

Abstract
Mutations in the Park2 gene, encoding the E3 ubiquitin-ligase parkin, are responsible for a familial form of Parkinson's disease (PD). Parkin-mediated ubiquitination is critical for the efficient elimination of depolarized dysfunctional mitochondria by autophagy (mitophagy). As damaged mitochondria are a major source of toxic reactive oxygen species within the cell, this pathway is believed to be highly relevant to the pathogenesis of PD. Little is known about how parkin-mediated ubiquitination is regulated during mitophagy or about the nature of the ubiquitin conjugates involved. We report here that USP8/UBPY, a deubiquitinating enzyme not previously implicated in mitochondrial quality control, is critical for parkin-mediated mitophagy. USP8 preferentially removes non-canonical K6-linked ubiquitin chains from parkin, a process required for the efficient recruitment of parkin to depolarized mitochondria and for their subsequent elimination by mitophagy. This work uncovers a novel role for USP8-mediated deubiquitination of K6-linked ubiquitin conjugates from parkin in mitochondrial quality control.
Keywords: deubiquitination, mitophagy, parkin, ubiquitin, USP8
See also: I Dikic & A Bremm (November 2014)
Introduction
Mutations in the Park2 gene are associated with a familial form of Parkinson's disease (PD) and are believed to impair the normal function of the parkin protein as an E3 ubiquitin (Ub)-ligase (Shimura et al, 2000). These loss-of-function mutations impede the function of parkin in a variety of cellular pathways, including mitochondrial quality control, a process believed to be central to the pathogenesis of PD (Narendra et al, 2008; Geisler et al, 2010; McLelland et al, 2014). In this pathway, parkin collaborates with another PD gene, PTEN-Induced Kinase I (PINK1), to promote the removal of dysfunctional mitochondria by autophagy (mitophagy) (Narendra et al, 2010b). The PINK1 protein is a mitochondrial kinase that is rapidly degraded upon import into healthy mitochondria (Jin et al, 2010; Greene et al, 2012). When the mitochondrial membrane potential is abolished, PINK1 import is stalled leading to its accumulation on the mitochondrial surface (Lazarou et al, 2012), where it phosphorylates the parkin Ub-like domain (Ubl) and Ub at serine 65 (S65), a residue conserved in both proteins (Kondapalli et al, 2012; Kane et al, 2014; Kazlauskaite et al, 2014; Koyano et al, 2014). S65 phosphorylation, in turn, promotes the activation and translocation of parkin from the cytosol to mitochondria (Kondapalli et al, 2012; Kane et al, 2014; Kazlauskaite et al, 2014; Koyano et al, 2014), where it ubiquitinates a number of mitochondrial proteins (Chan et al, 2011; Sarraf et al, 2013), in addition to ubiquitinating itself (Matsuda et al, 2010; Lazarou et al, 2013). Parkin E3 ubiquitin-ligase activity is critical for the efficient elimination of dysfunctional mitochondria by mitophagy (Geisler et al, 2010; Matsuda et al, 2010). However, parkin activity is auto-inhibited at baseline (Trempe et al, 2013; Wauer & Komander, 2013), and understanding how PINK1-mediated phosphorylation of Ub and parkin triggers its activation and recruitment to mitochondria is incomplete (Kondapalli et al, 2012; Kane et al, 2014; Kazlauskaite et al, 2014; Koyano et al, 2014).
A common mode of regulation used by E3 ubiquitin ligases involves auto-ubiquitination. Depending on which of the seven lysines within Ub is used for conjugation, E3 Ub-ligases can assemble poly-Ub chains on themselves and on substrates, with different topologies, to mediate distinct biological functions (Komander & Rape, 2012; Kulathu & Komander, 2012). Chains linked via Lys48 (K48) in Ub, the best-characterized type of Ub conjugation, typically lead to degradation of the substrate by the proteasome (Voges et al, 1999). In contrast, chains linked via one of the other six lysines in Ub can protect modified substrates from proteasomal degradation and divert them toward functions in a variety of cellular pathways including trafficking, signaling, and autophagy (Komander & Rape, 2012; Kulathu & Komander, 2012). Thus, regulation of parkin by auto-ubiquitination has the potential to profoundly affect its function. Auto-ubiquitination can be antagonized by deubiquitinating enzymes (DUBs), which remove Ub from the E3 (de Bie & Ciechanover, 2011). This is typically believed to protect the E3 from proteasomal degradation (Wu et al, 2004; Nathan et al, 2008; Mei et al, 2011). However, certain DUBs have been shown to exhibit specificity in hydrolyzing non-K48-linked Ub chains (Komander et al, 2009a; Reyes-Turcu et al, 2009; Mevissen et al, 2013). Whether or how this could affect the stability and function of a cognate E3 partner is only beginning to be explored (Marfany & Denuc, 2008; Ventii & Wilkinson, 2008; de Bie et al, 2010).
We recently reported that ataxin-3, a DUB responsible for Machado-Joseph's disease, can deubiquitinate parkin (Durcan et al, 2011, 2012). However, the effects of ataxin-3 on parkin-mediated mitophagy were not investigated. Moreover, as E3s can be regulated by multiple DUBs (Daviet & Colland, 2008; Nathan et al, 2008), we used an unbiased siRNA-based approach to ask whether deubiquitination of parkin regulates its function in mitophagy. We report here that USP8, a DUB previously associated with endosomal trafficking (Mizuno et al, 2005; Row et al, 2006), but not mitochondrial quality control, is required for the efficient recruitment of parkin to depolarized mitochondria and subsequent mitophagy. Moreover, we find that USP8 preferentially removes K6-linked Ub conjugates from parkin but has little effect on the ubiquitination or stability of other known mitochondrial substrates of parkin. Whereas K6-linked Ub conjugates on parkin appear to protect it from proteasomal degradation and to impede mitophagy, USP8-mediated removal of K6-linked Ub from parkin promotes parkin turnover and is required for mitophagy to proceed efficiently. Taken together, our work identifies USP8 and K6-linked Ub conjugates as key players in the regulation of parkin-mediated mitophagy.
Results
Reducing USP8 levels impairs recruitment of parkin to depolarized mitochondria
Given that DUBs can regulate the function of E3 Ub-ligases, we surmised that parkin might also collaborate with a DUB during mitophagy. Using an unbiased siRNA-based approach, we knocked down 87 putative DUBs encoded by the human genome (Supplementary Table S1) (Nijman et al, 2005) in U2OS cells stably expressing GFP-parkin and screened for effects on CCCP-induced parkin recruitment to mitochondria. Expression of endogenous parkin in these cells was undetectable relative to other cell lines (Supplementary Fig S1A). Silencing of USP8, but not any of the other DUBs, impaired parkin recruitment to mitochondria after 1 h of treatment with CCCP (Fig 1A–C, Supplementary Fig S1B–D). Immunoblotting confirmed that the USP8 protein was indeed reduced in these cells (Fig 1C, Supplementary Fig S1C). Time-lapse microscopy showed that knockdown of USP8 delayed but did not abolish parkin recruitment to depolarized mitochondria (Fig 1D and E, Supplementary Video S1). Indeed, after 2 h of CCCP treatment, parkin was ultimately recruited in USP8 siRNA-transfected cells (Fig 1D and E). This effect was not cell type-specific as knockdown of USP8 in HeLa cells transiently transfected with GFP-parkin caused a similar delay in parkin recruitment to mitochondria (data not shown). Furthermore, the delay does not appear related to impairment in the endocytic function of USP8, as knockdown of STAM1 and 2; two other core components of this pathway had no effect on parkin recruitment (Supplementary Fig S2).
Figure 1. USP8 siRNA delays parkin recruitment onto mitochondria.

A, B USP8 siRNA impedes parkin recruitment onto mitochondria following CCCP treatment. U2OS-GFP-parkin cells were transfected with non-targeting or USP8 siRNA (10 nM) for 60 h (A). Untreated cells or cells treated with CCCP for 1 h were fixed, and images were acquired after staining for the mitochondrial protein TOM20. After 1-h CCCP treatment, cells were analyzed for GFP-parkin co-localization onto TOM20-positive mitochondria (B). Experiments were blinded and performed in triplicate with 100 cells analyzed for each condition. The vertical bars represent SEM for three independent experiments. For statistical analysis, a two-way ANOVA with Tukey post-test was performed, **P < 0.01.
C Validation of USP8 siRNA knockdown. U2OS-GFP-parkin cells were transfected with non-targeting or USP8 siRNA oligos (10 nM) for 60 h. Cells were lysed and analyzed by immunoblotting for USP8, parkin (long and short exposure), and actin.
D, E A delay in parkin recruitment onto mitochondria is observed in cells transfected with USP8 siRNA by live-cell microscopy. U2OS-GFP-parkin cells were transfected with non-targeting or USP8 siRNA (10 nM) for 60 h (D). 16 h prior to imaging, cells were infected with CellLight® mitochondria-RFP (Mito-RFP) to visualize mitochondria. Live-cell imaging was initiated 5 min after CCCP treatment, and images were acquired every 5 min over a 140-min period. Parkin recruitment upon membrane depolarization is visualized by the appearance of punctate GFP fluorescence superposed onto mitochondrial RFP fluorescence. Quantification of GFP-parkin recruitment to mitochondria (E) is facilitated by calculating the percentage of cells showing recruitment of GFP-parkin onto mitochondria at 5-min intervals over a period of 145 min. Experiments were performed in triplicate with 350 cells analyzed in each condition. The vertical bars represent the mean ± SEM for three independent experiments (see also Supplementary Video S1).
F, G Expression of FLAG-HA-USP8 rescues the effects of USP8 siRNA. U2OS-GFP-parkin cells were co-transfected with USP8 RNAi (5 nM) and FLAG-USP8 (0.5 μg) for 60 h (F). Cells treated with CCCP for 1 h were fixed, and images were acquired after staining for the mitochondrial protein, TOM20. After 1-h CCCP treatment, cells were analyzed for GFP-parkin co-localization onto TOM20-positive mitochondria in cells either negative or positive for FLAG-USP8 (G). Experiments were blinded and performed in triplicate with 100 cells analyzed for each condition. For statistical analysis, a two-way ANOVA with Tukey post-test was performed, ****P < 0.0001. The vertical bars represent the mean ± SEM for three independent experiments (see also Supplementary Fig S3D and E).
Source data are available online for this figure.
