

Abstract
Signal peptide peptidase (SPP) catalyzes intramembrane proteolysis of signal peptides at the endoplasmic reticulum (ER), but has also been suggested to play a role in ER-associated degradation (ERAD). Here, we show that SPP forms a complex with the ERAD factor Derlin1 and the E3 ubiquitin ligase TRC8 to cleave the unfolded protein response (UPR) regulator XBP1u. Cleavage occurs within a so far unrecognized type II transmembrane domain, which renders XBP1u as an SPP substrate through specific sequence features. Additionally, Derlin1 acts in the complex as a substrate receptor by recognizing the luminal tail of XBP1u. Remarkably, this interaction of Derlin1 with XBP1u obviates the need for ectodomain shedding prior to SPP cleavage, commonly required for intramembrane cuts. Furthermore, we show that XBP1u inhibits the UPR transcription factor XBP1s by targeting it toward proteasomal degradation. Thus, we identify an ERAD complex that controls the abundance of XBP1u and thereby tunes signaling through the UPR.
Keywords: E3 ubiquitin-protein ligase, GxGD intramembrane protease, protein homeostasis, rhomboid pseudoprotease
Introduction
Intramembrane proteases cleave peptide bonds within cellular membranes and thereby control important processes ranging from transcription regulation to growth factor secretion (Lemberg, 2011). The largest and most diverse group of these unusual enzymes is formed by the GxGD aspartyl proteases including presenilin/γ-secretase as well as signal peptide peptidase (SPP) (Wolfe, 2009; Lichtenthaler et al, 2011). SPP localizes to the endoplasmic reticulum (ER) where it cleaves signal peptides that have been removed from precursors of secretory and membrane proteins (Weihofen et al, 2002). Like for most characterized intramembrane proteases, this release is part of a two-step mechanism: First signal peptidase cleaves off the substrate proteins' ectodomains, which enables the subsequent SPP-catalyzed intramembrane cut (Lemberg & Martoglio, 2002). So far, known functions of SPP include generation of signal peptide-derived bioactive peptides in immune surveillance and proteolytic maturation of hepatitis C virus core protein (Lemberg et al, 2001; McLauchlan et al, 2002). Moreover, recent studies suggest that SPP is part of ER-associated degradation (ERAD) (Loureiro et al, 2006; Stagg et al, 2009; Lee et al, 2010).
ERAD serves as an important cellular safeguard that recognizes incorrectly folded and unassembled proteins and directs them from the ER to the proteasome (Bagola et al, 2011). Moreover, the ERAD machinery also regulates the abundance of several important membrane proteins such as cholesterol synthesizing enzymes (Hampton & Garza, 2009). The ERAD network consists of a repertoire of substrate receptors, E3 ubiquitin ligases and one or several dislocon channels of unknown composition (Bagola et al, 2011). After being recruited to this dislocation machinery, ubiquitinated ERAD substrates are extracted from the ER by the AAA+-type ATPase p97 and delivered to cytosolic proteasomes (Ye et al, 2005). This energy-dependent dislocation reaction is modulated by cytosolic factors including deubiquitinases such as YOD1 (Ernst et al, 2009). A factor that links substrate recognition, ubiquitination, and extraction of ERAD substrates is Derlin1. Due to its role in dislocation, Derlin1 had been suggested to form a protein-conducting channel (Lilley & Ploegh, 2004; Ye et al, 2004). However, a recent study demonstrated that Derlin1 is a catalytically inactive homologue of rhomboid intramembrane proteases (Greenblatt et al, 2011), suggesting that it may serve as a receptor for ERAD substrates (Lemberg, 2013). The organization of the ERAD pathway is complex as vividly illustrated by the immune escape mechanisms employed by human cytomegalovirus (HCMV). Upon host cell infection, HCMV effects degradation of major histocompatibility complex (MHC) class I heavy chains by both a Derlin1-dependent and an SPP-dependent ERAD pathway, thereby avoiding detection by the immune system (Lilley & Ploegh, 2004; Ye et al, 2004; Loureiro et al, 2006). The emerging picture is that the ERAD machinery forms multiple parallel and partially redundant branches (Christianson et al, 2011).
It has been shown that SPP interacts with several quality control factors including the rhomboid pseudoprotease UBAC2 and the E3 ubiquitin ligase TRC8 (Stagg et al, 2009; Lee et al, 2010; Christianson et al, 2011), but the precise role of SPP in protein degradation is unclear. A proteolytic role for SPP in the turnover of the above-mentioned MHC class I heavy chains, which are type I membrane proteins, however, would be unexpected since it is commonly assumed that SPP only cleaves type II transmembrane (TM) segments (Weihofen et al, 2002). Additionally, no cleavage fragments have been observed for degradation of class I heavy chains, giving rise to the notion that SPP has a non-proteolytic function in assembling ERAD factors to functional units (Loureiro et al, 2006). By contrast, we recently showed a direct role for intramembrane proteolysis in the turnover of ERAD substrates. We found that the ER-resident ubiquitin-dependent rhomboid protease RHBDL4 cleaves type I and polytopic membrane proteins with unstable TM segments in order to trigger their degradation (Fleig et al, 2012). Thus, the extent to which the catalytic activity of intramembrane proteases is employed for ERAD is currently an open issue.
In response to ER stress, cells activate signal transduction mechanisms that are collectively called the unfolded protein response (UPR). In higher eukaryotes, three parallel UPR branches exist, namely proteolytic activation of the transcription factor ATF6, PERK-mediated translational control, and IRE1/XBP1 splicing. Taken together, these UPR factors transcriptionally upregulate the ER protein homeostasis network including chaperones and ERAD factors (Walter & Ron, 2011). IRE1 is an ER-resident RNase that triggers non-conventional splicing of the XBP1 pre-mRNA. This generated open reading frame encodes for the mature transcription factor XBP1s which comprises, besides a transcription activation domain, a basic leucine zipper (bZIP) dimerization domain (Yoshida et al, 2001). In contrast, the unspliced variant of XBP1 mRNA encodes XBP1u, which consists of a hydrophobic stretch in the C-terminal portion and lacks the transcription activation domain. When emerging from the translating ribosome, the C-terminal hydrophobic stretch targets its own mRNA-ribosome-nascent chain complex to the ER in order to facilitate IRE1-mediated XBP1 splicing (Yanagitani et al, 2009, 2011). Moreover, XBP1u negatively regulates the UPR by targeting XBP1s and activated ATF6 for degradation (Tirosh et al, 2006; Yoshida et al, 2006, 2009). However, the fate of XBP1u at the ER membrane and the mechanism of how it post-translationally influences the activity of XBP1s and ATF6 have not been resolved yet.
Here, we show that XBP1u is degraded by a 500-kDa ERAD complex consisting of SPP, Derlin1, and TRC8. This complex promotes intramembrane cleavage within an evolutionarily conserved, so far unrecognized, type II TM domain of XBP1u. We also observed that XBP1u, when not cleaved by SPP, targets the activated UPR transcription factor XBP1s toward proteasomal degradation. These findings have important implications for the mechanism underlying the modulation and inactivation of UPR transcription factors.
Results
Identification of an SPP-Derlin1-TRC8 complex
It has been reported previously that SPP forms distinct higher molecular weight complexes (Schrul et al, 2010), but their molecular composition is not known. We therefore decided to test whether known ERAD factors contribute to the formation of these assemblies. We generated a stable cell line expressing HA-tagged SPP under the control of a doxycycline-inducible promoter and performed immunoprecipitation experiments from detergent-solubilized cell extracts. By Western blot analysis of immunoisolated SPP complexes, we observed a robust interaction with Derlin1 (Fig 1A). By contrast, the structurally related rhomboid protease RHBDL4 and the ERAD E3 ligases Hrd1 and gp78 were not co-purified, indicating that the SPP-Derlin1 interaction is specific. Importantly, Derlin1 also co-purified with endogenous SPP (Fig 1B), suggesting that the assembly represents a physiological complex and is not a consequence of the overexpression conditions. This was further corroborated by co-purification of endogenous SPP with both, endogenous and ectopically expressed HA-tagged Derlin1 (Fig 1C and Supplementary Fig S1A). The relative low yield of co-purified SPP is consistent with numerous known alternative Derlin1-containing ERAD complexes (Christianson et al, 2011). Moreover, we confirmed that SPP interacts with TRC8 E3 ligase (Supplementary Fig S1B), as reported previously (Stagg et al, 2009). Since other major ERAD E3 ligases do not interact with SPP (Fig 1A), this result indicates that SPP forms a defined assembly with Derlin1 and TRC8.
Figure 1. SPP-Derlin1-TRC8 complex.

- Immunoprecipitation (IP) of CHAPS-solubilized HA-tagged SPP (SPP-HA) and Western blot (WB) analysis with indicated antibodies. Filled triangle, monomer of SPP-HA and endogenous (end.) SPP; open triangle, SDS-stable SPP dimer; asterisks, cross-reacting immunoglobulin chains.
- IP/WB analysis of endogenous SPP as in (A).
- IP/WB analysis of endogenous Derlin1 as in (A).
- BN-PAGE and WB analysis of immunoisolated digitonin-solubilized SPP-HA resolves a 200-kDa (SPP-C200) from a 500-kDa (SPP-C500) complex.
Source data are available online for this figure.
