

Abstract
Translational GTPases are universally conserved GTP hydrolyzing enzymes, critical for fidelity and speed of ribosomal protein biosynthesis. Despite their central roles, the mechanisms of GTP-dependent conformational switching and GTP hydrolysis that govern the function of trGTPases remain poorly understood. Here, we provide biochemical and high-resolution structural evidence that eIF5B and aEF1A/EF-Tu bound to GTP or GTPγS coordinate a monovalent cation (M+) in their active site. Our data reveal that M+ ions form constitutive components of the catalytic machinery in trGTPases acting as structural cofactor to stabilize the GTP-bound “on” state. Additionally, the M+ ion provides a positive charge into the active site analogous to the arginine-finger in the Ras-RasGAP system indicating a similar role as catalytic element that stabilizes the transition state of the hydrolysis reaction. In sequence and structure, the coordination shell for the M+ ion is, with exception of eIF2γ, highly conserved among trGTPases from bacteria to human. We therefore propose a universal mechanism of M+-dependent conformational switching and GTP hydrolysis among trGTPases with important consequences for the interpretation of available biochemical and structural data.
Keywords: catalytic mechanism, crystal structure, GTPase, monovalent cation, translation
Introduction
Translation—the ribosome-catalyzed synthesis of biologically functional polypeptides according to genetically encoded information—is one of the most fundamental and complex biochemical processes in extant cellular life. In all stages of protein biosynthesis, the ribosome depends on a set of auxiliary guanine nucleotide binding (G) proteins (termed translational GTPases or trGTPases) that include initiation factor 2 (IF2), which catalyzes ribosomal subunit joining and formation of the elongation-competent ribosome, elongation factor Tu (EF-Tu), which delivers aminoacyl-tRNA (aa-tRNA) to the ribosomal A site, as well as elongation factor G (EF-G) that catalyzes the translocation of the mRNA–tRNA complex on the ribosome (Marintchev & Wagner, 2004; Voorhees & Ramakrishnan, 2013). Despite the differences in their respective functions, trGTPases from bacteria to eukarya share a common evolutionarily conserved structural core composed of the G domain and domain II, which is supplemented with additional factor specific domains. The G domain forms the functional center in all trGTPases that couples GTP binding and hydrolysis to the specific biological function of the translation factor (Marintchev & Wagner, 2004). This present work focuses on so far unresolved aspects of the universal molecular mechanisms that govern the functional cycle of the G domain in trGTPases and that are therefore central to the understanding of the process of translation as well as its evolution.
In sequence and architecture, the G domain in trGTPases is related to small Ras-like G proteins and thus belongs to the superfamily of P-loop GTPases (Bourne et al, 1991). According to the classical view, G proteins are thought to act as molecular switches that oscillate between a GDP-bound “off” state and a structurally distinct GTP-bound “on” state (Bourne et al, 1991; Vetter & Wittinghofer, 2001). The exchange of GDP for GTP is accompanied by conformational changes in two conserved dynamic elements termed switch 1 and switch 2 that form specific interactions with the GTP-γ-phosphate. Conversion back to the GDP-bound state requires the hydrolysis of GTP and release of inorganic phosphate (Pi), allowing switch 1 and 2 to relax back into their inactive conformations. These structural changes ensure that only the GTP-bound form of a G protein is able to interact productively with effector molecules to carry out its biological function (Vetter & Wittinghofer, 2001).