USP8 knockdown also led to an increase in steady-state parkin levels (Fig 1C, Supplementary Fig S1C), with noticeable cell-to-cell variability based on the intensity of GFP fluorescence (Fig 1D, Supplementary Fig S1D). However, impaired parkin recruitment could not simply be explained by an increase in parkin expression, as cells with both high and low parkin levels displayed a comparable delay in mitochondrial recruitment (Supplementary Fig S1E). Nonetheless, our findings suggest that USP8 is required not only for the efficient recruitment of parkin to mitochondria but also for the efficient turnover of parkin itself. Indeed, we noticed that knocking down USP8 in cell lines and primary neurons increased both transfected and endogenous parkin levels (Fig 1C, Supplementary Figs S1C and S3A–C). Conversely, USP8 overexpression reduced steady-state parkin levels in a dose-dependent manner and rescued the effects of USP8 siRNA on parkin levels (Supplementary Fig S3D and E). Moreover, the delay in parkin recruitment onto mitochondria induced by USP8 siRNA was rescued in cells transduced with a FLAG-USP8 expression plasmid (Fig 1F and G). Thus, knockdown of USP8 increases parkin levels and delays its recruitment onto mitochondria.
Silencing USP8 impairs mitophagy
Prolonged treatment with CCCP ultimately leads to the elimination of depolarized mitochondria by parkin-dependent mitophagy (Narendra et al, 2008). Given that silencing USP8 delayed parkin recruitment to mitochondria, we asked whether it also affected mitophagy. U2OS-GFP-parkin cells were transfected with either non-targeting or USP8 siRNA and treated with CCCP for 24 h. As expected, with transfection of non-targeting siRNA, the majority of cells showed a loss of TOM20 (Fig 2A upper), TIM23, and COX1 (data not shown) staining, indicating their mitochondria had been efficiently cleared by autophagy. In contrast, fewer cells transfected with USP8 siRNA lost TOM20 staining, indicating that mitophagy was impaired (Fig 2A and B). Prolonged CCCP treatment also led to a marked reduction in parkin staining in cells transfected with non-targeting siRNA, as has been noted previously (Fig 2A upper) (Geisler et al, 2010; Tanaka et al, 2010; Rakovic et al, 2013). In contrast, in USP8 siRNA-transfected cells, GFP-parkin staining persisted after 24 h of CCCP treatment and formed puncta that co-localized with mitochondrial markers (TOM20, COX1, and Tim23), providing a robust readout of parkin-mediated mitophagy (Fig 2A and C, Supplementary Fig S4A and B). However, as with parkin recruitment, the effect on mitophagy appears to be delayed rather than abolished, as no differences were detectable between non-targeting and USP8 siRNA-treated cells by 48 h of CCCP treatment (Fig 2D and E).
Figure 2. USP8 siRNA impairs parkin-mediated mitophagy.

A–E USP8 siRNA impairs parkin-mediated mitophagy. U2OS-GFP-parkin cells were transfected with non-targeting or USP8 siRNA (10 nM) for 48 h (A). Transfected cells were either left untreated or were treated with CCCP for 24 or 48 h before fixation. Immunofluorescence images of cells were acquired after staining for TOM20. Cells were outlined in white as a result of low GFP-parkin levels. The percentage of all U2OS-GFP-parkin cells after 24 h (B, C) or 48 h (D, E) CCCP treatment lacking TOM20-positive mitochondria or containing GFP-parkin puncta co-localizing with TOM20-positive mitochondria was quantified in cells transfected with non-targeting or USP8 siRNA. Experiments were blinded and performed in triplicate with 100 cells analyzed for each condition. The vertical bars represent SEM. For statistical analysis, a two-way ANOVA with Tukey post-test was performed, *P < 0.05, **P < 0.01; NS, not significant.
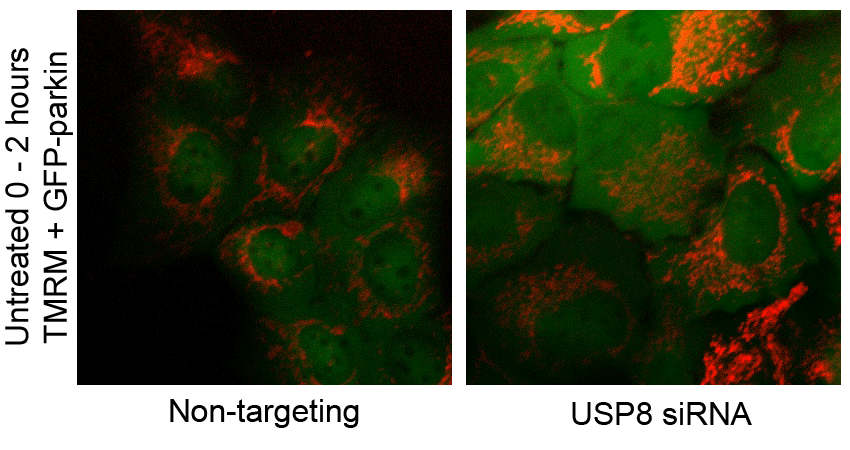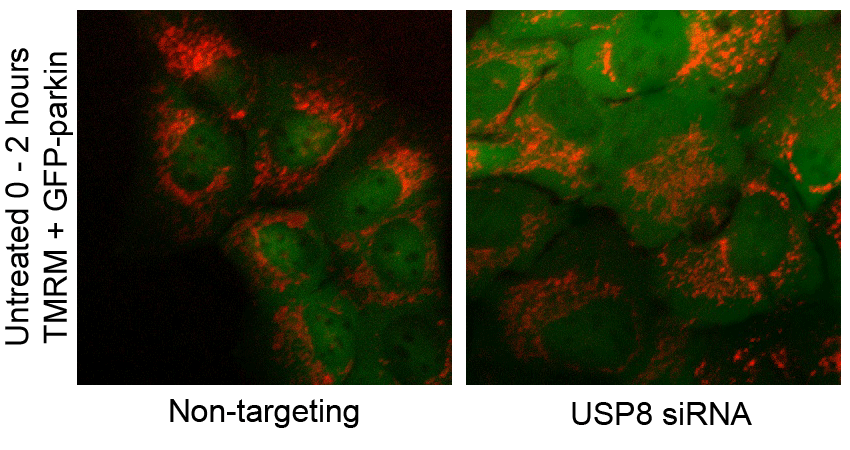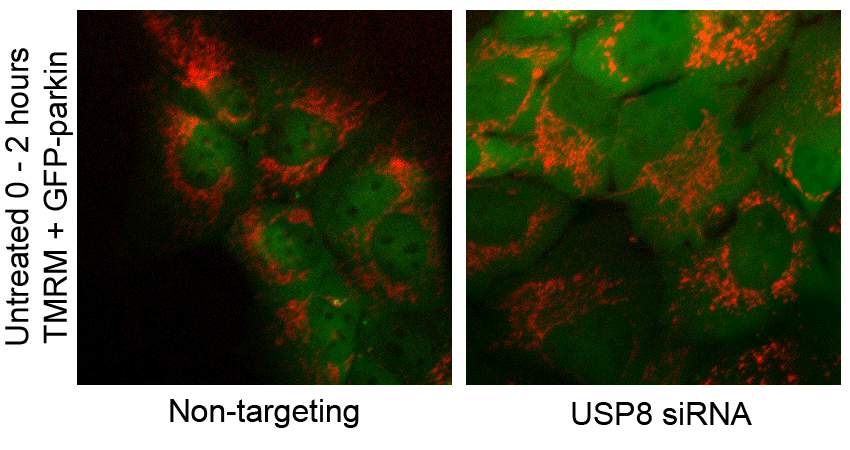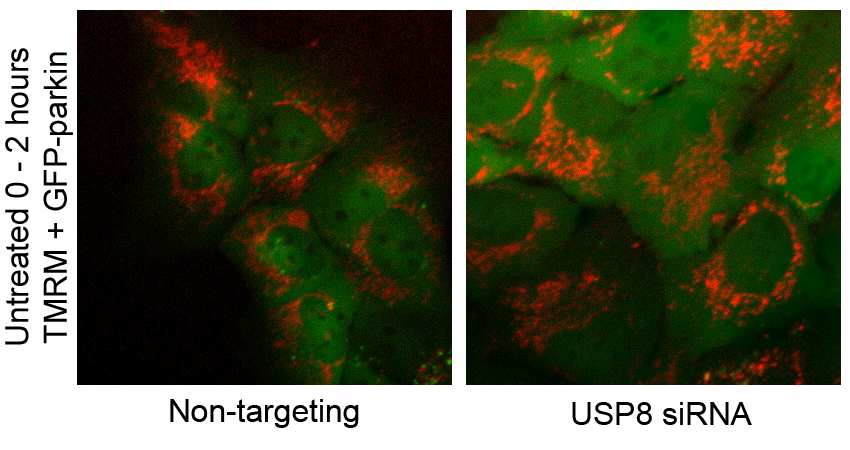
F USP8 siRNA has no effect on CCCP-induced mitochondrial depolarization. U2OS-GFP-parkin cells transfected with non-targeting or USP8 siRNA (10 nM) for 60 h were first incubated with the potentiometric dye TMRM (600 nM) 20 min prior to imaging. Prior to CCCP treatment, cells were imaged for 10 min to confirm TMRM staining. Following CCCP treatment, images of cells were acquired every minute for 10 min (see also Supplementary Video S2). The membrane potential in untreated cells was tested by imaging untreated cells for 1 h (see also Supplementary Video S3).
G USP8 siRNA has no discernible effect on CCCP-induced PINK1 accumulation. U2OS-GFP-parkin cells transfected with non-targeting, PINK1 siRNA, or USP8 siRNA (10 nM) for 60 h were either left untreated or were treated with CCCP for 1 or 3 h. Cells were lysed and immunoblotted for parkin, actin, and PINK1.
Source data are available online for this figure.
CCCP dissipates the proton gradient across the mitochondrial inner membrane, which blocks the import and degradation of PINK1 (Narendra et al, 2010b; Greene et al, 2012). This in turn leads to an accumulation of PINK1 at the mitochondrial outer surface, where it phosphorylates Ub and parkin at serine 65, thereby activating parkin and triggering its translocation from the cytosol (Kondapalli et al, 2012; Kane et al, 2014; Kazlauskaite et al, 2014; Koyano et al, 2014). Thus, we asked whether the effect of USP8 on parkin recruitment was upstream or downstream of mitochondrial depolarization and PINK1 accumulation. Staining with the potentiometric dye TMRM revealed that silencing USP8 affected neither basal mitochondrial membrane potential nor the ability of CCCP to abolish mitochondrial membrane potential (Fig 2F, Supplementary Videos S2 and S3). Similarly, USP8 knockdown affected neither the steady-state levels of endogenous PINK1 nor its accumulation in response to CCCP (Fig 2G).