Next, we analyzed SPP complexes by blue-native polyacrylamide gel electrophoresis (BN-PAGE). As shown before, distinct SPP complexes can be resolved from detergent-solubilized cell extracts (Schrul et al, 2010). Immunoisolated SPP formed two main complexes with apparent molecular weights of 200 kDa and 500 kDa (Fig 1D). Intriguingly, endogenous Derlin1 and TRC8 were specifically found in the 500-kDa complex (Fig 1D), suggesting functional diversification of SPP. This hypothesis is consistent with earlier findings showing that the 200-kDa complex represents an SPP homo-tetramer and is specific for signal peptides (Schrul et al, 2010; Miyashita et al, 2011).
XBP1u is a type II membrane protein and a substrate for SPP
Since SPP so far has been reported to cleave type II-oriented TM segments that harbor residues with a low propensity to form α-helices (Lemberg & Martoglio, 2002; Weihofen et al, 2002), we searched annotated short-lived proteins for these TM domain features. A candidate for such an unstable TM segment with a high content of putative helix-breaking residues is a conserved hydrophobic stretch in XBP1u (Fig 2A), which previously has been shown to target its own mRNA-ribosome-nascent chain complex to the ER (Yanagitani et al, 2011). Kohno and co-workers suggested that XBP1u stays on the cytosolic side of the ER membrane as a peripheral membrane protein (Yanagitani et al, 2009). Since the topology has not been experimentally investigated so far, we tested whether XBP1u is a type II ER membrane protein. To exclusively analyze the fate of XBP1u, we ectopically expressed a 3′-splice site mutant that is not processed by IRE1 (Yoshida et al, 2001). GFP-tagged XBP1u co-localized with the ER maker RFP-KDEL and only weak cytosolic or nuclear signals were detected (Fig 2B). Accordingly, subcellular fractionation showed FLAG-tagged XBP1u predominantly in the microsomal fraction (Fig 2C). Upon extraction of this fraction with high salt or sodium carbonate, approximately half of XBP1u remained membrane-associated, which is typical of a TM anchor with modest hydrophobicity. Consistent with a membrane-spanning type II topology, insertion of an N-glycosylation site, C-terminal of the hydrophobic stretch, leads to Endo H-sensitive glycosylation of a fraction of XBP1u (Fig 2D). We assume that XBP1uR232N is only partially modified because the glycosylation sequence we introduced is relatively inefficient. A type II topology was further supported by a fluorescence protease protection (FPP) assay of digitonin-permeabilized cells (Lorenz et al, 2006) expressing XBP1u constructs harboring GFP fused either to the N- or C-terminus (Fig 2E and Supplementary Fig S2A–C). Trypsin treatment of cells expressing XBP1u-N-GFP and the luminal ER maker RFP-KDEL showed rapid loss of GFP intensity after selective permeabilization of the plasma membrane, whereas the luminal ER marker was protected (Fig 2E and Supplementary Fig S2A). In contrast, GFP fused to the C-terminus of XBP1u was protected, whereas the cytosolic mCherry-tag of co-transfected ER-resident CD3δ was efficiently degraded (Fig 2E and Supplementary Fig S2B). Identical results were obtained by selective permeabilization and immunofluorescence analysis of cells transfected with XBP1u harboring a N-terminal FLAG-tag and a C-terminal HA-tag (Supplementary Fig S2D), ruling out that the GFP-tag was responsible for the observed topology. Finally, an in vitro translocation assay (Supplementary Fig S2E) strongly indicates that XBP1u is a type II membrane protein. We note that a fraction of XBP1u may be peripheral attached as stalled nascent chains and translocation intermediates, as has been suggested (Yanagitani et al, 2009). Nevertheless, we conclude that the predominant fraction of XBP1u has a membrane-spanning topology, as corroborated by quantification of FPP assays showing that the XBP1u C-terminal tail was protected in the range of the luminal control (Supplementary Fig S2C). Shifting the artificially introduced glycosylation acceptor site toward the TM domain showed that position 215 can be glycosylated, whereas more N-terminal sites were not modified (Supplementary Fig S2F). Since the oligosaccharyltransferase requires a minimal distance of 12 residues from substrate TM anchors (Nilsson & von Heijne, 1993), we predict the XBP1u TM segment to span residues 186–202 (Fig 2D).
Figure 2. XBP1u is a type II membrane protein that is cleaved by SPP.
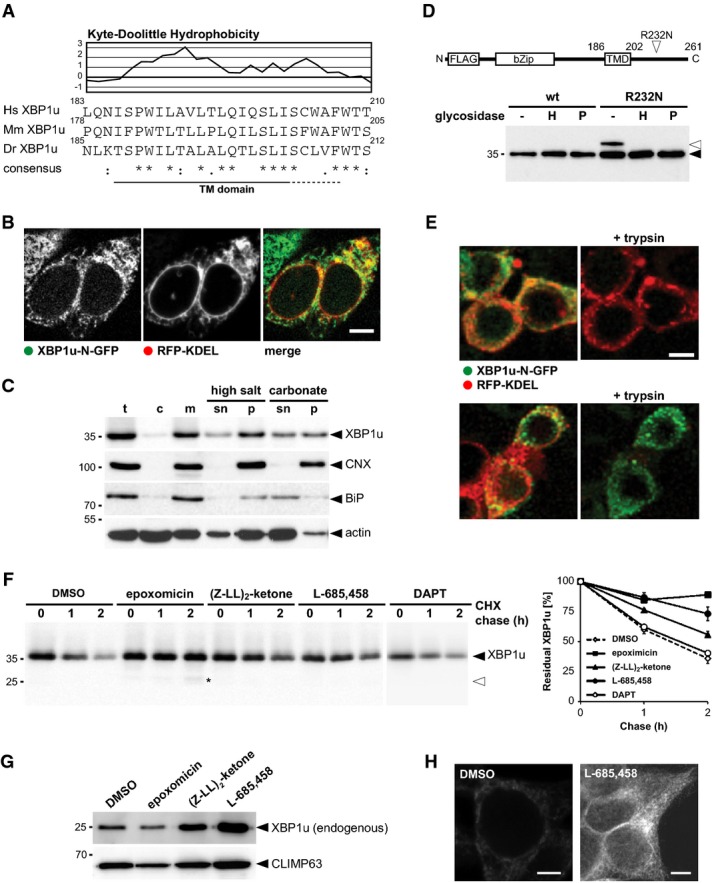
- Kyte-Doolittle hydrophobicity plot (window size of 5) of human (H. sapiens, Hs) XBP1u and comparison to its mouse (M. musculus, Mm) and zebrafish (D. rerio, Dr) orthologues reveals a conserved low-scoring TM domain.
- XBP1u N-terminally tagged with GFP co-localized with the ER marker RFP-KDEL. Scale bar, 5 μm.
- Subcellular fractionation, extraction of the microsomal membrane fraction (m) with high salt and sodium carbonate reveals that XBP1u-N-FLAG is an integral membrane protein. t, total cell extract; c, cytosol; sn, supernatant, p, pellet.
- XBP1u is a type II membrane protein as demonstrated by the sensitivity of the glycosylated R232N mutant (open triangle) to EndoH (H) and PNGaseF (P).
- FPP assay shows that GFP fused to the C-terminus of XBP1u is protected, whereas an N-terminal fusion is accessible for trypsin treatment. The luminal ER marker RFP-KDEL and CD3δ with mCherry fused to its cytosolic C-terminus were used as positive and negative controls, respectively. Of note, low-level nuclear GFP signal was not present when SPP-catalyzed cleavage of XBP1u was blocked (see Fig 4B and Supplementary Fig S3), indicating that it is a consequence of SPP-triggered release. Scale bar, 5 μm.
- Cycloheximide (CHX) chase in presence of 5 μM epoxomicin, 50 μM (Z-LL)2-ketone, 5 μM L-685,458, and 50 μM DAPT, respectively. Western blot quantification is shown (means ± SEM, n = 3). DMSO, vehicle control; asterisks, N-terminal XBP1u fragment.
- Treatment of HEK293T cells with 5 μM epoxomicin, 50 μM (Z-LL)2-ketone, and 5 μM L-685,458 for 16 h, respectively, and analysis of endogenous XBP1u in isolated microsomal membrane fractions. The unrelated structural ER protein CLIMP63 was used as a loading control.
- Immunofluorescence analysis of HEK293T cells shows that endogenous XBP1u accumulates in cells treated with SPP inhibitor L-685,458 (5 μM). Scale bar, 5 μm.
Source data are available online for this figure.