Despite its importance for the understanding of the translation process, the mechanism of GDP/GTP-dependent conformational switching has remained obscure for nearly all trGTPases. Over the years, high-resolution structures have become available for various trGTPases in their apo states as well as in complex with GDP and the nonhydrolyzable GTP analogs GDPNP or GDPCP (β-γ-imidoguanosine 5′-triphosphate and β-γ-methyleneguanosine 5′-triphosphate, respectively). Surprisingly, however, from these structures, a highly heterogeneous picture emerged for the mechanisms of conformational switching in the G domains of trGTPases that is at odds with their evolutionary and structural homology: While only EF-Tu exhibited clearly distinct GDP- and GDPNP-bound conformations in agreement with the classical concept (Berchtold et al, 1993; Polekhina et al, 1996), EF-G, aIF5B and eRF3 adopted virtually identical structures in their respective GDP- and GDPNP/GDPCP-bound forms, invoking suggestions about nonclassical mechanisms in these G proteins (Roll-Mecak et al, 2000; Kong et al, 2004; Hansson et al, 2005). SelB, finally, seemed to constitute an intermediate case with switch 2 undergoing conformational changes upon GDPNP binding, while switch 1 remained mainly flexible similar to the GDP-bound state (Leibundgut et al, 2005). In order to reconcile the structural data with the classical model of GTPase function, it was proposed that some of these apparently nonclassical trGTPases follow a mechanism of “conditional switching” (Hauryliuk et al, 2008a). According to this concept, the GTP alone is insufficient to induce the GTP-conformation in the G domain but requires the ribosome as additional cofactor for the efficient conformational switch to the “on” state. However, biochemical experiments as well as a recent crystal structure of eIF5B bound to GTP indicate that EF-G, SelB and eIF5B in fact do undergo significant structural rearrangements in the presence of true GTP instead of GDPNP or GDPCP, indicating that they conform to the classical model without the requirement of the ribosome or aa-tRNA for the conformational switch (Hauryliuk et al, 2008b; Paleskava et al, 2012; Kuhle & Ficner, 2014). These observations are paralleled by a large body of biochemical data, indicating that GDPNP is not an authentic GTP analog for nearly all trGTPases (Hauryliuk et al, 2006, 2008a; Wilden et al, 2006; Paleskava et al, 2010, 2012; Burnett et al, 2013) and that the use of nonhydrolyzable analogs in structural experiments most likely accounts for the apparent deviation from the classical switch mechanism (Hauryliuk et al, 2008a). Hence, it seems evident that the stable conformational switch is dependent on critical contributions by the GTP molecule that are, however, not provided by the structurally similar GDPNP/GDPCP. It is therefore a central conceptual assumption of this present work that the identification of this so far unknown contribution by the GTP molecule provides a key to a more unified view on the GDP/GTP cycle in trGTPases, in agreement with their common evolutionary descent.
Another central and yet unresolved problem in the functional cycle of trGTPases is the molecular mechanism of ribosome-induced GTP hydrolysis. Similar to other Ras-like G proteins, trGTPases possess a low intrinsic GTPase activity, which is accelerated by several orders of magnitude upon productive binding to the ribosome (Mesters et al, 1994; Rodnina et al, 2000). In analogy to the systems of the GTPase activating protein (RasGAP) of Ras or the regulators of G protein signaling (RGS) of Gα proteins, it was shown that the ribosome stimulates rapid GTP hydrolysis in trGTPases by the precise positioning of an invariant histidine (Hiscat) in switch 2 (corresponding to Gln61 in Ras) in its catalytically active conformation (Voorhees et al, 2010; Tourigny et al, 2013). However, the actual origin of the catalytic effect in the activated system has remained controversial (Voorhees et al, 2010; Adamczyk & Warshel, 2011; Liljas et al, 2011; Aleksandrov & Field, 2013; Wallin et al, 2013). That is, it remained unclear how the nucleophilic water (Wcat) is activated for its attack on the γ-phosphate and how the subsequent stabilization of the transition state (TS) is achieved. In the Ras system, RasGAPs further stimulate GTP hydrolysis by supplying an additional catalytic residue, the “arginine-finger”, into the active site, which is responsible for ˜2,000-fold acceleration of the GTPase reaction by direct electrostatic stabilization of developing negative charges in the TS (Scheffzek et al, 1997). Analogous catalytic elements provided in cis or in trans have been identified in P-loop GTPases from Gα proteins to MnmE, dynamin and the signal recognition particle (Scrima & Wittinghofer, 2006; Bos et al, 2007; Gasper et al, 2009; Chappie et al, 2010). However, up to now, trGTPases seemed to be an exception among Ras-related G proteins, as the search for an arginine-finger or an analogous catalytic element has been unsuccessful (Wieden et al, 2001; Mohr et al, 2002; Kubarenko et al, 2005; Rodnina, 2009).