Using time-lapse microscopy, we also demonstrate using mitochondrially targeted RFP that USP8 siRNA neither altered mitochondrial morphology, fusion, or fission at baseline (Supplementary Fig S4C, Supplementary Video S4) nor did it affect mitochondrial fragmentation in response to CCCP (Supplementary Video S5). Finally, steady-state levels of mitochondrial proteins appeared unchanged between non-targeting and USP8 knockdown cells (Supplementary Fig S4D). Thus, the effect of USP8 on parkin recruitment is downstream of mitochondrial depolarization and PINK1 accumulation and is not the result of off-target siRNA effects on PINK1 or on mitochondrial dynamics.
USP8 deubiquitinates parkin
We noticed an apparent increase in the levels of Ub conjugates on parkin in the absence of USP8 (Fig 1C). These findings were confirmed in HEK293T cells by overexpressing HA-Ub and FLAG-parkin, a paradigm that stimulates parkin auto-ubiquitination at steady state. Indeed, immunoprecipitation with FLAG followed by immunoblotting with HA showed that parkin Ub conjugates were significantly increased in cells transfected with USP8 siRNA (Fig 3A and B). Conversely, HA-Flag-USP8 overexpression reduced parkin auto-ubiquitination (Supplementary Fig S5A). To address whether USP8 could directly deubiquitinate parkin, we carried out in vitro ubiquitination reactions using recombinant and affinity-purified GST-parkin, Ub, ATP and E1 and E2 enzymes. Ubiquitinated GST-parkin, bound to glutathione sepharose beads, was then washed extensively to remove free Ub and other components of the reaction, followed by incubation with either full-length USP8 or one of several commercially available DUBs. These DUBs all exhibited enzymatic activity in Ub-AMC cleavage assays (Durcan et al, 2012). However, save for USP8 and USP2, none could hydrolyze Ub conjugates on parkin (Fig 3C). The DUB activity of USP8 appeared to be specific toward parkin, as it was much less efficient at deubiquitinating other E3s, including HHARI, an RBR family E3 Ub-ligase structurally very similar to parkin (Fig 3D) (Duda et al, 2013; Trempe et al, 2013). In addition to GST-parkin, USP8 could efficiently deubiquitinate untagged parkin, indicating that it targeted Ub conjugated to parkin per se rather than to the N-terminal GST-tag (Fig 3E). The effect of USP2 on parkin was not surprising, as it had been shown previously to disassemble Ub chains indiscriminately (Komander et al, 2009b). Indeed, USP2 was able to deubiquitinate several E3s non-selectively in addition to parkin (Supplementary Fig S5B) (Komander et al, 2009b). However, USP2 siRNA showed no effect on parkin levels or on parkin recruitment to mitochondria (Fig 3F, Supplementary Fig S6). Similarly, knockdown of ataxin-3, the first identified DUB partner of parkin (Durcan et al, 2011), had no effect on the mitochondrial recruitment of parkin (Fig 3F, Supplementary Fig S6). Recently, USP15 and USP30 were shown to inhibit mitophagy by counteracting parkin-mediated ubiquitination (Bingol et al, 2014; Cornelissen et al, 2014). However, in contrast to USP8, these two DUBs were believed to deubiquitinate substrates of parkin on mitochondria downstream of recruitment rather than deubiquitinating parkin itself. Consistent with this model, we find that USP15 cannot deubiquitinate parkin directly (Fig 3C) and that neither the knockdown of USP15 (not shown) nor USP30 (Fig 3F, Supplementary Fig S6) affects parkin recruitment to mitochondria. Taken together, the findings indicate that USP8 directly and specifically deubiquitinates parkin.
Figure 3. USP8 deubiquitinates parkin.

A, B USP8 knockdown causes Ub conjugates to accumulate on FLAG-parkin in HEK293T cells. HEK293T cells were co-transfected with FLAG-parkin (0.5 μg), HA-UbWT (0.5 μg) and non-targeting or USP8 siRNA (5 nM). Lysates were immunoprecipitated with FLAG resin and analyzed by immunoblotting for HA and FLAG. Input lysates (5% of total input) were analyzed by immunoblotting for USP8 and actin. The optical densities of the Ub-parkin conjugates were quantified using NIH ImageJ, and the data represent the mean ± SEM for three independent experiments. For statistical analysis, a two-way ANOVA with Tukey post-test was performed, **P < 0.01.
C The activity of USP8 toward parkin relative to other DUBs. GST-parkin bound to glutathione beads was left to ubiquitinate for 2 h alone at 37°C. After 2 h, the beads were washed to remove reaction components and ubiquitinated GST-parkin was then incubated in the presence or absence of the indicated DUB for 1 h at 37°C. Reactions were immunoblotted for Ub and Ponceau stained for GST-parkin.
D USP8 preferentially hydrolyzes preassembled parkin Ub conjugates. For these reactions, GST-parkin, GST-CHIP, GST-HHARI (RBR domain), or GST-cIAP1 bound to glutathione beads were left to ubiquitinate for 2 h alone at 37°C. After 2 h, the beads were washed to remove reaction components. The ubiquitinated E3 was then incubated in the presence or absence of the full-length His-USP8 for 1 h at 37°C. Reactions were analyzed by Ponceau S staining and immunoblotting for Ub.
E USP8 can hydrolyze preassembled Ub conjugates on untagged parkin. For these reactions, untagged parkin was left to ubiquitinate for 2 h alone at 37°C with UbcH7 as the E2. After 2 h, apyrase was added for 20 min to terminate the reaction. Ubiquitinated parkin was now incubated for 1 h at 37°C in the presence or absence of His-tagged full-length USP8. Reactions were immunoblotted for Ub and parkin.
F Knockdown of other DUBs compared to USP8 had no effect on the recruitment of parkin onto mitochondria following CCCP treatment. U2OS-GFP-parkin cells were transfected with either non-targeting, USP8, ataxin-3, USP30, or USP2 siRNA (10 nM) for 60 h. Cells treated with CCCP for 1 h were fixed, and images were acquired after staining for the mitochondrial protein, TOM20 (see also Supplementary Fig S6).
Source data are available online for this figure.
USP8 regulates parkin auto-ubiquitination during mitophagy
Next, we examined the effect of USP8 on the ubiquitination of parkin and mitochondrial proteins in cells during mitophagy. In cells transfected with non-targeting siRNA, CCCP treatment led to a transient increase followed by a profound reduction in parkin levels, as described previously (Fig 4A–C) (Chan et al, 2011; Rakovic et al, 2013). Concurrently with the changes in parkin levels, the electrophoretic mobility of parkin was transiently shifted upward, consistent with a CCCP-induced activation of parkin and parkin auto-ubiquitination, as reported previously (Matsuda et al, 2010; Lazarou et al, 2013). Indeed, in control conditions, Ub affinity chromatography using Ub binding Agarose (TUBEs) followed by immunoblotting for parkin confirmed that it was markedly but transiently ubiquitinated in response to CCCP (Fig 4B, upper). In comparison, the increase in parkin levels and ubiquitination after CCCP persisted several hours longer in USP8 knockdown cells (Fig 4A–C). No comparable differences were noted in total Ub levels, suggesting that the effect of USP8 on parkin ubiquitination was specific (Fig 4B, lower). Indeed, both steady-state levels (Supplementary Fig S4D) and CCCP-induced changes in levels and ubiquitination of other mitochondrial proteins, including known substrates of parkin such as the mitofusins and VDAC (Geisler et al, 2010; Lazarou et al, 2013; Sarraf et al, 2013), did not appear to be greatly affected by USP8 knockdown (Fig 4A, lower). Finally, consistent with our findings in GFP-parkin U2OS cells, knockdown of USP8 in HEK293 cells also caused a marked increase in the ubiquitination of untagged parkin following treatment with CCCP (Fig 4D).
Figure 4. Parkin auto-ubiquitination is enhanced following USP8 siRNA.

A–C USP8 siRNA prolongs parkin auto-ubiquitination following treatment with CCCP over 4 h. U2OS-GFP-parkin cells were transfected with non-targeting or USP8 siRNA (10 nM) for 60 h (A). Cells were either left untreated or were treated with CCCP for the indicated time intervals. Lysates were analyzed by immunoblotting for parkin, actin, USP8, Mfn1, Mfn2, PDH (E2), Tim23, TOM20, and VDAC1. From these lysates, ubiquitinated proteins were purified with Agarose-TUBEs (B). Bound proteins were analyzed by immunoblotting for parkin and Ub. The optical densities of ubiquitinated parkin (parkin smear excluding unmodified parkin) and total actin were quantified using NIH ImageJ (C), and the data represent the mean ± SEM for three independent experiments. For statistical analysis, a two-way ANOVA with Tukey post-test was performed, *P < 0.05, ***P < 0.001, ****P < 0.0001; NS, not significant.
D USP8 siRNA enhances the auto-ubiquitination of untagged parkin following treatment with CCCP. HEK293T cells were co-transfected with untagged parkin and either non-targeting or USP8 siRNA (10 nM) for 60 h. Cells were either left untreated or were treated with CCCP for the indicated time intervals. Lysates were analyzed by immunoblotting for parkin, actin, USP8, and Mfn1.
Source data are available online for this figure.
To determine which lysine residues in parkin were ubiquitinated in cells during mitophagy, we purified GFP-parkin using GFP-nAb Agarose, followed by mass spectrometry (Fig 5A). In untreated cells, no sites of ubiquitination were detected on parkin by LC-MS/MS, although phosphorylation at serine 131 was observed in both untreated and CCCP-treated cells (Fig 5B). Following treatment with CCCP, ubiquitination at three sites, K27, K48, and K76, and phosphorylation at S65 were identified by LC-MS/MS, all within the parkin Ubl domain (Fig 5B) and consistent with previous findings (Kondapalli et al, 2012; Sarraf et al, 2013). Next, we performed Parallel Reaction Monitoring (PRM) to determine the relative abundance of Ub conjugates at each site, dynamically during mitophagy. These were normalized to total parkin levels, using a control, unmodified parkin peptide (Supplementary Table S2). Whereas Ub conjugates were undetectable by PRM at baseline, CCCP treatment led to an increase in ubiquitination at each of the three sites (Fig 5C). In contrast, silencing USP8 led to a detectable accumulation of Ub conjugates at two of the three sites (K48 and K76) on parkin at baseline and to a further increase in parkin ubiquitination after 4 h of CCCP at all three sites (Fig 5D–F). Indeed, LC-MS/MS did not reveal any additional lysines on parkin that were ubiquitinated upon knockdown of USP8, either at baseline or after CCCP. Thus, USP8 opposes parkin auto-ubiquitination during mitophagy. In the absence of USP8, Ub conjugates accumulate after several hours of parkin activity on depolarized mitochondria, predominantly on these sites within the parkin Ubl. In the absence of USP8, low levels of parkin Ub conjugates also accumulate at steady state, possibly due to constitutive parkin activity and ongoing mitophagy in these cells.