Next, we asked whether XBP1u is a proteolytic substrate for SPP. Previous reports concluded that XBP1u is a cytosolic protein that is rapidly degraded by the ubiquitin-proteasome system (Tirosh et al, 2006; Yoshida et al, 2006). To decipher how XBP1u is degraded, we performed cycloheximide chase experiments in the presence of GxGD protease inhibitors as well as the proteasome inhibitor epoxomicin (Fig 2F). In addition to the previously reported sensitivity to proteasome inhibitors, XBP1u was stabilized when SPP activity was blocked by (Z-LL)2-ketone or L-685,458, whereas a high dose of the presenilin/γ-secretase-specific inhibitor DAPT did not show any effect (Fig 2F). Since we observed consistent results with two chemically distinct SPP inhibitors, which do not cross-react with the proteasome (Weihofen et al, 2000), this result strongly suggests that XBP1u is degraded in an SPP-dependent manner. Surprisingly, when the proteasome was blocked, we did detect stabilization of the full-length form and only traces of the XBP1u cleavage fragment (Fig 2F). Of note, similar feedback inhibition between the proteasome and the ERAD machinery has been observed for the classical dislocation pathways (Elkabetz et al, 2004; Lipson et al, 2008; Nakatsukasa et al, 2013). Moreover, we conclude that XBP1u fragments are difficult to detect and are subject to alternative proteases, as it has been observed for other intramembrane proteolysis events (Lemberg, 2011). Consistent with a physiological role of SPP in XBP1u degradation, using an XBP1-specific antibody, we detected accumulation of endogenous XBP1u in enriched ER membranes upon treatment of untransfected HEK293T cells with (Z-LL)2-ketone or L-685,458 (Fig 2G). In contrast, epoxomicin treatment of HEK293T cells reduced XBP1u levels, likely because of IRE1-mediated XBP1 splicing upon proteasome inhibition (Supplementary Fig S2G). Likewise, we observed stabilization of endogenous XBP1u by SPP inhibitors in two additional human cell lines, namely HeLa and U2OS (Supplementary Fig S2H). Moreover, immunofluorescence analysis of fixed HEK293T cells showed an accumulation of endogenous XBP1u in the ER upon SPP inhibition (Supplementary Fig S2H and I), whereas upon epoxomicin treatment, we observed accumulation of a fuzzy XBP1u signal adjacent to the ER (Supplementary Fig S2I). Taken together, these results show that endogenous XBP1u is substrate for a non-canonical branch of the ERAD pathway that links two proteolytic systems, the intramembrane protease SPP and the proteasome.
Turnover of XBP1u is mediated by specific TM domain features
Next, we asked how XBP1u is selected for degradation. Commonly, GxGD-type intramembrane proteases are believed to require substrate activation by a preceding cut close to the TM segment, although an exception from this rule has been described for the SPP-like protease 3 (SPPL3) (Voss et al, 2012). Since we did not detect any significant cleavage in the XBP1u luminal domain by signal peptidase or any other sheddase (Fig 2F), we searched the TM domain for other features that may target it for SPP-catalyzed cleavage. Our previous analysis of SPP revealed hydrophilic and small amino acid residues as primary determinant for signal peptide processing, pointing toward a conformational control of the protease's access to the substrate scissile peptide bond (Lemberg & Martoglio, 2002). To test the influence of such putative helix-destabilizing residues on XBP1u degradation, we mutated a characteristic serine–proline motif and alanine in the N-terminal portion of the XBP1u TM domain to leucine (XBP1umt1). Then, we analyzed the stability by a cycloheximide chase (Fig 3A) and investigated the structural properties of a synthetic peptide by circular dichroism (CD) spectroscopy (Fig 3B). Consistent with our previous results on SPP specificity, only a minor fraction of XBP1umt1 was degraded within a 4 h chase time (compared to a half-life of 1 h for the WT). As hypothesized, mutations increased the helicity of the XBP1umt1 TM domain by 20% (Fig 3B). Since topology of XBP1umt1 was unchanged as assessed by FPP assay of ectopically expressed GFP fusion proteins (Supplementary Fig S3A), we conclude that SPP-catalyzed cleavage of XBP1u is influenced by the secondary structure of the type II TM anchor. However, stabilization of the TM helix still allowed XBP1u turnover, albeit with a minimal rate. Therefore, we next investigated the role of conserved glutamine and serine residues in the C-terminal TM region of XBP1u, a motif that so far has not been studied in the context of SPP-catalyzed cleavage (Fig 3A). Interestingly, mutation of this characteristic TM signature to leucine completely blocked degradation of XBP1u (XBP1umt2). Since this mutation did not affect helix stability (Fig 3B) and type II membrane topology (Supplementary Fig S3B), this result indicates that in addition to overall substrate conformation, XBP1u degradation is also governed by more subtle structural features of its TM domain that may include its positioning within the enzyme. A similar impact of primary and secondary structure on substrate cleavability has been described for rhomboid proteases (Strisovsky et al, 2009; Dickey et al, 2013), suggesting that this interplay between TM helix stability and sequence determinants is a more general mechanism governing intramembrane proteolysis.
Figure 3. TM domain determines XBP1u for SPP-dependent degradation.
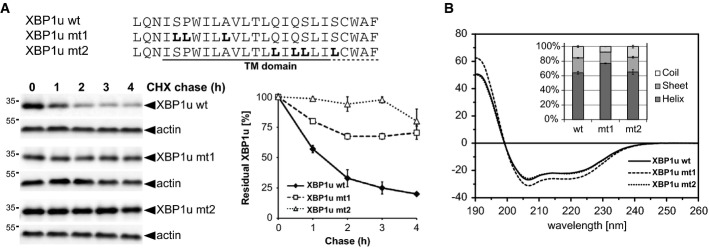
- The primary structure of the XBP1u TM domain determines degradation kinetics as assessed by cycloheximide (CHX) chase and Western blotting (means ± SEM, n = 3).
- CD spectra of XBP1u TM domain peptides tagged at both termini with KKK sequences (see Supplementary Materials and Methods) and were obtained in 40% trifluoroethanol in aqueous buffer which mimics the water-filled lumen of an intramembrane protease while preventing unspecific peptide aggregation (means ± SD, n = 3). Inset: secondary structure contents as calculated from the CD spectra.
Source data are available online for this figure.
SPP-catalyzed cleavage mediates p97-independent degradation
We next asked whether XBP1u degradation depends on p97-mediated pulling, commonly required for turnover of ERAD substrates (Ye et al, 2005), or whether SPP-catalyzed clipping directly releases peptide fragments. To test this, we generated an inducible cell line expressing a p97-specific shRNA knockdown construct and analyzed XBP1u turnover by cycloheximide chase. Strikingly, XBP1u degradation was not affected by p97 knockdown, whereas the classical ERAD dislocation substrate CD3δ was completely stabilized (Fig 4A). Likewise, dominant-negative mutants of the p97-associated deubiquitinases YOD1 and ataxin3, which are known to interfere with extraction of classical ERAD substrates (Wang et al, 2006; Ernst et al, 2009), did not show any effect on XBP1u turnover (Supplementary Fig S4A).
Figure 4. XBP1u is degraded by a p97-independent mechanism.

- Doxycycline-induced expression of p97-specific knockdown construct (shRNA) stabilized FLAG-tagged CD3δ but had no influence on XBP1u-N-FLAG as assessed by cycloheximide (CHX) chase. Right panel, Western blot quantification (means ± SEM, n = 3).
- Fluorescence microscopy of XBP1u-GFP- and RFP-KDEL-expressing cells that had been treated with 0.5 μM epoxomicin and 5 μM L-685,458, respectively. Scale bars, 5 μm.
- Co-expression of XBP1u-N-FLAG with SPP WT in presence of 5 μM epoxomicin increased levels of N-terminal fragment (open triangle), whereas the catalytically inactive SPPD265A mutant shows no activity.
Source data are available online for this figure.
Since it is difficult to observe XBP1u cleavage fragments by Western blotting and analysis of fixed cells due to technical limitations, we treated HEK293T cells expressing XBP1u-N-GFP with epoxomicin in order to better detect release of the N-terminal GFP moiety. Consistent with a proteolytic release of XBP1u, fluorescence microscopy showed robust cytosolic and nuclear levels upon inhibition of the proteasome (Fig 4B). Since inhibition of SPP with L-685,458 caused accumulation of XBP1u-N-GFP in the ER and no cytosolic or nuclear signal was detected, we conclude that the release was triggered by SPP. Despite that, upon inhibiting the proteasome, we also observed accumulation of XBP1u in the ER (Fig 4B). Likewise, Western blot analysis revealed stabilization of the full-length XBP1u (Figs 2F and 4C). Furthermore, in presence of epoxomicin, upon Western blot analysis of immunoisolated XBP1u, we detected several ubiquitinated species (Supplementary Fig S4B). This is consistent with a previous report showing that XBP1u is polyubiquitinated in HeLa cells (Lee et al, 2003). Taken together with the stabilization effect on the full-length form upon epoxomicin treatment, this result strengthens the hypothesis that under our experimental conditions there is a so far unrecognized feedback inhibition between the proteasome and the ER-resident SPP complex. Co-expression of XBP1u and SPP in epoxomicin-treated cells, however, increased formation of the N-terminal XBP1u cleavage fragment (Fig 4C), confirming that it is generated by SPP-catalyzed cleavage. In contrast, expression of the D265A active site mutant of SPP (SPPD265A) did not show any increase of this product, demonstrating that the observed effect depends on the catalytic activity of SPP. By comparison to custom-made reference peptides, the main SPP cleavage site was approximately mapped to residues leucine 194 and threonine 195 (Supplementary Fig S4C). Consistent with the function of SPP as an endoprotease, tagging XBP1u at the C-terminus allowed detection of a corresponding 15-kDa C-terminal fragment that was rapidly degraded by the proteasome (Supplementary Fig S4D). Interestingly, upon inhibition of the proteasome, in addition to polyubiquitination of full-length XBP1u, also a higher molecular weight adduct of the C-terminal cleavage fragment was detected. Taken together with the role of TRC8 in XBP1u turnover (see below), this result suggests that the luminal C-terminal fragment gets ubiquitinated and dislocated like soluble ERAD substrates (Bagola et al, 2011).