Here, we discuss the unresolved questions concerning the GTP-induced conformational switch and GTP hydrolysis in trGTPases in light of the novel assumption that trGTPases utilize a monovalent cation (M+ ion) as structural and catalytic cofactor. We show that eIF5B (eukaryal IF2 ortholog) as well as aEF1A (archaeal EF-Tu ortholog) coordinate an M+ ion in their active sites in the same position as known M+-dependent GTPases, thus placing a positive charge analogous to the guanidino group of the arginine-finger in the Ras–RasGAP complex. The coordination shell for the M+ ion is, with the notable exception of eIF2γ, universally conserved among trGTPases from bacteria to humans and directly involves the oxygen atom of GTP that is replaced in GDPNP and GDPCP but not in the slowly hydrolyzable GTPγS. In combination with mutational, biochemical and isothermal titration calorimetry (ITC) data, these findings provide the conceptual framework and a significant explanatory power for the interpretation of a large body of previously unexplained observations for trGTPases, resulting in the conclusions that: (i) the M+ ion acts as structural cofactor that stabilizes the “on” state of the G domain and thus contributes to the conformational switch in trGTPases; (ii) GDPNP and GDPCP are unable to coordinate the M+ ion and thereby destabilize the GTP-conformation; (iii) GTPγS is able to coordinate the M+ ion and thus a suitable GTP analog for trGTPases; (iv) the M+ ion participates in the GTP hydrolysis reaction, most likely by stabilizing its TS; and (v) with few exceptions, M+-dependency is universal among canonical trGTPases.
Results
GTP-bound eIF5B coordinates an M+ ion in its catalytic center
Recently, we reported the crystal structure of GTP-bound eIF5B from C. thermophilum, solved at 1.9 Å resolution (protein data bank (PDB): 4NCN) (Kuhle & Ficner, 2014). This structure revealed a Na+ ion next to the GTP-γ-phosphate in a catalytically relevant position of the active site, which has so far never been reported for a trGTPase (Fig 1A and Supplementary Fig S1A). In line with previously reported values (Harding, 2002), this Na+ ion is penta-coordinated with coordination distances between 2.3 and 2.5 Å by two oxygens from the α- and γ-phosphates, the β-γ-bridging oxygen, the carboxylate group of Asp533 in the P-loop (which we denote AspMC for aspartate involved in monovalent cation-binding) and the backbone CO from Gly555 in switch 1 (GlyMC). The latter is part of a short peptide backbone excursion of switch 1 (formed by Gly554 and Gly555) which approaches the Na+ ion opposite to the β-γ-bridging oxygen and is denoted “MC-loop” (Fig 1).
Figure 1. GTP-bound eIF5B coordinates an M+ ion in its GTPase center.

- Overview of G domain and domain II of GTP-bound eIF5B with a Na+ ion (purple sphere) in the active site. P-loop and switch regions are shown in cyan; GTP is shown as balls and sticks. The Na+ ion is bound by a pentameric coordination sphere (inset; indicated by purple lines) formed by GTP, AspMC and GlyMC. Mg2+ and water molecules are shown as spheres in light brown and gray, respectively; the catalytic histidine (His480) and highly conserved residues that interact with GTP or the cations are indicated; hydrogen bonds are shown as dashed lines.
- The active site of eIF5B·GTPγS with a K+ ion (yellow sphere) bound in a heptameric coordination sphere.
- Superposition of eIF5B·GTPγS structures with either Na+ (purple sphere) or K+ (yellow sphere) bound in the active site. Due to the shorter coordination distances to the Na+ ion, AspMC and GlyMC (purple sticks) are drawn closer to the GTP molecule than in the K+-structure (yellow sticks).
- Superposition of eIF5B·GTPγS·K+ and MnmE (purple; PDB: 2GJ8) bound to GDP, AlF4 and K+.
GTPγS but not GDPNP supports crystallization of eIF5B in its GTP-bound conformation
The position of the M+ ion in eIF5B·GTP suggests that it provides a direct contribution to the stabilization of the GTP-bound switch 1 by forming a stable link to the GTP molecule (Fig 1A). To test the relevance of this contribution, crystallization trials were performed with the construct cteIF5B(517–858) in the presence of GTP, GDPNP and GTPγS. If the M+ is relevant for the conformational switch in eIF5B, GTP and GTPγS but not GDPNP, in which the β-γ-bridging oxygen is replaced by an NH group (Fig 2A), should readily stabilize the GTP-conformation and allow crystallization of cteIF5B(517–858) in an optimization screen for the original crystallization condition [0.1 M HEPES/NaOH (pH 7); 13% PEG 4000; 0.1 M NaOAc] (see Supplementary Materials and Methods for details).
Figure 2. Biochemical characterization of nucleotide binding and GTP hydrolysis by eIF5B.