Figure 5. Knockdown of USP8 leads to an increase in Ub conjugates on parkin.

A–C Mass spectrometry analysis of GFP-parkin in untreated and CCCP-treated U2OS-GFP-parkin cells. U2OS-GFP-parkin cells were left untreated or treated with CCCP for 1 h (A). Lysates (400 μg) were immunoprecipitated with GFP-nAb Agarose. Lysates and beads (5% of total bead volume) were analyzed by immunoblotting for parkin and actin. Following on-bead digestion, GFP-parkin samples were analyzed by LC-MS/MS mass spectrometry (B). Identified sites of ubiquitination and phosphorylation on parkin and Ub are indicated. Peptides for each of the 3 sites of ubiquitination on GFP-parkin (K27, K48 and K76) and an unmodified control parkin peptide were selected for time-scheduled parallel reaction monitoring (PRM) (C). The relative abundance of each peptide between samples was calculated, and the relative amount of Ub conjugates present at each individual lysine in parkin was normalized relative to total parkin levels. The data represent the mean value of two technical replicates.
D–F PRM mass spectrometry analysis of GFP-parkin following USP8 knockdown U2OS-GFP-parkin cells were transfected with either non-targeting or USP8 siRNA (10 nM). After 60 h, cells were either left untreated or were treated with CCCP for 4 h. The abundance of Ub conjugates at the indicated sites in GFP-parkin was analyzed by parallel reaction monitoring. The relative abundance of each peptide between samples was calculated, and the relative amount of Ub conjugates present at each individual lysine in parkin was normalized relative to total parkin levels. The data represent the mean value of two technical replicates.
G, H AQUA analysis and quantification of Ub linkages in parkin poly-Ub conjugates. In vitro ubiquitination reactions were carried out with GST-parkin bound to glutathione beads for 2 h at 37°C in the presence or absence of Ub, E1, UbcH7, and GST alone (G). Ubiquitinated GST-parkin was eluted, trypsinized, and analyzed on an LC-ESI-TOF. Internal peptides for Ub and K6, K11, K33, K48, and K63 Ub linkages were used in the analysis. Ub conjugates were not detected on GST alone. K27, K29, or K33 linkages were not detected in parkin poly-Ub conjugates. Poly-Ub conjugates did not form on parkin when Ub, E1, or E2 were absent (data not shown). The data represent the mean ± SEM for two independent experiments. For statistical analysis, a one-way ANOVA with Tukey post-test was performed, **P < 0.01, *P < 0.05. AQUA analysis of parkin self-ubiquitination in the presence of different E2 Ub-conjugating enzymes (H). UbcH7, UbcH5b, and Ube7 were the E2s tested in the analysis.
Source data are available online for this figure.
USP8 selectively removes K6-linked ubiquitin conjugates from parkin
What types of Ub chains are conjugated to parkin during mitophagy? Modification of proteins with K48-linked Ub chains typically leads to degradation of the substrate by the proteasome. In contrast, chains linked via one of the other six lysines in Ub can protect modified substrates from proteasomal degradation and divert them toward functions in variety of cellular pathways including trafficking, signaling, and autophagy (Komander & Rape, 2012; Kulathu & Komander, 2012). Given that collectively our findings so far indicated that, paradoxically, repressing USP8-mediated deubiquitination appears to protect parkin from degradation (Fig 1C, Supplementary Figs S1C and S3A–C), we asked whether such alternative, non-K48-linked Ub modifications were involved. Indeed, affinity purification of GFP-parkin from CCCP-treated cells, followed by LC-MS/MS, revealed Gly-Gly modifications on K6, K11, K48, and K63 of Ub, indicating that Ub chains on parkin were linked via these residues (Fig 5B). We also noted phosphorylation at S65 of Ub, as recently reported (Fig 5B) (Kane et al, 2014; Kazlauskaite et al, 2014; Koyano et al, 2014). None of these modifications were present in the absence of CCCP. Thus, activation of parkin during mitophagy leads to the generation of K6-, K11-, K48-, and K63-linked Ub chains and the phosphorylation of Ub at S65. To determine the nature of the Ub conjugates on parkin more quantitatively, we carried out in vitro ubiquitination reactions followed by absolute quantification (AQUA) mass spectrometry (Kirkpatrick et al, 2006). Remarkably, approximately 60% of poly-Ub chains on parkin were linked via K6 (Fig 5G), irrespective of the E2 used in the assay (Fig 5H). K48-, K11-, and K63-linked poly-Ub chains were also present, albeit in much lower proportions than K6. None of the other linkages were detected, consistent with our LC-MS/MS findings in cells (Fig 5B). Thus, our findings indicate that a considerable proportion of parkin auto-ubiquitination, both in vitro and in cells during mitophagy, involves K6, a Ub chain linkage not previously associated with parkin function.
Given that K6 appears to represent the predominant Ub-linkage in parkin Ub conjugates, we next asked whether USP8 might preferentially target K6 linkages. We carried out sequential in vitro parkin/USP8 ubiquitination/deubiquitination reactions using either wild-type Ub or a Ub mutant, in which K6 was mutated to arginine (UbK6R) (Fig 6A and B, Supplementary Fig S5C). Whereas both full-length and isolated USP8 catalytic domain efficiently removed wild-type (presumably mostly K6-linked) Ub conjugates from parkin, USP8 failed to efficiently deubiquitinate parkin modified with non-K6-linked UbK6R (Fig 6A and B, Supplementary Fig S5C). These UbK6R conjugates were not inherently resistant to deubiquitination as USP2 efficiently removed them from parkin (Fig 6A, Supplementary Fig S5C). Moreover, USP8 was able to deubiquitinate parkin modified with UbK11R-, UbK48R-, and UbK63R-linked Ub, as these chains presumably still contained K6-linked Ub (Supplementary Fig S5D). However, in line with previous findings (Mizuno et al, 2005; Row et al, 2006), we found that USP8 did not appear to exhibit intrinsic linkage selectivity when the Ub chains were not attached to parkin, as it could efficiently cleave K6-, K11-, K48-, and K63-linked free di-Ub (Supplementary Fig S5E). Thus, our findings indicate that USP8 preferentially removes K6-containing Ub chains from parkin.
Figure 6. USP8 hydrolyzes K6 linkages in parkin Ub conjugates.

A, B USP8 is unable to hydrolyze preassembled parkin Ub conjugates when K6 linkages are absent. GST-parkin bound to glutathione beads was left to ubiquitinate for 2 h alone at 37°C in the presence of wild-type Ub or UbK6R (A). After 2 h, the beads were washed to remove reaction components. Ubiquitinated GST-parkin was then incubated in the presence or absence of His-tagged full-length USP8, the USP8 catalytic domain, or the untagged USP2 catalytic domain for 1 h at 37°C. Reactions were immunoblotted for Ub and GST-parkin was stained with Ponceau S. The optical densities of the Ub conjugates relative to total GST-parkin were quantified using NIH ImageJ (B), and the data represent the mean ± SEM for three independent experiments. For statistical analysis, a two-way ANOVA with Tukey post-test was performed, **P < 0.01; NS, not significant.
C, D Expression of HA-UbK6R rescues the delay in mitochondrial recruitment of parkin following USP8 siRNA. U2OS-GFP-parkin cells were co-transfected with USP8 siRNA (5 nM) and either HA-Ubwild-type, HA-UbK6 only, or HA-UbK6R (1 μg) for 60 h (C). Cells were treated with CCCP for the indicated time periods and fixed. Immunofluorescence images of cells were acquired after staining for HA and TOM20. After 1-h treatment with CCCP, cells were analyzed for the recruitment of GFP-parkin onto TOM20-positive mitochondria in HA-positive cells (D). Experiments were blinded and performed in triplicate with 100 cells analyzed for each condition. The vertical bars represent SEM. For statistical analysis, a two-way ANOVA with Tukey post-test was performed, *P < 0.05, **P < 0.01; NS, not significant.
Source data are available online for this figure.
K6-linked ubiquitin conjugation regulates parkin-mediated mitophagy
To determine whether USP8-mediated removal of K6-containing Ub chains from parkin is required for efficient parkin function in mitophagy, we manipulated the ratio of K6-linked Ub in cells. We transfected U2OS-GFP-parkin cells with USP8 siRNA and either wild-type HA-Ub, HA-UbK6 only, or HA-UbK6R and determined the effects on CCCP-induced mitochondrial recruitment of parkin and mitophagy. In USP8 knockdown cells, overexpression of either wild-type HA-Ub or HA-UbK6 only had little effect on the delay in parkin recruitment, with only approximately 40% cells showing co-localization with mitochondria after 1 h of CCCP (Fig 6C and D), similar to our findings with endogenous Ub (Fig 1, Supplementary Fig S1). In contrast, HA-UbK6R overexpression almost completely rescued parkin recruitment to levels seen in cells without USP8 knockdown. Thus, reducing the levels of K6-linked Ub conjugates in cells bypasses the need for USP8 to remove K6-linked Ub from parkin. To examine mitophagy, we again determined the loss of TOM20 staining and the presence of GFP-parkin puncta on mitochondria in USP8 knockdown cells after 24 h of CCCP (Supplementary Fig S7). Compared to wild-type HA-Ub, overexpression of HA-UbK6 only further impaired parkin-mediated mitophagy (Supplementary Fig S7). In contrast, HA-UbK6R overexpression again rescued mitophagy (Supplementary Fig S7). Taken together, these findings indicate that skewing Ub conjugation toward K6 potentiates the effects of USP8 knockdown on parkin function in mitophagy, whereas skewing it away from K6 rescues the effect of USP8 knockdown. Thus, we conclude that USP8-mediated removal of K6-linked Ub from parkin is required for mitophagy to proceed efficiently.