SPP controls XBP1u abundance but is not involved in its role as a negative UPR regulator
We next sought to assess the potential impact of SPP-mediated XBP1u cleavage on UPR signaling. XBP1u has previously been reported to heterodimerize with XBP1s and activated ATF6 via its bZIP domain, thereby triggering their proteasomal downregulation (Tirosh et al, 2006; Yoshida et al, 2006, 2009). We therefore compared the ability of XBP1u and the cleavage-deficient XBP1umt2 to interfere with the UPR using a stable cell line expressing doxycycline-inducible XBP1s transfected with an UPR transcription reporter construct (Yoshida et al, 1998). Induction of XBP1s with doxycycline leads to activation of the UPR luciferase reporter. Co-expression of either XBP1u or XBP1umt2 reduced this effect (Fig 5A). Although the drastic difference in the half-lives of XBP1u and XBP1umt2 prevented us from determining the stoichiometry of this effect, the phenocopy observed for XBP1umt2 suggests that SPP-catalyzed release of XBP1u is dispensable for downregulation of XBP1s. Consistent with a functional interaction of membrane-anchored XBP1u with XBP1s, immunoisolation of detergent-solubilized XBP1u and XBP1umt2 both co-purified XBP1s (Supplementary Fig S5A). Likewise, expression of XBP1umt2 in the doxycyclin-inducible XBP1s cells accelerated turnover of XBP1s in the similar range as XBP1u wild-type (WT) (Supplementary Fig S5B). In order to test how this mechanism interferes with endogenous UPR transcription factors, we measured the influence of both XBP1u constructs on tunicamycin-induced activation of the UPR reporter in inducible stable cell lines (Fig 5B). Both XBP1u WT and XBP1umt2 suppressed tunicamycin-induced activation of the UPR reporter when compared to the parental T-REx cells, indicating that membrane-anchored XBP1u is a physiological inhibitor of the UPR. SPP-catalyzed release of XBP1u, however, does not directly affect this function beyond the abundance control of this negative regulator.
Figure 5. Inhibition of UPR by XBP1u is independent of SPP-catalyzed cleavage.
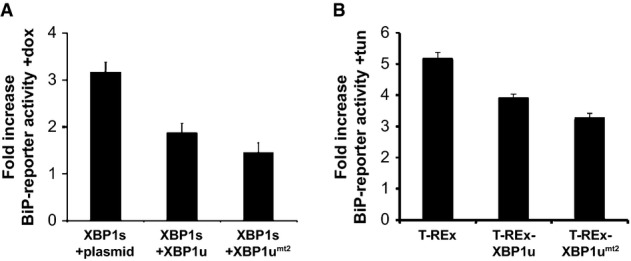
- Doxycycline (dox)-induced XBP1s expression in HEK293 T-REx cells induces BiP-reporter activity. Co-expression of XBP1u and the XBP1umt2 reduces this activation. Quantification of relative induction of luciferase activity is shown (means ± SEM, n = 3).
- Doxycycline-induced expression of XBP1u and XBP1umt2 in HEK293 T-REx cells reduces tunicamycin (tun)-induced activation of the endogenous UPR compared to the parenteral T-REx cells as assessed by a BiP-reporter luciferase assay (means ± SEM, n = 3).
XBP1u is targeted specifically by the SPP ERAD complex
Since XBP1u was sensitive to SPP inhibitors, we tested whether it interacts with SPPD265A, which acts as a dominant-negative mutant by binding but not cleaving its substrates (Dev et al, 2006; Schrul et al, 2010). Consistent with the experiments using SPP inhibitors, pulse-chase analysis revealed that co-expression of SPPD265A blocked degradation of XBP1u (Fig 6A). In contrast, co-expression of the dominant-negative mutants of YOD1 (YOD1C160S) or the SPP-related Golgi protease SPPL3 fused to an ER retention signal (SPPL3DAKK) showed no effect, indicating that the effect was specific. Since co-expression of SPPD265A preserved topology of XBP1u as a type II membrane protein (Supplementary Fig S6A), this result suggests that substrate trapping causes the dominant-negative effect. Consistent with this, XBP1u was co-immunoprecipitated with SPPD265A, whereas the unrelated membrane protein RAMP4 was not (Fig 6B). Unexpectedly, also SPP WT co-purified XBP1u although with a slightly reduced efficiency (Supplementary Fig S6B). This observation indicates that the SPP-reaction cycle is slow, which is consistent with a recent kinetic analysis of rhomboid intramembrane proteases (Dickey et al, 2013). The substrate trapping by SPPD265A was specific for the 500-kDa SPP complex, as XBP1u co-purified only with this SPP complex (Fig 6C). This selective interaction is different from the previously reported interaction of a scrambled opsin-derived TM fragment (OP91*) with all SPP complexes (Fig 6C) (Crawshaw et al, 2004; Schrul et al, 2010). Taken together these results show that XBP1u has to interact with the 500-kDa SPP complex in order to get degraded.
Figure 6. XBP1u is specifically recognized by SPP-Derlin1-TRC8 complex.
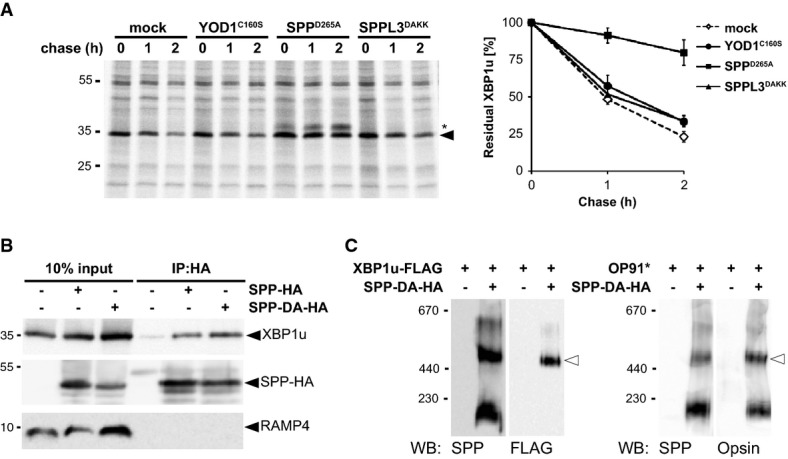
- Co-expression of SPPD265A stabilized XBP1u, whereas YOD1C160S and SPPL3DAKK had no effect. Right panel, quantification of the relative amount of 35S-pulse-labeled XBP1u-N-FLAG (means ± SEM, n = 3). Asterisks, unidentified co-immunoprecipitated protein.
- HA-tagged SPP (SPP-HA) and the catalytic mutant (SPP-DA-HA) co-immunoprecipitate with XBP1u but not with RAMP4.
- Substrate-trapping SPP-DA-HA was co-expressed with XBP1u (left panel) or OP91* (right panel) and analyzed by BN-PAGE as shown (Fig 1D).
Source data are available online for this figure.
TRC8 and Derlin1 promote XBP1u degradation
Next, we aimed to decipher the molecular mechanism of SPP-triggered degradation of XBP1u and tested whether interfering with TRC8 and Derlin1 can block its degradation. To this end, we co-expressed a dominant-negative mutant of TRC8 lacking the active site RING-domain (TRC8ΔR) (Brauweiler et al, 2007), and the Derlin1G180V mutant which, by an unknown mechanism, blocks dislocation of the model ERAD substrate NHK (Greenblatt et al, 2011). To test the effect on XBP1u degradation, we performed a pulse-chase analysis and saw that both mutants prolonged the half-life of XBP1u in a range comparable to co-expression of SPPD265A (Fig 7A). Since Derlin1G180V still assembles in the 500-kDa SPP ERAD complex (Supplementary Fig S7A), this result indicates that SPP-triggered XBP1u degradation relies on the rhomboid pseudoprotease. Likewise, XBP1u specifically co-purified with immunoisolated TRC8 (Supplementary Fig S7B), and co-expression of TRC8ΔR reduced ubiquitination of XBP1u (Supplementary Fig S7C), indicating the required presence of an enzyme–substrate interaction. Taken together, our results suggest that both Derlin1 and TRC8 are required to prepare XBP1u for SPP-catalyzed clipping. Given that cleavage of signal peptides by SPP requires prior ectodomain shedding by signal peptidase (Lemberg & Martoglio, 2002), we asked to what extent XBP1u substrate selection is affected by its ectodomain. First, to more generally test the role of the XBP1u C-terminus on protein stability, we generated an XBP1u deletion construct lacking the entire luminal ectodomain (XBP1uΔ) and compared its turnover to that of full-length protein. Strikingly, truncated XBP1uΔ showed a shorter half-life than the full-length protein (10 min relative to 55 min, respectively) (Fig 7A and B), indicating that the XBP1u ectodomain has a stabilizing effect. Since XBP1uΔ was found in the microsomal fraction (Supplementary Fig S7D) and was cleaved by ectopically expressed SPP (Supplementary Fig S7E), we concluded that the XBP1u ectodomain is not required for ER targeting but affects the interplay with the SPP-Derlin1-TRC8 complex. Consistent with this, turnover of XBP1uΔ was still reduced by co-expression of SPPD265A and TRC8ΔR (Fig 7B). The relatively high turnover rate compared to the full-length substrate indicates that, upon blocking the SPP-dependent pathway, other ERAD branches are capable of degrading XBP1u (Christianson et al, 2011). In contrast to blocking SPP and TRC8, co-expression of the dominant-negative Derlin1G180V did not show any effect on XBP1uΔ turnover. Since co-expression of XBP1uΔ did not affect association of Derlin1 with the 500-kDa ERAD complex (Supplementary Fig S7F), we suggest that the dominant-negative effect of this mutant is mediated through interaction with the XBP1u ectodomain. Consistent with this conclusion, Derlin1G180V physically interacts with full-length XBP1u, whereas no co-immunoprecipitation was observed for XBP1uΔ (Fig 7C).