A GDPNP disrupts the coordination sphere for the M+ ion (blue sphere) formed by AspMC, GlyMC and three oxygens from GTP by replacing the β-γ-bridging oxygen (O) with an NH group.
B Temperature dependency of binding enthalpy changes (ΔH) upon eIF5B(517–858) interaction with GDP (•), GDPNP (○) or GTPγS (▾) measured by ITC.
C Intrinsic GTPase activity of eIF5B(517–858) [wild-type (wt) or the D533A mutant] determined at 35°C in the presence of 200 mM of the indicated salts, 25 μM protein and 300 μM GTP and subsequent analysis by HPLC. The order in which the combinations are given on the right corresponds to the relative rates of GTP hydrolysis.
D Intrinsic GTPase activity of eIF5B(517–858) wild-type (black), D533A (red), D533N (green) and D533R (green) in the presence of NaCl under conditions as in (C).
E, F Dependency of the intrinsic GTPase activity of eIF5B(517–858) wild-type (•), D533A (○) and D533R (▾) on the concentration of KCl (E) or NaCl (F).
Data information: Experiments were repeated two to three times; standard deviations are given by error bars (in some cases not visible because they are smaller than the symbol size).
In line with our assumption, eIF5B(517–858)·GTP and eIF5B(517–858)·GTPγS readily crystallized, while no crystals grew in the presence of GDPNP. The structure of eIF5B(517–858)·GTP, solved at 1.55 Å resolution, is very similar to that reported previously (Kuhle & Ficner, 2014) (see Table1 for details of data collection and refinement). The electron densities for both Na+ ions in the asymmetric unit are well defined with occupancies of 100%.
Table 1.
Crystallization, X-ray data collection and refinement statistics for structures of wild-type elF5B(517–858)
| elF5B(517–858)·GTP·Na+ | elF5B(517–858)·GTPγS·Na+ | elF5B(517–858)·GTPγS·K+ | |
|---|---|---|---|
| Crystallization | |||
| Condition | 100 mM HEPES (pH 7.25), 12% PEG 4000, 100 mM NaOAc | 100 mM HEPES (pH 7), 13% PEG 4000, 125 mM NaOAc | 11% PEG 8000, 6% glycerol, 50 mM KCI |
| Temperature (°C) | 20 | 20 | 20 |
| Data collection | |||
| Space group | P21 | P21 | P41212 |
| Unit cell | a = 55.6 Å | a = 55.6 Å | a = 116.1 Å |
| b = 116.5 Å | b = 115.9 Å | b = 116.1 Å | |
| c = 66.2 Å | c = 66.1 Å | c = 120.3 Å | |
| α = 90° | α = 90° | α = 90° | |
| β = 101.1° | β = 101 4° | β = 90° | |
| γ = 90° | γ = 90° | γ = 90° | |
| Molecules/asym. unit | 2 | 2 | 2 |
| Resolution (Å) | 1.55 (1.64–1.55) | 1.53 (1.63–1.53) | 2.28 (2.42–2.28) |
| Observed reflections | 405,616 (64,352) | 510,862 (86,738) | 307,464 (49,685) |
| Unique reflections | 118,431 (18,849) | 121,606 (20,942) | 38,004 (6,021) |
| Completeness (%) | 98.8 (97.7) | 98.5 (98.4) | 99.9 (99.7) |
| <I>/σ | 15.5 (2.3) | 18.8 (2.8) | 19.3 (3.8) |
| Rsym (%) | 4.6 (50.5) | 3.7 (49.6) | 6.3 (54.2) |
| CC (1/2) (%) | 99.9 (75.1) | 99.9 (82.2) | 99.9 (99.1) |
| Refinement | |||
| Rwork (%) | 15.7 | 16.3 | 20.0 |
| Rfree (%) | 18.4 | 18.5 | 23.9 |
| Rmsd from standard stereochemistry | |||
| Bond length (Å) | 0.019 | 0.02 | 0.005 |
| Bond angles (°) | 1.8 | 1.9 | 1.0 |
| Ramachandran plot statistics | |||
| Most favored (%) | 96.9 | 98.1 | 98.5 |
| Allowed regions (%) | 1.1 | 1.9 | 1.5 |
| Disallowed regions (%) | 0 | 0 | 0 |
Values in parentheses refer to the highest resolution shell.