Finally, to test whether enhancing K6-linked Ub conjugation would be sufficient to impair parkin function in mitophagy, we overexpressed the HA-Ub mutants in the absence of USP8 siRNA. Overexpression of HA-UbK6 only impaired parkin recruitment, with only approximately 40% of HA-positive cells showing co-localization with mitochondria after 1 h of CCCP (Fig 7A and B). Remarkably, the degree of impairment was similar to the one seen with USP8 knockdown (Fig 1, Supplementary Fig S1). In contrast, neither wild-type HA-Ub nor HA-UbK6R had comparable effects on parkin recruitment (Fig 7A and B). Furthermore, HA-UbK6 only increased the number of HA-positive cells that had lost TOM20 staining and that contained GFP-parkin puncta after 24 h of CCCP (Fig 7C and D). In contrast, HA-UbK6R had the opposite effect. Thus, skewing Ub conjugation specifically toward K6 impairs parkin function in mitophagy, in effect phenocopying the effects of USP8 knockdown. Taken together, the findings further support a model whereby USP8-mediated removal of K6-linked Ub from parkin is critical for mitophagy to proceed efficiently (Fig 7E).
Figure 7. Increasing K6-linked Ub conjugates impairs parkin-mediated mitophagy.

A, B Expression of HA-UbK6 only delays the mitochondrial recruitment of parkin following treatment with CCCP. U2OS-GFP-parkin cells were transfected with either HA-Ubwild-type, HA-UbK6 only, or HA-UbK6R (2 μg) for 60 h (A). Cells were treated with CCCP for indicated time periods and fixed. Immunofluorescence images of cells were acquired after staining for HA and TOM20. After 1-h treatment with CCCP, cells overexpressing either HA-Ubwild-type, HA-UbK6 only, or HA-UbK6R (2 μg) were analyzed for the recruitment of GFP-parkin onto TOM20-positive mitochondria in HA-positive cells (B). Experiments were blinded and performed in triplicate with 100 cells analyzed for each condition. The vertical bars represent SEM. For statistical analysis, a two-way ANOVA with Tukey post-test was performed, **P < 0.01.
C, D Expression of HA-UbK6 only impairs parkin-mediated mitophagy. U2OS-GFP-parkin cells were transfected with either HA-Ubwild-type, HA-UbK6 only, or HA-UbK6R (2 μg) for 60 h (C). Cells were treated with CCCP for 24 h and fixed. Immunofluorescence images of cells were acquired after staining for HA and TOM20. After 24 h treatment with CCCP, the percentage of HA-positive cells negative for TOM20 staining was quantified (D). Experiments were blinded and performed in triplicate with 100 cells analyzed for each condition. For statistical analysis, a two-way ANOVA with Tukey post-test was performed, **P < 0.01.
E Schematic for how USP8 knockdown delays parkin-mediated mitophagy. At steady state, parkin is auto-inhibited with minimal levels of auto-ubiquitination observed. Ub chains that do form can be removed by USP8 acting upon the K6 linkages within these conjugates. However, when USP8 is absent, Ub conjugates persist on parkin, whose presence appears to delay parkin recruitment onto depolarized mitochondria by impeding the interaction of parkin with either PINK1/phosphorylated Ub or another parkin activating protein. During mitophagy, a robust increase in parkin auto-ubiquitination occurs, followed by removal of Ub conjugates and subsequent clearance of damaged mitochondria. When USP8 is knocked down, these chains are now left intact. Thus, their presence delays parkin-mediated clearance by potentially impeding parkin from interacting with p62, LC3, or another autophagic adaptor protein required for the successful completion of mitophagy.
Discussion
In this study, we identified USP8 as a novel DUB partner for parkin and demonstrated that it is required for parkin-mediated mitophagy to proceed efficiently. As dysfunctional mitochondria are an important source of reactive oxygen species to which neurons are particularly vulnerable, understanding the mechanisms that govern mitochondrial quality control will be essential to developing therapies for neurodegenerative diseases such as PD. Like parkin, USP8 is expressed throughout the brain and in particular, in the substantia nigra (Bruzzone et al, 2008), a region that is affected in PD. Silencing USP8 delays parkin recruitment to mitochondria and impairs the turnover of parkin itself, which accumulates on aggregated clusters of mitochondria that have failed to be degraded by autophagy. Remarkably, these defects are attributable to a lack of USP8-mediated removal of K6-linked Ub conjugates from parkin. This underscores several important implications of our study. First, our findings reveal that along with ubiquitination, deubiquitination is important for mitochondrial quality control. Second, we show that parkin auto-ubiquitination is critical for its function in mitophagy and that auto-ubiquitination occurs predominantly via K6-linked conjugates. Finally, we discover that USP8 preferentially cleaves K6-linked Ub chains on parkin and implicate it for the first time in mitochondrial quality control.
To date, most studies on USP8 have focused on its role in endosomal trafficking in the ESCRT pathway and in regulating the endocytosis of the EGFR and other receptors (Mizuno et al, 2005; Row et al, 2006; Berlin et al, 2010; Ali et al, 2013). Parkin has also been implicated in EGFR internalization and endosomal trafficking via mono-ubiquitination of Eps15, an adaptor protein that is also deubiquitinated by USP8 (Fallon et al, 2006; Mizuno et al, 2006). Thus, in addition to regulating parkin in mitophagy, USP8 may also affect parkin function in this pathway. Considering the prominent role of USP8 in the ESCRT complex, it was possible that impaired mitophagy could be associated with a defect in the ESCRT pathway, caused by knockdown of USP8. However, delayed recruitment was not observed following knockdown of two other core ESCRT proteins, STAM1 and 2, strongly implying that the effect of USP8 on mitophagy was not associated with its function in the ESCRT pathway. Moreover, we found no evidence that USP8 knockdown affected mitochondrial dynamics, mitochondrial depolarization, and PINK1 accumulation, nor did it affect the ubiquitination or turnover of mitochondrial GTPases involved in dynamics, strongly implying that USP8 is regulating parkin activity via deubiquitination.
We reported previously that ataxin-3, a DUB responsible for Machado-Joseph's disease, deubiquitinates parkin and that parkin turnover was increased by overexpressing the polyglutamine repeat-expanded, Machado-Joseph's disease-linked mutant form of ataxin-3 (Durcan & Fon, 2011; Durcan et al, 2011). Moreover, ataxin-3-mediated deubiquitination was functionally coupled to parkin ubiquitination such that ataxin-3 was unable to remove pre-existing Ub conjugates from parkin (Durcan et al, 2012). This explains why ataxin-3 was unable to deubiquitinate parkin in the sequential ubiquitination/deubiquitination assays used in this study. More importantly, ataxin-3 siRNA did not impact parkin recruitment onto mitochondria. Similarly, knockdown of USP2, a DUB we found to efficiently deubiquitinate parkin in vitro, had no effect on parkin recruitment. However, our findings do not exclude a role for other DUBs in the regulation of parkin function. Indeed, two recent studies identified a role for USP15 and the mitochondrial DUB USP30 in parkin-mediated mitophagy (Bingol et al, 2014; Cornelissen et al, 2014). Neither of these was identified in our screen for DUBs that regulate parkin recruitment to depolarized mitochondria nor do they appear to deubiquitinate parkin directly. Instead, overexpression of these DUBs leads to deubiquitination of parkin substrates on mitochondria, which in turn slowed the rate of mitochondrial clearance by autophagy. Conversely, knockdown of these DUBs increased the rate of mitophagy but had no discernible effect on parkin recruitment. Thus, our findings with USP8 and recent findings with USP15 and USP30 establish that along with ubiquitination (parkin), phosphorylation (PINK1 and Ub), and GTPase activity (mitofusins and DRP1) (Twig et al, 2008; Narendra et al, 2010b; Tanaka et al, 2010; Chan et al, 2011; Kane et al, 2014; Koyano et al, 2014), deubiquitination plays a central role in mitophagy. Moreover, our work shows that DUBs can affect mitophagy at multiple levels, controlling both the extent of ubiquitination of parkin substrates and of parkin itself, with opposing effects on the turnover of damaged mitochondria by autophagy.
Using several complementary unbiased approaches including LC-MS/MS, PRM, and AQUA mass spectrometry, we show that parkin directly assembles K6-, K11- K48-, and K63-linked Ub conjugates on itself both in vitro and in cells. These chains appear to be conjugated to K27, K48, and K76 in the parkin Ubl domain, consistent with recent work (Sarraf et al, 2013). In the absence of USP8, these Ub chains accumulate to high levels during mitophagy and persist on parkin at these three sites. Removal of these chains, and especially those linked via K6 in Ub, appears critical for mitophagy to proceed efficiently. Parkin has been reported previously to conjugate Ub linked via a number of different lysines including K48, K63, K27, and K29 as well as linear Ub chains and mono-Ub (Lim et al, 2005; Matsuda et al, 2006; Geisler et al, 2010; Muller-Rischart et al, 2013). Most of these studies relied exclusively on overexpression of Ub mutants or on chain-specific antibodies. Remarkably, using quantitative AQUA mass spectrometry, we find that parkin preferentially assembles K6-linked Ub conjugates on itself, with K11-, K48-, and K63-linked Ub, together accounting for only 40% of all chain linkages. This predilection for assembling K6 chains is likely an intrinsic property of parkin as it occurred regardless of the E2 enzyme used in the assay.
Increasing K6-linked auto-ubiquitination (i.e. by silencing USP8) is likely to stabilize parkin by protecting it from degradation in the proteasome or lysosome (Shang et al, 2005; McLelland et al, 2014), in line with the role of K6 in stabilizing RING1b and BRCA1/BARD (Nishikawa et al, 2004; Ben-Saadon et al, 2006; de Bie et al, 2010), the only other mammalian E3s known to assemble K6-linked Ub chains. These E3s are believed to function in histone modification and DNA repair (Ben-Saadon et al, 2006). Interestingly, a recent study reported that parkin collaborates with Rad6a/Ube2a, an E2 involved in DNA repair, although K6 ubiquitination was not examined (Haddad et al, 2013; Zou et al, 2013). These findings do not preclude a role for the ubiquitination of parkin or parkin substrates via other linkages in distinct cellular pathways such as NF-κB signaling (K63, N-terminal/linear) (Henn et al, 2007; Muller-Rischart et al, 2013), endocytosis and trafficking (mono-Ub) (Fallon et al, 2006; Trempe et al, 2009), autophagy (K27, K48, K63) (Geisler et al, 2010; Narendra et al, 2010a; Okatsu et al, 2010; Chan et al, 2011; van Wijk et al, 2012), or proteasomal degradation (K11, K48) (Matsumoto et al, 2010; Chan et al, 2011). Recent work showing that parkin is auto-inhibited and uses a HECT-like conjugation mechanism now makes it possible to examine the structural basis for the types of Ub chains assembled by parkin and the specific E2(s) involved (Riley et al, 2013; Spratt et al, 2013; Trempe et al, 2013; Wauer & Komander, 2013).