Figure 7. TRC8 and Derlin1 mediate degradation of XBP1u.
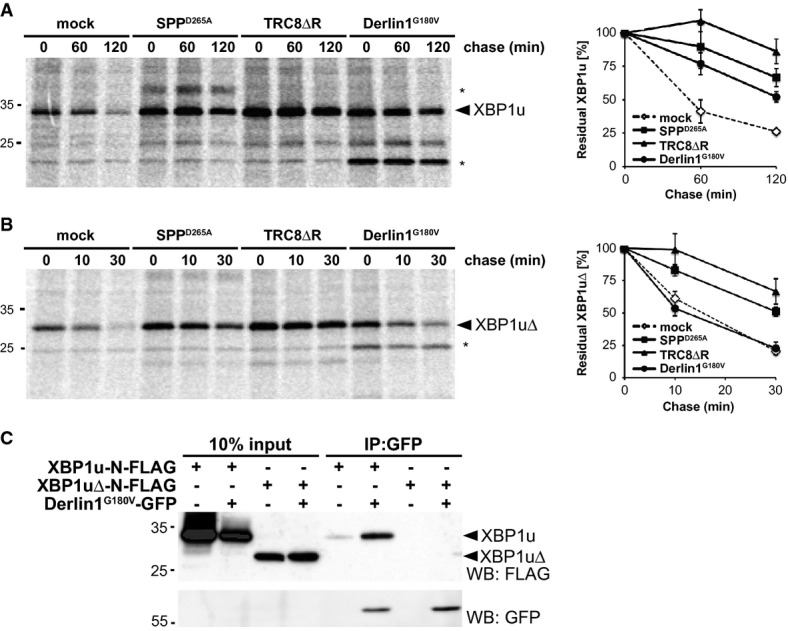
- Cells transfected with XBP1u-N-FLAG were co-transfected either with empty vector (mock), SPPD265A, TRC8ΔR or Derlin1G180V as indicated and subjected to 35S-pulse-label chase analysis (means ± SEM, n = 3). Asterisks, unidentified co-immunoprecipitated protein.
- Pulse-chase analysis of XBP1uΔ turnover as in (A).
- GFP-tagged Derlin1G180V co-immunoprecipitates with FLAG-tagged XBP1u but not with XBP1uΔ.
Source data are available online for this figure.
Luminal domain of XBP1u mediates recruitment to the SPP-dependent ERAD
Since Derlin1 is specific for ERAD substrates, probably acting as receptor for unfolded luminal domains (Lemberg, 2013), we asked whether in addition to the XBP1u TM determinants specific features of its C-terminal tail are required for SPP-triggered turnover. To this end, we performed a domain swap experiment between XBP1u and a stable type II membrane protein (Fig 8A). Since we aimed to specifically investigate the role of the luminal domain, we choose invariant chain (CD74), a substrate for the related intramembrane protease SPPL2a, which upon targeting to lysosomes is subject to regulated intramembrane proteolysis (Bergmann et al, 2013; Schneppenheim et al, 2013; Beisner et al, 2013). Interestingly, a chimera comprising the N-terminal portion and TM domain of CD74 and the luminal tail of XBP1u (CD74-XBP1u) was rapidly degraded with a half-life below 20 min (Fig 8B), whereas CD74 WT has a half-life in the range of several hours (Supplementary Fig S8A). This result shows that the luminal XBP1u domain targets a bona fide type II membrane protein for degradation. Co-expression of CD74-XBP1u with SPPD265A slowed degradation of this chimera, indicating that it is substrate for SPP-mediated turnover. Consistent with this, also TRC8ΔR and Derlin1G180V reduced the decay of the CD74-XBP1u chimera (Fig 8B). Thus, the XBP1u tail is sufficient to target the CD74 TM domain for degradation by the SPP-TRC8-Derlin1 complex. Accordingly, the CD74-XBP1u chimera was co-purified with Derlin1G180V, although the yield was lower than observed for XBP1u WT (Supplementary Fig S8B). However, fusing the CD74 luminal domain to the cytosolic portion and TM domain of XBP1u created an unstable protein that was not affected by co-expression of SPPD265A, TRC8ΔR, or Derlin1G180V (Fig 8A and C), suggesting that this chimera is folding-deficient and subsequently targeted to a canonical ERAD dislocation pathway (Christianson et al, 2011). Taken together these results strongly suggest that in the context of the SPP complex, Derlin1 recognizes specific features of the XBP1u tail and is not involved in degradation of bulk unfolded proteins. To test the physiological relevance of Derlin1 as XBP1u substrate receptor, we applied siRNA to target endogenous Derlin1 and measured degradation rate by cycloheximide chase (Fig 8D). Strikingly, Derlin1 knockdown caused a mild but significant stabilization of full-length XBP1u with half-life increasing from 55 to 120 min (Fig 8D). Consistent with the result using the dominant-negative Derlin1G180V mutant, XBP1uΔ was not significantly stabilized by the Derlin1 knockdown (Supplementary Fig S8C). Taken together, these results show that Derlin1 targets full-length XBP1u into SPP-dependent ERAD without prior ectodomain shedding.
Figure 8. XBP1u tail targets a type II membrane protein for SPP-, TRC8-, and Derlin1-dependent degradation.
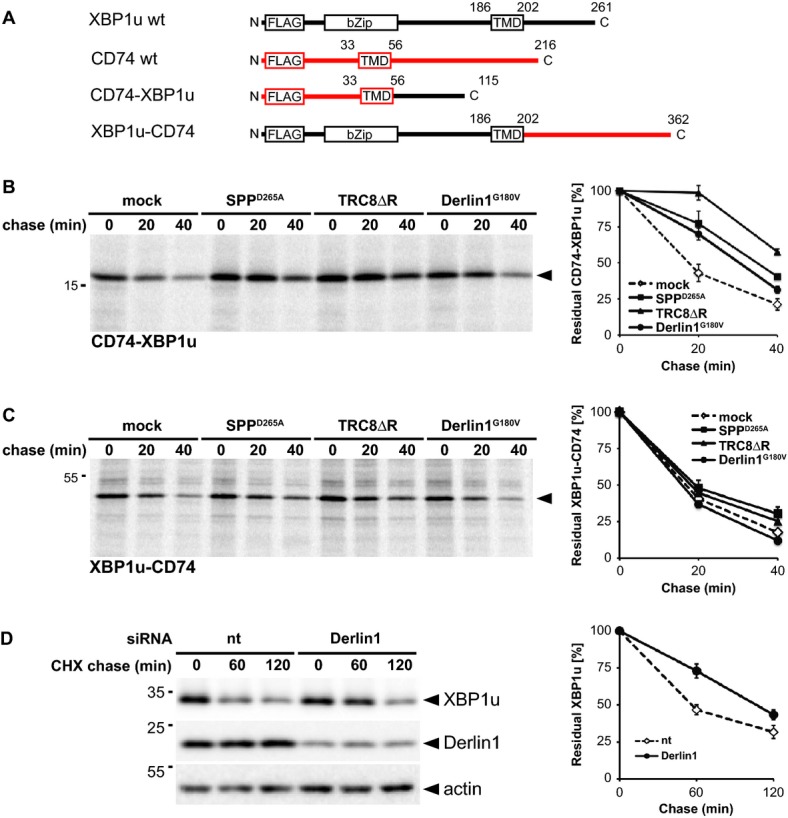
- Outline of domain fusions between CD74 (indicated in red) and XBP1u.
- CD74-XBP1u chimera was co-transfected either with empty vector (mock), SPPD265A, TRC8ΔR or Derlin1G180V as indicated and subjected to 35S-pulse-label chase analysis (means ± SEM, n = 3).
- Pulse-chase analysis of XBP1u-CD74 turnover as in (B).
- Knockdown of Derlin1 by siRNA reduces XBP1u degradation (means ± SEM, n = 3). CHX, cycloheximide chase; nt, non-targeting control siRNA.
Source data are available online for this figure.
Discussion
We have investigated the interaction of SPP with ERAD factors and identify XBP1u as a proteolytic substrate for a SPP-dependent degradation route. We show that XBP1u is a type II membrane protein and define intramembrane cleavage as a trigger for its p97-independent release for proteasomal degradation. As previously it has been suggested that XBP1u is a peripheral membrane protein (Yanagitani et al, 2009), our analysis revealing a type II signal anchor for XBP1u extends the number of membrane-anchored UPR regulators and puts SPP at a hub of ER membrane protein homeostasis.
SPP forms a functional complex with Derlin1 and TRC8
We show that SPP forms a 500-kDa assembly with the ERAD factors Derlin1 and TRC8. Likewise, presenilin, the active subunit of the γ-secretase, assembles with non-proteolytic cofactors to form a 230-kDa complex (Osenkowski et al, 2009), indicating that association with substrate receptors and regulators is a general principle in the control of intramembrane proteolysis. While earlier work showed that the 200-kDa SPP complex is specific for signal peptides (Schrul et al, 2010), we now demonstrate that in the 500-kDa SPP complex TRC8 and Derlin1 prime the intramembrane protease for XBP1u (Fig 9). Previous analysis of HCMV-induced degradation of MHC class I molecules had suggested an SPP-dependent ERAD mechanism that does not require Derlin1 (Loureiro et al, 2006). In our experimental system using non-infected cells, however, there is functional interplay between SPP and Derlin1. Our results using SPP-specific inhibitors and the substrate-trapping SPPD265A mutant strongly indicate that the proteolytic activity of SPP is required for XBP1u turnover. Moreover, XBP1u was bound and stabilized by ectopic expression of a dominant-negative TRC8 mutant, suggesting that ubiquitination is required for SPP-catalyzed cleavage and subsequent proteasomal degradation. However, we emphasize that this remains speculative until the precise role of TRC8 in the SPP-reaction cycle has been elucidated.