Rwork and Rfree factors are calculated using the formula 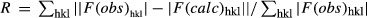 , where f(obs)hkl and f(calc)hkl are observed and measured structure factors, respectively. Rwork and Rfree differ in the set of reflections they are calculated from: Rfree is calculated for the test set, whereas Rwork is calculated for the working set.
, where f(obs)hkl and f(calc)hkl are observed and measured structure factors, respectively. Rwork and Rfree differ in the set of reflections they are calculated from: Rfree is calculated for the test set, whereas Rwork is calculated for the working set.
The structure of eIF5B(517–858)·GTPγS was solved at 1.53 Å resolution, exhibiting an overall structure and nucleotide binding virtually identical to that of the GTP-bound form (Supplementary Fig S1B and D). The sulfur atom of the γ-phosphate points outward and thus replaces the nonbridging oxygen atom that is involved in the coordination of the M+ ion in eIF5B·GTP. Nonetheless, both eIF5B·GTPγS complexes in the asymmetric unit contain the Na+ ion in the pentameric coordination shell with coordination distances of 2.2–2.6 Å to the four oxygen ligands from the GTP molecule, AspMC and GlyMC, and 2.83 Å to the sulfur atom [in agreement with the theoretical distance of 2.8 Å, calculated from the van der Waals radius of sulfur (1.8 Å) and the effective ion radius of Na+ (1.0 Å for coordination number 5) (Bondi, 1964; Shannon, 1976)].
These results demonstrate that GTPγS is a suitable structural GTP analog that is able to coordinate the M+ ion and to stabilize the GTP-conformation in eIF5B. In contrast, GDPNP seems unable to stabilize efficiently the same conformation (and thus to provide the same surfaces for crystal contacts) as GTP, resulting in the inability of the eIF5B·GDPNP complex to crystallize under the same conditions as eIF5B·GTP. The likely explanation for this observation is that, while GTP and GTPγS provide all the ligands required for the coordination of the Na+ ion and thus allow the stable association of switch 1, GDPNP is unable to provide the M+ ion as structural cofactor due to the replacement of the β-γ-bridging oxygen of GTP with a β-γ-bridging imido (NH) group (Fig 2A).
GTPγS but not GDPNP is able to substitute for GTP to induce the conformational switch of eIF5B in solution
GTP binding to apo eIF5B induces a substantial rearrangement of the switch regions, involving an approximately 180° flip of switch 1 and the burial of ˜1,800 Å2 of solvent accessible surface area (ASA) within G domain and domain II, accompanied by significant changes in heat capacity (ΔCp = −553 cal mol−1 K−1) (Kuhle & Ficner, 2014). Our structural data argue for a scenario in which the M+ ion provides a direct contribution as structural cofactor to stabilize the “on” state of eIF5B. In order to test this assumption for free eIF5B, we probed the conformational changes in cteIF5B(517–858) upon binding of GDP, GTPγS or GDPNP using ITC under conditions reported earlier for GTP binding (Kuhle & Ficner, 2014) (Supplementary Table S1, Fig 2B and Supplementary Fig S2).
The affinities of GDP, GTPγS and GDPNP to cteIF5B(517–858) were measured at different temperatures between 10 and 30°C. At 30°C, GDP binds to cteIF5B(517–858) with an equilibrium dissociation constant (Kd) of 10.4 μM, 2.5-fold weaker than GTP (4.1 μM). GDPNP binds with a Kd of 20.8 μM (at 30°C), approximately 5-fold weaker than GTP. GTPγS has an about 4-fold higher affinity to cteIF5B(517–858) (0.92 μM at 30°C) than GTP. Comparable values were reported from fluorescence experiments with mammalian eIF5B and mant-nucleotides (Pisareva et al, 2007).
The interactions of cteIF5B(517–858) with GTPγS, GDPNP and GDP result in significant exothermic heat effects (ΔH = −15.8, −9.7 and −11.5 kcal mol−1, respectively, at 30°C). As for GTP, their binding is driven by favorable changes in binding enthalpy and opposed by unfavorable entropic contributions (TΔS = −7.4, −3.2 and −4.6 kcal mol−1 for GTPγS, GDPNP and GDP at 30°C, respectively) (Supplementary Table S1).