Whether the specificity for removing K6-linked Ub from parkin extends to other USP8 substrates is unknown. However, as USP8 cleaves K6-, K11-, K48-, and K63-linked free di-Ub chains in vitro, our results suggest that the interaction with parkin may help restrict USP8's activity toward K6. Indeed, no USP-family member DUB tested has shown K6-specificity toward free Ub chains (Hospenthal et al, 2013). Moreover, whether parkin also uses K6 for substrate ubiquitination or only for auto-ubiquitination is not known. However, as USP8 does not affect the ubiquitination or stability of the parkin substrates we tested during mitophagy, we suspect that these are not linked via K6. This point further highlights the idea that parkin auto-ubiquitination may serve a distinct role from substrate ubiquitination.
Beyond the role of parkin, our work manipulating the ratios of different Ub linkages in cells suggests, for the first time, a role for K6 ubiquitination per se in mitochondrial quality control. While we are not certain of the mechanisms, it must be kept in mind that parkin does not make K6-linked chains exclusively, as seen in our AQUA analysis. It is therefore plausible that other DUBs could eventually cleave mixed chains at other sites (i.e. K11, K48, and K63) or even at K6, albeit perhaps with lower efficiency. Such a process might explain the observed delay in recruitment and mitophagy, with an increased presence of these conjugates impeding parkin from interacting with substrates, PINK1, and/or phosphorylated Ub. The latter two have been shown to be essential for parkin activation and translocation onto the mitochondria (Kondapalli et al, 2012; Kane et al, 2014). As parkin auto-ubiquitination appears to occur predominantly on the Ubl domain (K48, K76), it is also possible that these conjugates interfere with binding to Ubl-interacting proteins (i.e. endophilin, Eps15, ataxin-3) (Fallon et al, 2006; Durcan & Fon, 2011; Trempe et al, 2013) or PINK1-mediated phosphorylation of the Ubl at S65 (Kondapalli et al, 2012). Moreover, the presence of increased levels of Ub chains linked primarily via K6 appears to impair parkin-mediated mitophagy, perhaps by interfering with ongoing parkin E3 Ub-ligase activity or because of a reduced capacity of these chains to recruit p62, LC3, or other autophagy proteins. Regardless of the precise mechanism, our work uncovers a novel layer of regulation, mediated by USP8 and K6-linked auto-ubiquitination, critical for parkin-dependent mitophagy.
Materials and Methods
Chemicals, antibodies, and plasmids
Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (Sigma) was used at a final concentration of 20 μM. Tetramethylrhodamine, methyl ester (TMRM) (Invitrogen) was used at a final concentration of 600 nM. Antibodies in Western blots were used to detect: parkin (1:20K), USP8 (1:2K), TOM20 (1:20K), STAM1 (1:5K), USP30 (1:2K), mitofusin 1 and 2 (1:2K) (Santa Cruz); Ub (1:10K) (Covance); COX1 (1:5K), Complex 1 (NDUFS3) (1:10K), DRP1 (1:5K), VDAC1 (1:10K) (Abcam); Tim23 (1:10K), Opa1 (1:5K) (BD Biosciences); PINK1 (1:2K) (Novus Biologicals); anti-HA (1:20K) (Roche); FLAG (1:20K) (Sigma); STAM2 (1:5K), ataxin-3 (1:20K), anti-His, actin (Millipore); and USP2 (kind gift from Dr. Simon Wing, McGill). For immunofluorescence microscopy, TOM20 (Santa Cruz); PDH (E2), COX1 (Abcam), and anti-HA (Roche) antibodies were used at a 1:1K dilution. GST-parkin, FLAG-parkin, and HA-Ub plasmids were described previously (Durcan et al, 2011). FLAG-HA-USP8 (#22608) and His-USP8Catalytic domain (#28115) were purchased from Addgene. Di-ubiquitin conjugates, UbcH7, UbcH5b, His-USP8 (full-length), and all commercial DUBs were purchased from Boston Biochem, with the exception of USP15 and USP18 (Enzo Life Sciences). GST-CHIP was a kind gift from Dr. L. Beitel (Lady Davis Institute). GST-HHARI (RBR domain only) was kind gift of Dr. R. Klevit, (U. Washington). GST-cIAP1 was kind gift of Dr. P. Barker (McGill).
siRNA oligos and libraries
A human ON-TARGETplus Smartpool siRNA library was used to knockdown 87 putative DUBs (G-104705-01, Thermo Scientific). The AllStars negative control siRNA (1027280, Qiagen) was used for non-targeting siRNA. The siRNAs used were USP8.1: SI00073017, USP8.2: SI00073024, USP8.3: SI00073031, USP8.4: SI03103604. SI00073024, ataxin-3: SI00071113, USP30: SI03122714, and USP2: SI03246698 (Qiagen). USP8.2 was the USP8 siRNA used in assays to knockdown USP8. The siRNA oligos used to knockdown STAM 1 and 2 were as follows:
STAM1 siRNA: 5′-CAGCAAUGAUUAAGAACCUUA-3′
STAM2 siRNA: 5′-CUGCUCAAACUUCAUAUUUUA-3′
Cell culture
U2OS-GFP-parkin cells were a kind gift of Dr. R. Screaton, U. Ottawa. This cell line was created by stably infecting cells with a GFP-parkin viral vector, generating a pool of stable cells expressing similar levels of GFP-parkin. All U2OS, HEK293T, HeLa, and SH-SY5Y cell lines were maintained at 37°C and 5% CO2 in DMEM with 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Wisent Life Sciences).
siRNA and plasmid transfection
For siRNA transfections, cells were passed into wells/plates containing the siRNA (final concentration of 10 nM), HiPerFect (Qiagen), and OptiMEM (Life Technologies) and incubated for 24 h. Fresh media was added the next day. At 60 h post-transfection, cells were either lysed, fixed on coverslips, or were used in live-cell microscopy experiments.
For co-transfection of siRNA with a plasmid, reactions containing the siRNA (final concentration of 5 nM), plasmid (1 μg), JetPrime (PolyPlus Transfections), and transfection buffer were added to cells in dishes plated 24 h prior to transfection and incubated overnight. For transfection of GFP or FLAG-parkin with other plasmids, 0.5 μg of the parkin construct was incubated with other plasmids for a final DNA amount of 2 μg in reactions. Reactions containing DNA, JetPrime, and transfection buffer were added to cells in dishes plated 24 h prior to transfection and incubated overnight.
Control and USP8 microRNA constructs:
MicroRNA targeting sequences were designed using the Life Technologies algorithm Block-iT Designer.
-
USP-8 (sense – 2911)
5′-TGCTGAGTACATGAAGGCCTCGAAGGGTTTTGGCCACTGACTGACCCTTCGAGCTTCATGTACT-3′
-
USP-8 (anti-sense – 2911)
5′-CCTGAGTACATGAAGCTCGAAGGGTCAGTCAGTGGCCAAAACCCTTCGAGGCCTTCATGTACTC-3′
Oligonucleotides encoding the shRNA were first cloned into a pcDNA6.2/GW-EmRFP.miR vector (Life Technologies). The USP-8- shRNAmiR expression cassette was next PCR-amplified using the following primers:
-
miRGFP BglII sense
5′-GCGCAGATCTACCGGTCGCCACCATGGTGAGCAAGGGCGAGGAGC-3′
-
miRXFP XhoI antisense
5′-GCGCCTCGAGTGCGGCCGGATCTGGGCCATTTGTTCCATGTGAGTGC-3′
The fragments generated were digested with BglII and XhoI and cloned into the pRRLsinPPTeGFP vector (Gift of Dr. Brigitte Ritter and Dr. Peter McPherson). The control construct was generated in an identical manner containing the scrambled sequence outlined below:
-
Control (sense)
5′-TGCTGACGTGACACGTTCGGAGAATTGTTTTGGCCACTGACTGACAATTCTCCACGTGTCACGT-3′
-
Control (anti-sense)
5′-CCTGACGTGACACGTGGAGAATTGTCAGTCAGTGGCCAAAACAATTCTCCGAACGTGTCACGTC-3′
MiRNA viruses were produced in HEK293T cells using the Miami system (Addgene), a lentiviral-based procedure requiring either the control or USP8 miRNA pRRLsinPPTeGFP vectors described above and a packaging mix composed of pRSV-Rev, pMD2.g, and pMDLg/pRRE.
Viral-mediated infection and lysis of cortical neurons
Mouse cortical neurons were dissected from day 15 C57 Black mouse embryos (Jackson Labs) and plated onto poly-P-lysine (Sigma) coated plates. Cells were maintained in Neurobasal medium supplemented with penicillin/streptomycin, B27, N2, and L-glutamate (Life Technologies). After 3 days, neurons were infected with viruses at a MOI of 10 or 15 for 3 h. After 3 h, virus-containing media was replaced with culture media supplemented with fresh Neurobasal medium. Neuronal cultures were harvested 6 days post-viral transduction in ice-cold lysis buffer (20 mM Hepes, pH 7.4, 1 mM EDTA, 1% NP-40, and protease inhibitors).
Protein expression, binding and in vitro assays
GST and His fusion proteins were expressed in E. coli strain BL21 (DE3) at 23°C for 3 h and affinity-purified using Glutathione Sepharose 4B beads (GE Life Sciences), or Ni-NTA Agarose (Qiagen), respectively.
Binding assays with Agarose-TUBEs (Life Sensors), FLAG-parkin immunoprecipitations, and in vitro ubiquitination/deubiquitination assays were performed as described previously (Durcan et al, 2011, 2012). For GFP-parkin immunoprecipitations, cells were lysed in ice-cold RIPA buffer, with 10 μl of GFP-nAb Agarose beads (Allele Biotech) incubated with 400 μg of protein lysate for 2 h at 4°C. GFP-parkin bound on beads was now washed with 50 mM ammonium bicarbonate, pH 8 (AMBIC) solution prior to mass spectrometry analysis. The E2 enzyme used in our ubiquitination assays was UbcH7, except where indicated as UbcH5b or Ubc7.