Figure 9. Model for SPP-Derlin1-TRC8-mediated proteolytic release of the type II membrane protein XBP1u.
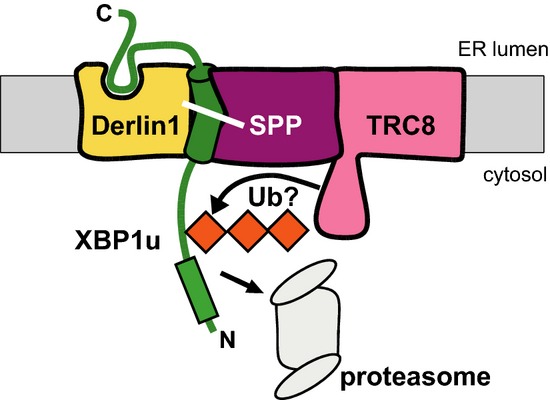
The rhomboid pseudoprotease Derlin1 may act as a receptor for the luminal XBP1u domain thereby uncoupling SPP-catalyzed cleavage from shedding events. TRC8 may ubiquitinate (Ub) XBP1u leading to degradation by ER-associated proteasomes.
Specific TM domain features and Derlin1-mediated ectodomain contact determine XBP1u turnover
Specificity of intramembrane proteases follows different principles than that of soluble proteases (Strisovsky et al, 2009; Dickey et al, 2013; Lemberg, 2013). Our previous work on SPP revealed that residues with a low propensity to form TM helices are required for efficient signal peptide processing (Lemberg & Martoglio, 2002). Moreover, we showed that signal peptides have to be liberated from the pre-protein by signal peptidase in order to get cleaved. Here, we demonstrate that a helix-break and a conserved TM sequence motif are needed for SPP-catalyzed cleavage of a type II membrane anchor. However, different to signal peptides, SPP can cleave the full-length protein without preceding ectodomain shedding. During revision of our paper, Lehner and co-workers independently reported a proteolytic role of SPP in ERAD of selected tail-anchored membrane proteins (Boname et al, 2014). While substrate recognition of tail-anchored proteins with a short C-terminus is closer to previously known cleavage of signal peptides, identification of Derlin1 as a substrate adaptor puts an additional layer to this non-canonical proteolysis driven ERAD branch. Together with a recent report that the SPPL3-catalyzed cleavage of the foamy virus envelope protein is not dependent on ectodomain size (Voss et al, 2012), our result on SPP-catalyzed cleavage of a type II membrane protein broadens the spectrum of potential substrates for GxGD-type proteases. For SPP-triggered degradation of membrane proteins, Derlin1 emerges as a factor that recognizes specific features in the XBP1u luminal portion. Although the exact mechanism of Derlin1 is unknown, we suggest that in the SPP complex it is potentially responsible for uncoupling intramembrane proteolysis from shedding events by recognizing the luminal substrate tail (Fig 9). By assisting recruitment of a type II membrane protein for SPP-catalyzed cleavage, Derlin1 fulfills a different role as the non-proteolytic γ-secretase subunit nicastrin, which acts as a molecular ruler in preventing productive recognition of full-length membrane proteins (Shah et al, 2005). Consistent with the proposed function of Derlin1 as a substrate receptor for SPP-catalyzed release of a type II membrane protein, it has been shown to interact with both, luminal but also membrane-anchored ERAD substrates (for review, see Lemberg, 2013).
SPP controls the abundance of the negative UPR regulator XBP1u
The role of the Golgi-resident intramembrane-cleaving site-2 protease in activation of ATF6 is well established (Ye et al, 2000), but so far no link of ER-resident proteases to the UPR has been described. Our results indicate a possible role of SPP in tuning the abundance of the UPR regulator XBP1u. Since C-terminally truncated XBP1u lacking the hydrophobic domain is transcriptionally inert (Yoshida et al, 2006), fragments released by SPP-catalyzed cleavage are predicted to be inactive. Instead, the SPP-catalyzed intramembrane cleavage and release of XBP1u from the membrane triggers its proteasomal degradation and thereby provides a mechanism to regulate XBP1u abundance. This then indirectly modulates the inhibitory role of XBP1u during UPR signaling. It will be interesting to study whether SPP-triggered degradation of XBP1u itself is regulated, be it by stress-induced activation of SPP or by a conformational switch in the XBP1u luminal domain. In this study, we detect endogenous XBP1u, indicating that, despite its constant turnover by the SPP ERAD complex, it serves as a physiological inhibitor of UPR signaling. Using a stable XBP1u mutant that cannot be cleaved by SPP, we provide evidence that XBP1u does not itself need to be cleaved for inactivation of XBP1s to occur. Instead, we suggest that it acts as a tether for soluble transcription factors triggering their degradation by the proteasome. Since XBP1u has been shown to interfere with the transcription factors XBP1s (Tirosh et al, 2006; Yoshida et al, 2006), activated ATF6 (Yoshida et al, 2009), and the autophagy regulator FoxO1 (Zhao et al, 2013), it will be interesting to study what the physiological function of this crosstalk is. Since misfunction of the UPR and autophagy are both linked to diseases ranging from cancer to neurodegenerative disorders (Wang & Kaufman, 2012; Nixon, 2013), inhibition of SPP and the subsequent rise in XBP1u levels emerge as a promising new therapeutic intervention strategy to suppress unwanted transcriptional activation.
Regulated intramembrane proteolysis is a widely used paradigm in cell biology, which controls trafficking of fundamentally important bioactive molecules. We have now further extended this range by demonstrating that SPP-dependent degradation controls the abundance of a negative transcriptional regulator. Taken together with our earlier work on the ER rhomboid protease RHBDL4 that degrades unstable type I and polytopic membrane proteins (Fleig et al, 2012), clipping of ERAD substrates emerges as a more generally used principle. Further insights into the substrate spectrum of this, so far unrecognized, SPP-dependent degradation route and how its activity is regulated will likely reveal additional unexpected roles for the ERAD machinery.
Materials and Methods
Cell lines and transfection
HEK293T cells were grown in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS) at 37°C in 5% CO2. Transient transfections were performed using 25 kDa linear polyethylenimine (Polysciences) (Durocher et al, 2002) as has been described (Fleig et al, 2012). Typically, 1 μg plasmid encoding substrate candidate and 300 ng plasmid encoding SPP or ERAD factors were used per well of a 6-well plate (see Supplementary Materials and Methods for plasmids). Total transfected DNA (2 μg/well) was held constant by the addition of empty plasmid. If not otherwise stated, cells were harvested 24 h after transfection. For inhibition of the proteasome and SPP, 16 h post-transfection epoxomicin (Calbiochem), (Z-LL)2-ketone (Calbiochem), and L-658,485 (Calbiochem) were added from 2,000× stock solutions in dimethyl sulfoxide (DMSO). As a vehicle control, the same amount of DMSO was used. Subsequently, cells were further incubated and harvested 16 h later.
To prepare inducible stably transfected cells expressing pcDNA5/FRT/TO/SPP-HA, pcDNA5/FRT/TO/FLAG-XBP1s, pcDNA5/FRT/TO/FLAG-XBP1u, pcDNA5/FRT/TO/FLAG-XBP1umt2, and pcDNA5/FRT/TO/miRNA-p97, Flp-In HEK293 T-REx cells were co-transfected with pOG44 (Invitrogen), respectively, followed by selection with blasticidin (10 μg/ml) and hygromycin B (125 μg/ml). For transfection of siRNA, 2 × 105 HEK293T cells were seeded per 6-well. After 24 h, cells were transfected with 20 nM siRNA-oligonucleotide using Oligofectamine (Invitrogen). After 48 h incubation with siRNA, cells were transfected with DNA as described above and harvested 24 h later.
Cycloheximide and pulse-label chase experiments
Cycloheximide (100 μg/ml) chase was conducted 24 h after transfection of HEK293T cells, and cell extracts were subjected to Western blot analysis. For pulse-label chase experiments, cells were metabolically labeled for 10 min with 55 μCi/ml 35S-methionine/cysteine protein labeling mix (Perkin Elmer) and chased in cold medium as had been described (Fleig et al, 2012). Radiolabeled proteins were isolated by immunoprecipitation and analyzed by SDS-PAGE and autoradiography.
Subcellular fractionation
Cells transfected with FLAG-XBP1u were detached by cold PBS-EDTA and resuspended in hypotonic buffer (10 mM HEPES–KOH pH 7.4, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 10 μg/ml phenylmethylsulfonyl fluoride (PMSF), and each of 10 μg/ml chymostatin, leupeptin, antipain, and pepstatin). After 10 min incubation on ice, cells were lysed by passing five times through a 27-gauge needle. Cellular debris and nuclei were discarded after centrifugation at 1,000 g for 5 min at 4°C. The supernatant was spun at 100,000 g for 30 min at 4°C. The membrane pellet was further resuspended in RM buffer (250 mM sucrose, 50 mM HEPES–KOH pH 7.4, 50 mM KOAc, 2 mM Mg(OAc)2, 1 mM DTT) and extracted either by 400 mM KOAc or 100 mM Na2CO3 (pH 11.3) as had been described (McLauchlan et al, 2002). After centrifugation at 140,000 g for 10 min at 4°C, the pellet fraction was directly resuspended in SDS sample buffer, whereas the supernatant was precipitated with 10% (w/v) trichloroacetic acid, washed with acetone, and resuspended in SDS sample buffer.
