS, VB, VDL, AnP, MG, AlP and CDM did the experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Batchelder EL, Hollopeter G, Campillo C, Mezanges X, Jorgensen EM, Nassoy P, Sens P, Plastino J. Membrane tension regulates motility by controlling lamellipodium organization. Proc Natl Acad Sci USA. 2011;108:11429–11434. doi: 10.1073/pnas.1010481108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt HJ, Cappella B, Kappl M. Force measurements with the atomic force microscope: technique, interpretation and applications. Surf Sci Rep. 2005;59:1–152. [Google Scholar]
- Czuchra A, Meyer H, Legate KR, Brakebusch C, Fassler R. Genetic analysis of beta1 integrin “activation motifs” in mice. J Cell Biol. 2006;174:889–899. doi: 10.1083/jcb.200604060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgrosellier JS, Barnes LA, Shields DJ, Huang M, Lau SK, Prevost N, Tarin D, Shattil SJ, Cheresh DA. An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat Med. 2009;15:1163–1169. doi: 10.1038/nm.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitriadis EK, Horkay F, Maresca J, Kachar B, Chadwick RS. Determination of elastic moduli of thin layers of soft material using the atomic force microscope. Biophys J. 2002;82:2798–2810. doi: 10.1016/S0006-3495(02)75620-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith CG, Yamada KM, Galbraith JA. Polymerizing actin fibers position integrins primed to probe for adhesion sites. Science. 2007;315:992–995. doi: 10.1126/science.1137904. [DOI] [PubMed] [Google Scholar]
- Gardsvoll H, Ploug M. Mapping of the Vitronectin-binding Site on the Urokinase Receptor: involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region. J Biol Chem. 2007;282:13561–13572. doi: 10.1074/jbc.M610184200. [DOI] [PubMed] [Google Scholar]
- Houk AR, Jilkine A, Mejean CO, Boltyanskiy R, Dufresne ER, Angenent SB, Altschuler SJ, Wu LF, Weiner OD. Membrane tension maintains cell polarity by confining signals to the leading edge during neutrophil migration. Cell. 2012;148:175–188. doi: 10.1016/j.cell.2011.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PE, Diaz-Gonzalez F, Leong L, Wu C, McDonald JA, Shattil SJ, Ginsberg MH. Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J Biol Chem. 1996;271:6571–6574. doi: 10.1074/jbc.271.12.6571. [DOI] [PubMed] [Google Scholar]
- Indrieri M, Podesta A, Bongiorno G, Marchesi D, Milani P. Adhesive-free colloidal probes for nanoscale force measurements: Production and characterization. Rev Sci Instrum. 2011;82:023708. doi: 10.1063/1.3553499. [DOI] [PubMed] [Google Scholar]
- Kjøller L, Hall A. Rac mediates cytoskeletal rearrangements and increased cell motility induced by urokinase-type plasminogen activator receptor binding to vitronectin. J Cell Biol. 2001;152:1145–1157. doi: 10.1083/jcb.152.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunigal S, Lakka SS, Gondi CS, Estes N, Rao JS. RNAi-mediated downregulation of urokinase plasminogen activator receptor and matrix metalloprotease-9 in human breast cancer cells results in decreased tumor invasion, angiogenesis and growth. International journal of cancer Journal international du cancer. 2007;121:2307–2316. doi: 10.1002/ijc.22962. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lebeau AM, Duriseti S, Murphy ST, Pepin F, Hann B, Gray JW, Vanbrocklin HF, Craik CS. Targeting uPAR with antagonistic recombinant human antibodies in aggressive breast cancer. Cancer Res. 2013;73:2070–2081. doi: 10.1158/0008-5472.CAN-12-3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Mitra N, Gratkowski H, Vilaire G, Litvinov R, Nagasami C, Weisel JW, Lear JD, DeGrado WF, Bennett JS. Activation of integrin alphaIIbbeta3 by modulation of transmembrane helix associations. Science. 2003;300:795–798. doi: 10.1126/science.1079441. [DOI] [PubMed] [Google Scholar]
- Lieber AD, Yehudai-Resheff S, Barnhart EL, Theriot JA, Keren K. Membrane tension in rapidly moving cells is determined by cytoskeletal forces. Curr Biol. 2013;23:1409–1417. doi: 10.1016/j.cub.2013.05.063. [DOI] [PubMed] [Google Scholar]