Mass spectrometry analysis
For AQUA analysis of ubiquitinated GST-parkin, samples were washed twice for 5 min at 4°C with AMBIC solution. Following these washes, GST-parkin was eluted from the beads by eluting in 0.1% Rapigest (Waters) dissolved in AMBIC solution for 30 min at 50°C. AQUA analysis of Ub chain linkage was performed as described previously (Kirkpatrick et al, 2006).
For LC-MS/MS and PRM analysis, samples were diluted to 2 M urea/60 mM AMBIC and on-bead trypsin digestion was performed overnight at 37°C. The supernatant was reduced with 7.5 mM DTT at 37°C and, after cooling for 10 min, alkylated with 15 mM chloroacetamide at room temperature for 20 min in the dark. Chloroacetamide was used in order to avoid artifacts isobaric to di-glycine on lysine residues associated with iodoacetamide (Nielsen et al, 2008). Samples were dried under vacuum, reconstituted in 1% acetonitrile (ACN)/1% formic acid (FA), and acidified with trifluoroacetic acid for desalting and removal of residual NP-40 by MCX (Waters Oasis MCX 96-well Elution Plate). After elution in 10% ammonium hydroxide/90% methanol, samples were once again dehydrated and reconstituted in 1%ACN/1%FA.
A Thermo EASY-nLC system was coupled to a Q-Exactive mass spectrometer (Thermo Fisher Scientific) equipped with a Proxeon nanoelectrospray ionization source (Thermo Fisher Scientific). The LC column was a PicoFrit fused silica capillary column (15 cm × 75 μm i.d; New Objective, Woburn, MA), self-packed with C-18 reverse-phase material (Jupiter 5 μm particles, 300 Å pore size; Phenomenex, Torrance, CA) using a high pressure packing cell. For LC-MS/MS identification of ubiquitination sites on GFP-parkin, peptides were loaded on-column and eluted with a three-slope gradient at a flow rate of 600 nl/min. Solvent B (0.1% FA in ACN) first increased from 1 to 5% in 6 min, then from 5 to 35% in 50 min, and finally from 35 to 80% B in 9 min. Solvent A was 0.1% FA in water. MS data were acquired using a data-dependent top16 method with a dynamic exclusion window of 3 s. The scan range was from 300 to 1,800 m/z with an isolation window of 2.0 Th and resolution of 70,000 at m/z 200. The automatic gain control (AGC) target was set at 1e6, the maximum ion fill time (IT) at 100 ms, the intensity threshold at 1.4e4, and the underfill ratio at 0.7%. For MS2, the resolution was defined at 17,500, the AGC target at 1e5, and maximum IT at 50 ms. The normalized collision energy used was 27 eV. Nanospray and S-lens voltages were set to 1.2–1.8 kV and 50 V, respectively. Capillary temperature was 250°C. Protein database searches were performed using Mascot 2.3 (Matrix Science). The mass tolerance for precursor ions was set to 10 ppm and for fragment ions to 0.52 Da. The enzyme specified was trypsin; four missed cleavages were allowed. Cysteine carbamidomethylation was specified as a fixed modification, and methionine oxidation, serine and threonine phosphorylation, and lysine LeuArg-Gly-Gly and Gly-Gly modifications as variable modifications.
For Parallel Reaction Monitoring (PRM), peptides were also loaded on-column and separated using a three-slope gradient at a 600 nl/min flow rate. Solvent B (0.1% FA in ACN) first increased from 0 to 3% in 4 min, then from 3 to 30% in 20 min, and finally from 30 to 80% B in 4 min. Solvent A was 0.1% FA in water. All parkin peptides (K27, K48 and K76) observed in the LC-MS/MS analysis of GFP-parkin, as well as a control unmodified peptide for parkin, were selected for time-scheduled parallel reaction monitoring (targeted MS2 mode). Precursor ion m/z, normalized collision energies, and retention time windows are listed in Supplementary Table S2. The maximum IT was set to 100 ms and the AGC at 2e5. Nanospray and S-lens voltages and capillary temperature were as described above. Raw data were analyzed using Skyline (64 bit) 2.5.0.5675 (MacLean et al, 2010). Peaks were manually verified to be free of interference, for the overlap of fragment ion traces for a given precursor, and for peak shape. Each peptide was quantified by integrating the area under the curve for the four most intense fragment ions.
Fixed-cell immunofluorescence
Cells grown on coverslips were fixed with 4% formaldehyde-PBS (prepared from 16% formaldehyde stock, Thermo Fisher Scientific) for 15 min at 37°C. After fixation, cells were washed four times with 1× PBS, permeabilized with 1× PBS containing 0.25% Triton X-100 for 10 min, and blocked with 3% BSA + PBST (1× PBS + 0.1% Tween-20) for 1 h at RT. Cells were labeled with primary antibodies diluted in PBST with 3% BSA overnight at 4°C and then with Alexa Fluor (Life Technologies) secondary antibodies (1:1,000 dilution) in PBST with 3% BSA for 3 h at RT. Cells were washed three times in PBS, and coverslips were mounted with fluorescent mounting medium (Dako). Confocal images were acquired on an LSM 510 Meta confocal microscope (Zeiss) using a 25×, 0.8NA or 63×, 1.4 NA objective. Excitation wavelengths of 488 nm, 543 nm, and 633 nm were used.
Time-lapse fluorescence microscopy
U2OS-GFP-parkin cells were seeded onto 35-mm glass-bottom microwell dishes (MatTek Corporation) and transfected with non-targeting or USP8 siRNA using HiPerFect. After 48 h, cells were transferred to a heated stage maintained at 37°C and at 5% CO2 using a Zeiss temperature controller and cell perfusion system (Zeiss). To visualize mitochondria, cells were transduced with CellLight® mitochondria-RFP (Life Technologies) 16 h prior to imaging. Microscopy was performed on a Zeiss AxioObserver.Z1 inverted fluorescent microscope. Fully automated multidimensional acquisition was controlled using the Zen 2012 software (Zeiss). Images were acquired using a 20× objective (Plan-Apochromat) with a side-mounted AxiocamMRm camera. GFP or mito-RFP was excited using the Zeiss XBO75 Xenon illumination system and detected using the appropriate filters (62HE ExGFP in combination with a Hyper GFP reflector or CY3, respectively). Fixed exposure times were as follows: GFP 70 ms and RFP 180 ms. Fluorescent images were acquired at 5-min intervals for a total of 145 min and compiled into movie files using the Zen 2012 software.
For the live-cell analysis of GFP-parkin recruitment onto mitochondria, 350 cells were examined over three separate experiments to ascertain the time required for GFP-parkin to be recruited onto mito-RFP-positive mitochondria. Parkin recruitment upon membrane depolarization is visualized by the appearance of punctate GFP fluorescence superposed onto mitochondrial RFP fluorescence. Quantification of GFP-parkin recruitment to mitochondria is facilitated by calculating the percentage of cells showing recruitment of GFP-parkin on mitochondria at 5-min intervals over a period of 145 min. For measuring relative parkin levels in an individual cell, in the first recorded frame, the pixel intensity of GFP-parkin fluorescence in a region of interest (ROI) of defined size was recorded. Following the quantification of relative parkin pixel intensity, the cell was now followed over time following CCCP treatment until the initial appearance of GFP-parkin puncta that are present on the mitochondria in the cell of interest. For this analysis, 20 cells transfected with non-targeting siRNA and 15 cells transfected with USP8 siRNA were tested. Relative parkin pixel intensity was plotted against the initial time of parkin recruitment on mitochondria.
To visualize mitochondria membrane potential, cells were labeled with the membrane potential-dependent cationic fluorophore probe Tetramethylrhodamine, methyl ester (TMRM) (600 nM) (Life Technologies) for 20 min at 37°C. The labeling medium was removed and replaced with 150 nM TMRM to maintain the equilibrium distribution of the fluorophore. Cells were imaged as described above. Fixed exposure times were as follows: GFP 70 ms and RFP 20 ms. Fluorescent images were acquired at 1-min intervals for a total of 5 min and compiled using the Zen 2012 software.
Statistical analysis
For analysis of immunofluorescence coverslips, slides were blinded for three independent experiments. 100 cells were counted per experiment. Cells were analyzed for the number of cells with TOM20 staining (CCCP 24 h), the number of cells with GFP-parkin co-localization on TOM20-positive mitochondria (CCCP 1–2 h), or for the number of cells containing GFP-parkin puncta co-localizing with TOM20-positive mitochondria (CCCP 24 h). All statistical analysis was run on data obtained from three independent experiments, with the exception of the AQUA mass spectrometry that was analyzed from two independent experiments. A one-way ANOVA followed by a post hoc Tukey test was performed on AQUA quantification to determine significance. A two-way ANOVA followed by a post hoc Tukey test was performed on densitometry and immunofluorescence quantification to determine significance. In immunofluorescence experiments comparing non-targeting and USP8 siRNA, Student's t-tests were performed. Findings were considered significant as follows: *P < 0.05, **P < 0.01, ***P < 0.001, or ****P < 0.0001.
Acknowledgments
We thank Dr. Jean-Francois Trempe, Karl Grenier, Dr. Kalle Gehring, and Dr. Heidi McBride for critical reading of the manuscript. We would like to thank Josée Champagne, Sylvain Tessier, and Marguerite Boulos for their assistance with LC-MS/MS and PRM analyses. We thank Steve Kaiser and Ron Kopito for their assistance with the AQUA analysis. Funding for this project was provided by the Parkinson's Society of Canada and a CIHR Operating grant (grant # MOP-62714).