Cell-free translocation and in vitro protease protection assay
In vitro transcription of FLAG-tagged XBP1uR232N using T7 RNA polymerase and translation in rabbit reticulocyte lysate (Promega) containing 35S-methionine/cysteine was performed as described (Lemberg & Martoglio, 2002). Where indicated, nuclease-treated rough microsomes prepared from dog pancreas were added (Martoglio et al, 1998). For protease protection, the reactions were treated with proteinase K for 30 min on ice. As a control, proteinase K was added in the presence of 1% Triton X-100. Subsequently, 10 μg/ml PMSF was added, and samples were analyzed by Tris–Bicine acrylamide gels (15% T, 5% C, 8 M urea) and autoradiography as described previously (Lemberg & Martoglio, 2002).
Microscopy and fluorescence protease protection assay
For detection of XBP1u-N-GFP, HEK293T cells transfected as described above were grown on 8-well ibidi microscopy chambers. 4 h post-transfection medium was changed, and cells were treated 18 h with 0.5 μM epoxomicin and 5 μM L-685,458 from a 2,000× stock solutions in DMSO. As a vehicle control, the same amount of DMSO was used. For live cell analysis, medium was buffered with 50 mM HEPES–KOH pH 7.4 and cells were kept at 37°C. Confocal microscopy was performed on a Zeiss LSM confocal microscope. Images were taken with a Plan-APOCHROMAT 63×/1.4 Oil objective lens with a pinhole setting of 1.0 Airy unit. Image processing was performed using ImageJ (http://rsb.info.nih.gov/ij/). For detection of endogenous XBP1u, untransfected HEK293T cells were grown on glass coverslides and fixed in PBS containing 4% paraformaldehyde for 30 min. Permeabilization was performed in PBS containing 0.5% Triton X-100 for 10 min. After blocking cells for 30 min in PBS containing 20% FBS and 0.5% Tween-20, cells were incubated in PBS/FBS/Tween-20 with primary antibodies for 1 h at room temperature (see Supplementary Materials and Methods for antibodies). Subsequently, cells were washed in PBS/FBS/Tween-20, incubated with fluorescent secondary antibodies, washed and mounted for analysis by confocal microscopy as described above. For selective permeabilization of PFA-fixed cells fixed on glass coverslides were treated with either 10 μg/ml digitonin or 0.5% Triton X-100 in PBS for 15 min at 4°C and analyzed by indirect immunofluorescence analysis as described above. The fluorescence protease protection assay was performed as described previously (Lorenz et al, 2006). In brief, cells were first permeabilized with 80 μM digitonin in KHM buffer (110 mM potassium acetate, 20 mM HEPES–KOH pH 7.4, 2 mM MgCl2), then washed with KHM buffer to remove digitonin and finally treated with 0,05% Trypsin-EDTA (Gibco) for the indicated time.
Immunoprecipitation
For immunoprecipitation, cells were solubilized with 1% CHAPS in IP buffer (50 mM HEPES–KOH, pH 7.4, 150 mM NaCl, 2 mM MgOAc2, 10% glycerol, 1 mM EGTA), containing 10 μg/ml PMSF and each of 10 μg/ml chymostatin, leupeptin, antipain, and pepstatin. Non-solubilized proteins were removed by centrifugation, and the indicated antibody was added together with protein A/G beads for overnight incubation at 4°C. For anti-GFP immunoprecipitation of Derlin1G180V-GFP, 2 mM cross-linker dithiobis(succinimidylpropionate) (Pierce) was added on ice for 30 min, quenched with 20 mM Tris–Cl pH 7.5 for 15 min on ice, and solubilized as described above. Immunoisolation with GFP was performed using a GFP-specific single chain antibody fragment (Rothbauer et al, 2008) coupled to NHS-activated sepharose beads as described (Fleig et al, 2012). Immunoprecipitates were washed three times in IP buffer containing 0.1% CHAPS and eluted in SDS sample buffer.
SDS-PAGE and Western blotting
Proteins were solubilized in SDS sample buffer (50 mM Tris–Cl pH 6.8; 10 mM EDTA, 5% glycerol, 2% SDS, 0.01% bromphenol blue) containing 5% β-mercaptoethanol. All samples were incubated for 15 min at 65°C. For deglycosylation, solubilized proteins were treated with EndoH and PNGaseF (New England Biolabs) according to the manufacturer's protocol. For Western blot analysis, proteins were separated by SDS-PAGE using Tris–glycine acrylamide gels and transferred to PVDF membrane followed by enhanced chemiluminescence analysis (Pierce) on X-ray films or the LAS-4000 system (Fuji). Data shown are representative of three independent experiments. For quantification, we used the Multi Gauge software (Fuji) and data acquired from the LAS-4000.
BN-PAGE
Cells were solubilized with 1% digitonin, and non-solubilized proteins were removed by centrifugation. After incubation with HA specific antibody and protein G beads, native SPP-HA complexes were eluted by HA peptide (300 μg/ml, Sigma). 1/40 volume of BN-sample buffer (500 mM 6-aminohexanoic acid, 100 mM Bis–Tris pH 7.0, and 5% (w/v) Coomassie Blue G250) was added before subjection to BN-PAGE (Wittig et al, 2006). Samples were loaded on 5–9% gradient BN-acrylamide gel and run at 4°C for 15 min at 10 mA with anode buffer (50 mM Bis–Tris pH 7) and cathode buffer B (50 mM Tricine pH 7, 15 mM Bis–Tris, and 0.02% Coomassie Blue G250). Then, the cathode buffer was exchanged for cathode buffer B/10 (buffer B but containing only 0.002% Coomassie Blue G250). For detection of protein complexes, acrylamide gel was soaked in Tris–glycine SDS electrophoresis buffer, and proteins were transferred onto PVDF membrane and analyzed by Western blotting.
Peptide synthesis and circular dichroism spectroscopy
See Supplementary Materials and Methods.
Luciferase reporter assay
See Supplementary Materials and Methods.
Acknowledgments
We thank Bernd Bukau, Bernhard Dobberstein, and Sebastian Schuck for critical reading of the manuscript. We are grateful Lina Fleig and Laura Woodward for reagents and unpublished data. The work was supported by the Baden–Württemberg Stiftung within the Network of Aging Research (NAR, University of Heidelberg) and the Deutsche Forschungsgemeinschaft (SFB 1036, TP 12) to MKL, and a fellowship from the Boehringer Ingelheim Fonds to NSM.