- Loftus JC, O'Toole TE, Plow EF, Glass A, Frelinger AL, 3rd, Ginsberg MH. A beta 3 integrin mutation abolishes ligand binding and alters divalent cation-dependent conformation. Science. 1990;249:915–918. doi: 10.1126/science.2392682. [DOI] [PubMed] [Google Scholar]
- Luo BH, Strokovich K, Walz T, Springer TA, Takagi J. Allosteric beta1 integrin antibodies that stabilize the low affinity state by preventing the swing-out of the hybrid domain. J Biol Chem. 2004;279:27466–27471. doi: 10.1074/jbc.M404354200. [DOI] [PubMed] [Google Scholar]
- Madsen CD, Ferraris GM, Andolfo A, Cunningham O, Sidenius N. uPAR-induced cell adhesion and migration: vitronectin provides the key. The Journal of cell biology. 2007a;177:927–939. doi: 10.1083/jcb.200612058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen DH, Engelholm LH, Ingvarsen S, Hillig T, Wagenaar-Miller RA, Kjoller L, Gardsvoll H, Hoyer-Hansen G, Holmbeck K, Bugge TH, Behrendt N. Extracellular collagenases and the endocytic receptor, urokinase plasminogen activator receptor-associated protein/Endo180, cooperate in fibroblast-mediated collagen degradation. The Journal of biological chemistry. 2007b;282:27037–27045. doi: 10.1074/jbc.M701088200. [DOI] [PubMed] [Google Scholar]
- Martin-Bermudo MD, Brown NH. Uncoupling integrin adhesion and signaling: the betaPS cytoplasmic domain is sufficient to regulate gene expression in the Drosophila embryo. Genes Dev. 1999;13:729–739. doi: 10.1101/gad.13.6.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14:325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- Moser M, Legate KR, Zent R, Fassler R. The tail of integrins, talin, and kindlins. Science. 2009;324:895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- Pirazzoli V, Ferraris GM, Sidenius N. Direct evidence of the importance of vitronectin and its interaction with the urokinase receptor in tumor growth. Blood. 2013;121:2316–2323. doi: 10.1182/blood-2012-08-451187. [DOI] [PubMed] [Google Scholar]
- Raucher D, Sheetz MP. Cell spreading and lamellipodial extension rate is regulated by membrane tension. The Journal of cell biology. 2000;148:127–136. doi: 10.1083/jcb.148.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S, Friedl P. Interstitial cell migration: integrin-dependent and alternative adhesion mechanisms. Cell Tissue Res. 2010;339:83–92. doi: 10.1007/s00441-009-0892-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HW, Marra P, Marshall CJ. uPAR promotes formation of the p130Cas-Crk complex to activate Rac through DOCK180. The journal of cell biology. 2008;182:777–790. doi: 10.1083/jcb.200712050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- Takada Y, Ylanne J, Mandelman D, Puzon W, Ginsberg MH. A point mutation of integrin beta 1 subunit blocks binding of alpha 5 beta 1 to fibronectin and invasin but not recruitment to adhesion plaques. J Cell Biol. 1992;119:913–921. doi: 10.1083/jcb.119.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarui T, Akakura N, Majumdar M, Andronicos N, Takagi J, Mazar AP, Bdeir K, Kuo A, Yarovoi SV, Cines DB, Takada Y. Direct interaction of the kringle domain of urokinase-type plasminogen activator (uPA) and integrin alpha v beta 3 induces signal transduction and enhances plasminogen activation. Thromb Haemost. 2006;95:524–534. doi: 10.1160/TH05-06-0457. [DOI] [PubMed] [Google Scholar]
- Tse JR, Engler AJ. Preparation of hydrogel substrates with tunable mechanical properties. Curr Protoc Cell Biol. 2010;40:10.16.1–10.16.16. doi: 10.1002/0471143030.cb1016s47. [DOI] [PubMed] [Google Scholar]
- Wei Y, Waltz DA, Rao N, Drummond RJ, Rosenberg S, Chapman HA. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J Biol Chem. 1994;269:32380–32388. [PubMed] [Google Scholar]
- Wei Y, Lukashev M, Simon DI, Bodary SC, Rosenberg S, Doyle MV, Chapman HA. Regulation of integrin function by the urokinase receptor. Science. 1996;273:1551–1555. doi: 10.1126/science.273.5281.1551. [DOI] [PubMed] [Google Scholar]
- Wei Y, Czekay RP, Robillard L, Kugler MC, Zhang F, Kim KK, Xiong JP, Humphries MJ, Chapman HA. Regulation of alpha5beta1 integrin conformation and function by urokinase receptor binding. J Cell Biol. 2005;168:501–511. doi: 10.1083/jcb.200404112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.