Author contributions
TMD and EAF conceived and planned the experiments, and interpreted the data. MYT acquired and analyzed all the live-cell analysis. JRP, DF, and BC performed and interpreted all the LC-MS/MS and PRM mass spectrometry analysis. MAA and TAS performed and interpreted all the AQUA mass spectrometry analysis. EAD generated USP8 miRNA viruses and cultured mouse cortical neurons, in addition to performing the USP8 knockdown experiments in neurons. PG assisted TMD in performing and interpreting data from Figs 6 and 7. TMD and EAF wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Supplementary information for this article is available online: http://emboj.embopress.org
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Ali N, Zhang L, Taylor S, Mironov A, Urbe S, Woodman P. Recruitment of UBPY and ESCRT exchange drive HD-PTP-dependent sorting of EGFR to the MVB. Curr Biol. 2013;23:453–461. doi: 10.1016/j.cub.2013.02.033. [DOI] [PubMed] [Google Scholar]
- Ben-Saadon R, Zaaroor D, Ziv T, Ciechanover A. The polycomb protein Ring1B generates self atypical mixed ubiquitin chains required for its in vitro histone H2A ligase activity. Mol Cell. 2006;24:701–711. doi: 10.1016/j.molcel.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Berlin I, Schwartz H, Nash PD. Regulation of epidermal growth factor receptor ubiquitination and trafficking by the USP8.STAM complex. J Biol Chem. 2010;285:34909–34921. doi: 10.1074/jbc.M109.016287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bie P, Zaaroor-Regev D, Ciechanover A. Regulation of the Polycomb protein RING1B ubiquitination by USP7. Biochem Biophys Res Commun. 2010;400:389–395. doi: 10.1016/j.bbrc.2010.08.082. [DOI] [PubMed] [Google Scholar]
- de Bie P, Ciechanover A. Ubiquitination of E3 ligases: self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death Differ. 2011;18:1393–1402. doi: 10.1038/cdd.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- Bruzzone F, Vallarino M, Berruti G, Angelini C. Expression of the deubiquitinating enzyme mUBPy in the mouse brain. Brain Res. 2008;1195:56–66. doi: 10.1016/j.brainres.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, De Strooper B, Verstreken P, Vandenberghe W. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet. 2014;23:5227–5242. doi: 10.1093/hmg/ddu244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daviet L, Colland F. Targeting ubiquitin specific proteases for drug discovery. Biochimie. 2008;90:270–283. doi: 10.1016/j.biochi.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Duda DM, Olszewski JL, Schuermann JP, Kurinov I, Miller DJ, Nourse A, Alpi AF, Schulman BA. Structure of HHARI, a RING-IBR-RING ubiquitin ligase: autoinhibition of an ariadne-family E3 and insights into ligation mechanism. Structure. 2013;21:1030–1041. doi: 10.1016/j.str.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Fon EA. Mutant ataxin-3 promotes the autophagic degradation of parkin. Autophagy. 2011;7:233–234. doi: 10.4161/auto.7.2.14224. [DOI] [PubMed] [Google Scholar]
- Durcan TM, Kontogiannea M, Thorarinsdottir T, Fallon L, Williams AJ, Djarmati A, Fantaneanu T, Paulson HL, Fon EA. The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum Mol Genet. 2011;20:141–154. doi: 10.1093/hmg/ddq471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Kontogiannea M, Bedard N, Wing SS, Fon EA. Ataxin-3 deubiquitination is coupled to parkin ubiquitination via E2 ubiquitin-conjugating enzyme. J Biol Chem. 2012;287:531–541. doi: 10.1074/jbc.M111.288449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon L, Belanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, Voortman J, Haber M, Rouleau G, Thorarinsdottir T, Brice A, van Bergen En Henegouwen PM, Fon EA. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol. 2006;8:834–842. doi: 10.1038/ncb1441. [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad DM, Vilain S, Vos M, Esposito G, Matta S, Kalscheuer VM, Craessaerts K, Leyssen M, Nascimento RM, Vianna-Morgante AM, De Strooper B, Van Esch H, Morais VA, Verstreken P. Mutations in the intellectual disability gene Ube2a cause neuronal dysfunction and impair parkin-dependent mitophagy. Mol Cell. 2013;50:831–843. doi: 10.1016/j.molcel.2013.04.012. [DOI] [PubMed] [Google Scholar]
- Henn IH, Bouman L, Schlehe JS, Schlierf A, Schramm JE, Wegener E, Nakaso K, Culmsee C, Berninger B, Krappmann D, Tatzelt J, Winklhofer KF. Parkin mediates neuroprotection through activation of IkappaB kinase/nuclear factor-kappaB signaling. J Neurosci. 2007;27:1868–1878. doi: 10.1523/JNEUROSCI.5537-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospenthal MK, Freund SM, Komander D. Assembly, analysis and architecture of atypical ubiquitin chains. Nat Struct Mol Biol. 2013;20:555–565. doi: 10.1038/nsmb.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460:127–139. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick DS, Hathaway NA, Hanna J, Elsasser S, Rush J, Finley D, King RW, Gygi SP. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol. 2006;8:700–710. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009a;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- Komander D, Reyes-Turcu F, Licchesi JD, Odenwaelder P, Wilkinson KD, Barford D. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 2009b;10:466–473. doi: 10.1038/embor.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, Knebel A, Alessi DR, Muqit MM. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- Kulathu Y, Komander D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–523. doi: 10.1038/nrm3394. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol. 2013;200:163–172. doi: 10.1083/jcb.201210111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci. 2005;25:2002–2009. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfany G, Denuc A. To ubiquitinate or to deubiquitinate: it all depends on the partners. Biochem Soc Trans. 2008;36:833–838. doi: 10.1042/BST0360833. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Kitami T, Suzuki T, Mizuno Y, Hattori N, Tanaka K. Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J Biol Chem. 2006;281:3204–3209. doi: 10.1074/jbc.M510393200. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto ML, Wickliffe KE, Dong KC, Yu C, Bosanac I, Bustos D, Phu L, Kirkpatrick DS, Hymowitz SG, Rape M, Kelley RF, Dixit VM. K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell. 2010;39:477–484. doi: 10.1016/j.molcel.2010.07.001. [DOI] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33:282–295. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y, Hahn AA, Hu S, Yang X. The USP19 deubiquitinase regulates the stability of c-IAP1 and c-IAP2. J Biol Chem. 2011;286:35380–35387. doi: 10.1074/jbc.M111.282020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen TE, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, Ekkebus R, Kulathu Y, Wauer T, El Oualid F, Freund SM, Ovaa H, Komander D. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. 2013;154:169–184. doi: 10.1016/j.cell.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno E, Iura T, Mukai A, Yoshimori T, Kitamura N, Komada M. Regulation of epidermal growth factor receptor down-regulation by UBPY-mediated deubiquitination at endosomes. Mol Biol Cell. 2005;16:5163–5174. doi: 10.1091/mbc.E05-06-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno E, Kobayashi K, Yamamoto A, Kitamura N, Komada M. A deubiquitinating enzyme UBPY regulates the level of protein ubiquitination on endosomes. Traffic. 2006;7:1017–1031. doi: 10.1111/j.1600-0854.2006.00452.x. [DOI] [PubMed] [Google Scholar]
- Muller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, Peis R, Deinlein A, Schweimer C, Kuhn PH, Lichtenthaler SF, Motori E, Hrelia S, Wurst W, Trumbach D, Langer T, Krappmann D, Dittmar G, Tatzelt J, Winklhofer KF. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell. 2013;49:908–921. doi: 10.1016/j.molcel.2013.01.036. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010a;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010b;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan JA, Sengupta S, Wood SA, Admon A, Markson G, Sanderson C, Lehner PJ. The ubiquitin E3 ligase MARCH7 is differentially regulated by the deubiquitylating enzymes USP7 and USP9X. Traffic. 2008;9:1130–1145. doi: 10.1111/j.1600-0854.2008.00747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen ML, Vermeulen M, Bonaldi T, Cox J, Moroder L, Mann M. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat Methods. 2008;5:459–460. doi: 10.1038/nmeth0608-459. [DOI] [PubMed] [Google Scholar]
- Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Nishikawa H, Ooka S, Sato K, Arima K, Okamoto J, Klevit RE, Fukuda M, Ohta T. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J Biol Chem. 2004;279:3916–3924. doi: 10.1074/jbc.M308540200. [DOI] [PubMed] [Google Scholar]
- Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, Tanaka K, Matsuda N. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakovic A, Shurkewitsch K, Seibler P, Grunewald A, Zanon A, Hagenah J, Krainc D, Klein C. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem. 2013;288:2223–2237. doi: 10.1074/jbc.M112.391680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BE, Lougheed JC, Callaway K, Velasquez M, Brecht E, Nguyen L, Shaler T, Walker D, Yang Y, Regnstrom K, Diep L, Zhang Z, Chiou S, Bova M, Artis DR, Yao N, Baker J, Yednock T, Johnston JA. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun. 2013;4:1982. doi: 10.1038/ncomms2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Row PE, Prior IA, McCullough J, Clague MJ, Urbe S. The ubiquitin isopeptidase UBPY regulates endosomal ubiquitin dynamics and is essential for receptor down-regulation. J Biol Chem. 2006;281:12618–12624. doi: 10.1074/jbc.M512615200. [DOI] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang F, Deng G, Liu Q, Guo W, Haas AL, Crosas B, Finley D, Taylor A. Lys6-modified ubiquitin inhibits ubiquitin-dependent protein degradation. J Biol Chem. 2005;280:20365–20374. doi: 10.1074/jbc.M414356200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Spratt DE, Julio Martinez-Torres R, Noh YJ, Mercier P, Manczyk N, Barber KR, Aguirre JD, Burchell L, Purkiss A, Walden H, Shaw GS. A molecular explanation for the recessive nature of parkin-linked Parkinson's disease. Nat Commun. 2013;4:1983. doi: 10.1038/ncomms2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe JF, Chen CX, Grenier K, Camacho EM, Kozlov G, McPherson PS, Gehring K, Fon EA. SH3 domains from a subset of BAR proteins define a Ubl-binding domain and implicate parkin in synaptic ubiquitination. Mol Cell. 2009;36:1034–1047. doi: 10.1016/j.molcel.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, Nagar B, Fon EA, Gehring K. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013;340:1451–1455. doi: 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventii KH, Wilkinson KD. Protein partners of deubiquitinating enzymes. Biochem J. 2008;414:161–175. doi: 10.1042/BJ20080798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- Wauer T, Komander D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013;32:2099–2112. doi: 10.1038/emboj.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk SJ, Fiskin E, Putyrski M, Pampaloni F, Hou J, Wild P, Kensche T, Grecco HE, Bastiaens P, Dikic I. Fluorescence-based sensors to monitor localization and functions of linear and K63-linked ubiquitin chains in cells. Mol Cell. 2012;47:797–809. doi: 10.1016/j.molcel.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Yen L, Irwin L, Sweeney C, Carraway KL., III Stabilization of the E3 ubiquitin ligase Nrdp1 by the deubiquitinating enzyme USP8. Mol Cell Biol. 2004;24:7748–7757. doi: 10.1128/MCB.24.17.7748-7757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Rezvani K, Wang H, Lee KS, Zhang D. BRCA1 downregulates the kinase activity of Polo-like kinase 1 in response to replication stress. Cell Cycle. 2013;12:2255–2265. doi: 10.4161/cc.25349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.