Author contributions
CC and NSM designed and performed most experiments and wrote the manuscript. BH carried out experiments. DA helped with experiments and wrote the manuscript. WS carried out the CD spectroscopy. DL designed the CD spectroscopy experiment and analyzed data. MKL guided the project, designed experiments, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Bagola K, Mehnert M, Jarosch E, Sommer T. Protein dislocation from the ER. Biochim Biophys Acta. 2011;1808:925–936. doi: 10.1016/j.bbamem.2010.06.025. [DOI] [PubMed] [Google Scholar]
- Beisner DR, Langerak P, Parker AE, Dahlberg C, Otero FJ, Sutton SE, Poirot L, Barnes W, Young MA, Niessen S, Wiltshire T, Bodendorf U, Martoglio B, Cravatt B, Cooke MP. The intramembrane protease Sppl2a is required for B cell and DC development and survival via cleavage of the invariant chain. J Exp Med. 2013;210:23–30. doi: 10.1084/jem.20121072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann H, Yabas M, Short A, Miosge L, Barthel N, Teh CE, Roots CM, Bull KR, Jeelall Y, Horikawa K, Whittle B, Balakishnan B, Sjollema G, Bertram EM, Mackay F, Rimmer AJ, Cornall RJ, Field MA, Andrews TD, Goodnow CC, et al. B cell survival, surface BCR and BAFFR expression, CD74 metabolism, and CD8- dendritic cells require the intramembrane endopeptidase SPPL2A. J Exp Med. 2013;210:31–40. doi: 10.1084/jem.20121076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boname JM, Bloor S, Wandel MP, Nathan JA, Antrobus R, Dingwell KS, Thurston TL, Smith DL, Smith JC, Randow F, Lehner PJ. Cleavage by signal peptide peptidase is required for the degradation of selected tail-anchored proteins. J Cell Biol. 2014;205:847–862. doi: 10.1083/jcb.201312009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauweiler A, Lorick KL, Lee JP, Tsai YC, Chan D, Weissman AM, Drabkin HA, Gemmill RM. RING-dependent tumor suppression and G2/M arrest induced by the TRC8 hereditary kidney cancer gene. Oncogene. 2007;26:2263–2271. doi: 10.1038/sj.onc.1210017. [DOI] [PubMed] [Google Scholar]
- Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Wade Harper J, Kopito RR. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2011;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawshaw SG, Martoglio B, Meacock SL, High S. A misassembled transmembrane domain of a polytopic protein associates with signal peptide peptidase. Biochem J. 2004;384:9–17. doi: 10.1042/BJ20041216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev KK, Chatterjee S, Osinde M, Stauffer D, Morgan H, Kobialko M, Dengler U, Rueeger H, Martoglio B, Rovelli G. Signal peptide peptidase dependent cleavage of type II transmembrane substrates releases intracellular and extracellular signals. Eur J Pharmacol. 2006;540:10–17. doi: 10.1016/j.ejphar.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Dickey SW, Baker RP, Cho S, Urban S. Proteolysis inside the membrane is a rate-governed reaction not driven by substrate affinity. Cell. 2013;155:1270–1281. doi: 10.1016/j.cell.2013.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30:E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkabetz Y, Shapira I, Rabinovich E, Bar-Nun S. Distinct steps in dislocation of luminal endoplasmic reticulum-associated degradation substrates: roles of endoplamic reticulum-bound p97/Cdc48p and proteasome. J Biol Chem. 2004;279:3980–3989. doi: 10.1074/jbc.M309938200. [DOI] [PubMed] [Google Scholar]
- Ernst R, Mueller B, Ploegh HL, Schlieker C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol Cell. 2009;36:28–38. doi: 10.1016/j.molcel.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleig L, Bergbold N, Sahasrabudhe P, Geiger B, Kaltak L, Lemberg MK. Ubiquitin-dependent intramembrane rhomboid protease promotes ERAD of membrane proteins. Mol Cell. 2012;47:558–569. doi: 10.1016/j.molcel.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Greenblatt EJ, Olzmann JA, Kopito RR. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat Struct Mol Biol. 2011;18:1147–1152. doi: 10.1038/nsmb.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Garza RM. Protein quality control as a strategy for cellular regulation: lessons from ubiquitin-mediated regulation of the sterol pathway. Chem Rev. 2009;109:1561–1574. doi: 10.1021/cr800544v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci USA. 2003;100:9946–9951. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SO, Cho K, Cho S, Kim I, Oh C, Ahn K. Protein disulphide isomerase is required for signal peptide peptidase-mediated protein degradation. EMBO J. 2010;29:363–375. doi: 10.1038/emboj.2009.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK, Bland FA, Weihofen A, Braud VM, Martoglio B. Intramembrane proteolysis of signal peptides: an essential step in the generation of HLA-E epitopes. J Immunol. 2001;167:6441–6446. doi: 10.4049/jimmunol.167.11.6441. [DOI] [PubMed] [Google Scholar]
- Lemberg MK, Martoglio B. Requirements for signal peptide peptidase-catalyzed intramembrane proteolysis. Mol Cell. 2002;10:735–744. doi: 10.1016/s1097-2765(02)00655-x. [DOI] [PubMed] [Google Scholar]
- Lemberg MK. Intramembrane proteolysis in regulated protein trafficking. Traffic. 2011;12:1109–1118. doi: 10.1111/j.1600-0854.2011.01219.x. [DOI] [PubMed] [Google Scholar]
- Lemberg MK. Sampling the membrane: function of rhomboid-family proteins. Trends Cell Biol. 2013;23:210–217. doi: 10.1016/j.tcb.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler SF, Haass C, Steiner H. Regulated intramembrane proteolysis – lessons from amyloid precursor protein processing. J Neurochem. 2011;117:779–796. doi: 10.1111/j.1471-4159.2011.07248.x. [DOI] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- Lipson C, Alalouf G, Bajorek M, Rabinovich E, Atir-Lande A, Glickman M, Bar-Nun S. A proteasomal ATPase contributes to dislocation of endoplasmic reticulum-associated degradation (ERAD) substrates. J Biol Chem. 2008;283:7166–7175. doi: 10.1074/jbc.M705893200. [DOI] [PubMed] [Google Scholar]
- Lorenz H, Hailey DW, Wunder C, Lippincott-Schwartz J. The fluorescence protease protection (FPP) assay to determine protein localization and membrane topology. Nat Protoc. 2006;1:276–279. doi: 10.1038/nprot.2006.42. [DOI] [PubMed] [Google Scholar]
- Loureiro J, Lilley BN, Spooner E, Noriega V, Tortorella D, Ploegh HL. Signal peptide peptidase is required for dislocation from the endoplasmic reticulum. Nature. 2006;441:894–897. doi: 10.1038/nature04830. [DOI] [PubMed] [Google Scholar]
- Martoglio B, Hauser S, Dobberstein B. Cotranslational translocation of proteins into microsomes derived from the rough endoplasmic reticulum of mammalian cells. In: Celis JE, editor. Cell Biology: A Laboratory Handbook. San Diego: Academic Press; 1998. pp. 265–274. Vol. 2. [Google Scholar]
- McLauchlan J, Lemberg MK, Hope G, Martoglio B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 2002;21:3980–3988. doi: 10.1093/emboj/cdf414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita H, Maruyama Y, Isshiki H, Osawa S, Ogura T, Mio K, Sato C, Tomita T, Iwatsubo T. Three-dimensional structure of the signal peptide peptidase. J Biol Chem. 2011;286:26188–26197. doi: 10.1074/jbc.M111.260273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Brodsky JL, Kamura T. A stalled retrotranslocation complex reveals physical linkage between substrate recognition and proteasomal degradation during ER associated degradation. Mol Biol Cell. 2013;24:1765–1775. doi: 10.1091/mbc.E12-12-0907. S1761-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson IM, von Heijne G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J Biol Chem. 1993;268:5798–5801. [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Osenkowski P, Li H, Ye W, Li D, Aeschbach L, Fraering PC, Wolfe MS, Selkoe DJ, Li H. Cryoelectron microscopy structure of purified gamma-secretase at 12 A resolution. J Mol Biol. 2009;385:642–652. doi: 10.1016/j.jmb.2008.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, Leonhardt H. A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics. 2008;7:282–289. doi: 10.1074/mcp.M700342-MCP200. [DOI] [PubMed] [Google Scholar]
- Schneppenheim J, Dressel R, Huttl S, Lullmann-Rauch R, Engelke M, Dittmann K, Wienands J, Eskelinen EL, Hermans-Borgmeyer I, Fluhrer R, Saftig P, Schroder B. The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J Exp Med. 2013;210:41–58. doi: 10.1084/jem.20121069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrul B, Kapp K, Sinning I, Dobberstein B. Signal peptide peptidase (SPP) assembles with substrates and misfolded membrane proteins into distinct oligomeric complexes. Biochem J. 2010;427:523–534. doi: 10.1042/BJ20091005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE, III, Sudhof T, Yu G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell. 2005;122:435–447. doi: 10.1016/j.cell.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Stagg HR, Thomas M, van den Boomen D, Wiertz EJ, Drabkin HA, Gemmill RM, Lehner PJ. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J Cell Biol. 2009;186:685–692. doi: 10.1083/jcb.200906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strisovsky K, Sharpe HJ, Freeman M. Sequence-specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol Cell. 2009;36:1048–1059. doi: 10.1016/j.molcel.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh B, Iwakoshi NN, Glimcher LH, Ploegh HL. Rapid turnover of unspliced Xbp-1 as a factor that modulates the unfolded protein response. J Biol Chem. 2006;281:5852–5860. doi: 10.1074/jbc.M509061200. [DOI] [PubMed] [Google Scholar]
- Voss M, Fukumori A, Kuhn PH, Kunzel U, Klier B, Grammer G, Haug-Kroper M, Kremmer E, Lichtenthaler SF, Steiner H, Schroder B, Haass C, Fluhrer R. Foamy virus envelope protein is a substrate for signal peptide peptidase-like 3 (SPPL3) J Biol Chem. 2012;287:43401–43409. doi: 10.1074/jbc.M112.371369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li L, Ye Y. Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J Cell Biol. 2006;174:963–971. doi: 10.1083/jcb.200605100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen A, Lemberg MK, Ploegh HL, Bogyo M, Martoglio B. Release of signal peptide fragments into the cytosol requires cleavage in the transmembrane region by a protease activity that is specifically blocked by a novel cysteine protease inhibitor. J Biol Chem. 2000;275:30951–30956. doi: 10.1074/jbc.M005980200. [DOI] [PubMed] [Google Scholar]
- Weihofen A, Binns K, Lemberg MK, Ashman K, Martoglio B. Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science. 2002;296:2215–2218. doi: 10.1126/science.1070925. [DOI] [PubMed] [Google Scholar]
- Wittig I, Braun HP, Schagger H. Blue native PAGE. Nat Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- Wolfe MS. Intramembrane proteolysis. Chem Rev. 2009;109:1599–1612. doi: 10.1021/cr8004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagitani K, Imagawa Y, Iwawaki T, Hosoda A, Saito M, Kimata Y, Kohno K. Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol Cell. 2009;34:191–200. doi: 10.1016/j.molcel.2009.02.033. [DOI] [PubMed] [Google Scholar]
- Yanagitani K, Kimata Y, Kadokura H, Kohno K. Translational pausing ensures membrane targeting and cytoplasmic splicing of XBP1u mRNA. Science. 2011;331:586–589. doi: 10.1126/science.1197142. [DOI] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci USA. 2005;102:14132–14138. doi: 10.1073/pnas.0505006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Oku M, Suzuki M, Mori K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol. 2006;172:565–575. doi: 10.1083/jcb.200508145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Uemura A, Mori K. pXBP1(U), a negative regulator of the unfolded protein response activator pXBP1(S), targets ATF6 but not ATF4 in proteasome-mediated degradation. Cell Struct Funct. 2009;34:1–10. doi: 10.1247/csf.06028. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Li X, Cai MY, Ma K, Yang J, Zhou J, Fu W, Wei FZ, Wang L, Xie D, Zhu WG. XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell Res. 2013;23:491–507. doi: 10.1038/cr.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.