In the temperature range between 10 and 30°C, ΔH is temperature dependent. When ΔH was plotted against the temperature, straight lines were obtained with the slopes representing the changes in heat capacity (ΔCp) upon complex formation (Table2 and Fig 2B) (Jelesarov & Bosshard, 1999). GTPγS causes a ΔCp of −539 cal mol−1 K−1, very similar to the value observed for GTP (−553 cal mol−1 K−1) (Kuhle & Ficner, 2014). Significantly smaller changes in heat capacity were obtained for GDPNP and GDP binding (ΔCp = −197 cal mol−1 K−1 and −228 cal mol−1 K−1, respectively). ΔCp can be used as an estimate for the change in solvent accessible surface area (ΔASA) upon complex formation. The burial of surface area was shown to be associated with a negative change in heat capacity, with ΔCp being proportional to the size of the surface area involved in the ligand binding process (Spolar & Record, 1994; Gomez et al, 1995; Perozzo et al, 2004). According to this correlation, GTPγS binding induces major structural rearrangements in cteIF5B(517–858) similar to those observed for GTP binding (Table2), suggesting nearly identical structures for cteIF5B(517–858)·GTP and cteIF5B(517–858)·GTPγS in solution. In contrast, GDPNP induces significantly smaller changes which are similar to those for GDP (Table2), indicating a GDP-like conformation for the cteIF5B(517–858)·GDPNP complex. These results are in line with the observations from the crystal structures of GTP-, GTPγS- and GDP-bound eIF5B and GDPNP-bound aIF5B (Roll-Mecak et al, 2000), as well as the assumption of a specific inability of GDPNP to efficiently stabilize the conformational switch of the G domain by repulsion of the M+ ion (Fig 2A).
Table 2.
Changes in heat capacity and solvent accessible surface area for cteIF5B(517–858) binding to GTP, GTPγS, GDPNP and GDP
| Ligand | ΔCp [cal mol−1 K−1] | ΔASAmin [Å2] (with Δcmin = 0.45) | ΔASAmax [Å2] (with Δcmax = 0.24) |
|---|---|---|---|
| GTP | −553 ± 11 | 1,229 | 2,304 |
| GTPγS | −539 ± 30 | 1,198 | 2,254 |
| GDPNP | −197 ± 26 | 433 | 820 |
| GDP | −228 ± 10 | 507 | 950 |
ΔCp, heat capacity change; obtained from ΔH/dT.
Δcmin and Δcmax area coefficients in cal K−1 (mol Å2)−1 for calculation of ΔASA.
ΔASAmin and ΔASAmax, changes in solvent accessible surface areas assuming that all changes were conferred by either apolar or 70% apolar and 30% polar surfaces, respectively.
Obtained from (Kuhle & Ficner, 2014).
Obtained from the slope of the linear fit to ΔH measured at different temperatures between 10 and 30°C.
The structure of eIF5B bound to GTPγS and potassium
Most M+-dependent enzymes show a preference for potassium (K+) over other M+ ions as cofactor. A preference for K+ is known also for the translation apparatus in general and the function of trGTPases in particular (Conway, 1964; Conway & Lipmann, 1964; Parmeggiani & Sander, 1981; Fasano et al, 1982). We therefore assumed that K+ is able to substitute for Na+ as cofactor in the GTP-bound form of eIF5B. Consistently, we were able to obtain crystals of eIF5B(517–858)·GTPγS in space group P41212 that grew within 2 weeks at 20°C under a condition containing 50 mM KCl (Table1). The structure was solved at 2.28 Å resolution and contains two eIF5B·GTPγS complexes in the asymmetric unit. The two water molecules in the coordination shell of the Mg2+ ion and Wcat are weakly defined in the electron density, with the latter lying 3.1–3.2 Å from the outward pointing sulfur atom of GTPγS. On the other side of the sulfur atom, opposite to Wcat, a strong electron density peak was observed that was assigned to a K+ ion with a coordination shell nearly identical to that of the Na+ ion in eIF5B·GTPγS (Fig 1B and Supplementary Fig S1C). However, additional weak densities were observed on the solvent-exposed side of the cation, suggesting that two water molecules contribute to a heptameric coordination shell. As expected for a K+ ion (Harding, 2002), the coordination distances to most oxygen ligands lie between 2.7 and 3.0 Å and are thus clearly different from those observed for the Na+ ions. The distance to the β-γ-bridging oxygen is slightly increased (˜3.47 Å), most likely due to the large sulfur atom that coordinates the K+ ion at a distance of 3.2 Å [in good agreement with the theoretical distance of 3.26 Å, calculated from the van der Waals radius of sulfur (1.8 Å) and effective ion radius of K+ (1.46 Å for coordination number 7) (Bondi, 1964; Shannon, 1976)].
Despite the different crystallization conditions and a different set of crystal contacts, the overall structure of eIF5B·GTPγS bound to K+ is nearly identical to that of eIF5B·GTP/GTPγS bound to Na+ with the switch regions stabilized in their typical GTP-conformation. Significant differences are limited to GlyMC and the MC-loop of switch 1 which is moved approximately 2 Å away from the guanine nucleotide, owing to the increased coordination distances to the K+ ion (Fig 1C). This demonstrates that the position of the MC-loop in switch 1 is directly influenced by the species of the M+ ions coordinated next to the nucleotide, indicating a direct contribution of the M+ ion to the stabilization of the activated switch 1 conformation.
eIF5B coordinates the M+ ion in the same position as known M+-dependent GTPases
Superposition of the eIF5B·GTP/GTPγS structures with MnmE and dynamin reveals that the K+ and Na+ ions in eIF5B are coordinated in the same position as the catalytic K+ and Na+ ions in the presence of GDP-AlFx in the two known M+-dependent GTPases (Scrima & Wittinghofer, 2006; Chappie et al, 2010) (Fig 1D and Supplementary Fig S1E). AspMC replaces an identically positioned and conserved Asn or Ser, and the backbone CO of GlyMC replaces two backbone COs from the “K-loop” in switch 1 which circles around the position of the MC-loop of eIF5B. The contacts formed between the K+ and Na+ ions and GTP in eIF5B are virtually identical to those of the K+ ion in MnmE or the Na+ ion in dynamin with the transition state mimic GDP-AlFx, where a β-phosphate oxygen of GDP represents the former β-γ-bridging oxygen and the outward pointing nonbridging γ-phosphate oxygen is mimicked by a fluoride ion.
The structural elements required for M+ coordination in eIF5B are highly conserved among trGTPases
The G domains of trGTPases are highly conserved in sequence and structure. In order to investigate whether M+ ion binding may be a common characteristic among trGTPases, we analyzed sequences and available structures with regard to key features involved in M+ ion binding in eIF5B. The key determinants for M+ ion binding are AspMC in the P-loop and GlyMC in the MC-loop of switch 1. Sequence analysis reveals that both residues, AspMC and GlyMC, are universally conserved among orthologs of eIF5B, EF-Tu, SelB, aIF2γ, eRF3, EF-G, RF3 and LepA from bacteria to human (Fig 3A). The only notable exception among trGTPases is eIF2γ, where AspMC and GlyMC are replaced by Ala and Asn, respectively.
Figure 3. Structural elements required for M+-coordination in eIF5B are universally conserved among trGTPases.

- Excerpt of a multiple sequence alignment of different trGTPases (orthologs of RF3, eIF5B, eIF2γ, SelB, eRF3, EF-Tu, LepA, EF-G) showing P-loop and switch 1. The upper and lower numbering corresponds to C. thermophilum eIF5B (Cthe) and Escherichia coli EF-Tu (Ecoli), respectively. Highly conserved residues are highlighted in dark blue, conserved residues in light blue. AspMC and GlyMC are highlighted in red; residues in eIF2γ that replace AspMC and GlyMC are highlighted in yellow.
- Superposition of eIF5B·GTPγS·K+ (colored as in Fig 1) with ribosome-bound EF-G·GDPCP (brown; PDB: 4JUW). Ribosome-bound EF-G provides all structural elements to bind the M+ ion; however, its coordination is prevented by the CH2 group of GDPCP in lieu of the β-γ-bridging oxygen (arrow). A water molecule (red sphere) is bound next to the M+-binding site instead.
- Similarly, EF-Tu·GDPNP (purple; PDB: 2C78) provides all structural elements to bind the M+ ion; however, its coordination is prevented by the NH group of GDPNP (arrow). A water molecule (red sphere) is bound next to the M+ binding site instead.
Structurally, the P-loop is well conserved among trGTPases. In contrast, switch 1 shows a high degree of variability in sequence and structure, which is, however, limited to regions lying N-terminally of the MC-loop. In a superposition of eIF5B·GTP with EF-Tu·GDPNP or the ribosome-bound EF-G·GDPCP (Fig 3B and C), the nucleotide binding motifs occupy virtually identical positions including all residues directly involved in the coordination of the nucleotide and Mg2+ ion. Importantly, the structural homology extends to AspMC as well as GlyMC, the latter of which invariably forms part of the characteristic MC-loop excursion of switch 1 at the end of helix A″ in EF-Tu and EF-G, placing its carbonyl group in the correct position to coordinate the cation.
GTP-bound aEF1A coordinates an M+ ion in its catalytic center
Most known structures of trGTPases that were reported to be in the GTP-conformation do not contain GTP but GDPNP or GDPCP that contain either an NH or a CH2 group in lieu of the β-γ-bridging oxygen. Both prevent the coordination of the M+ ion as observed in structures of known M+-dependent GTPases (Ash et al, 2011; Chappie et al, 2011) (Fig 2A and Supplementary Fig S4). We therefore searched the PDB for structures of trGTPases that were co-crystallized with GTP and found two structures, both of the archaeal EF-Tu ortholog aEF1A either in complex with pelota (PDB: 3AGJ) or with release factor RF1 (PDB: 3VMF).
Crystals of the aEF1A/pelota complex contained four aEF1A molecules (chains A, C, E, G) per asymmetric unit, each bound to GTP. The structure had been determined at 2.3 Å resolution (Kobayashi et al, 2010). Each of the aEF1A molecules contains a water molecule modeled in the position occupied by the M+ ion in eIF5B·GTP. Accordingly, the supposed water molecules are coordinated by five hydrogen bond acceptors with the sphere formed by the outward pointing α- and γ-phosphate oxygens and the β-γ-bridging oxygen of GTP, Asp16 in the P-loop (AspMC) and the CO of Gly69 (GlyMC) in switch 1. The MC-loop lies at the end of helix A″ in switch 1 and adopts the same conformation found in eIF5B (Figs1A and 4A and B). The coordination distances of the supposed water molecules lie between 2.2 and 2.7 Å with an overall average of 2.45 Å. Thus, the coordination pattern as well as the distances is in better agreement with those expected for a Na+ ion than for water (Harding, 2002). Reevaluation of the experimental X-ray diffraction data [downloaded from the PDB (3AGJ)] revealed positive difference electron density for the supposed water molecules in aEF1A molecules C and E in the asymmetric unit, indicating a higher density of electrons in these positions than provided by H2O (Supplementary Fig S3A). Replacement of the four water molecules by Na+ and subsequent refinement results in occupancies of 92 and 94% for the Na+ ions in molecules A and G, respectively, and 100% in molecules C and E. Since the crystals used for structure analysis were grown in the presence of NaCl (Kobayashi et al, 2010), these observations indicate that the supposed water molecules next to Asp16 of aEF1A are most likely Na+ ions (Fig 4A and Supplementary Fig S3C).
Figure 4. The GTP-bound EF-Tu ortholog aEF1A coordinates an M+ ion in its GTPase center.

- Overview of domains I (G) to III of GTP-bound aEF1A with a Na+ ion (purple sphere) in the active site. P-loop and switch regions are shown in yellow. The inset shows a detailed view on the active site with the coordination sphere of the Na+ ion (indicated by purple lines) formed by GTP, AspMC and GlyMC in the MC-loop.
- Superposition of GTP-bound aEF1A with either a Na+ (purple sphere) or NH4+ ion (blue sphere) in the active site. Both ions are coordinated by the identical sphere, however, with different coordination distances.
- Superposition of EF-Tu·GDPNP (switch 1 in red; PDB: 1EXM) with EF-Tu·GDPNP in the ternary complex (TC) with aa-tRNA (blue; PDBs: 1TTT, 1OB2) and aEF1A·GTP·M+ (M+ in blue; switch 1 in yellow). The M+ ion stabilizes a conformation of helix A″ that is required for stable TC formation.
- Dependency of the intrinsic GTPase activity of E. coli EF-Tu wild-type (•) or the D21A mutant (○) on the concentration of K+ ions, determined in the presence of 0–1 M KCl under single turnover conditions (20 μM GTP-bound EF-Tu) at 30°C and subsequent analysis by HPLC.
- Intrinsic GTPase activity of E. coli EF-Tu determined in the presence of 200 mM of the indicated salts under single turnover conditions. The order in which the combinations are given on the right corresponds to the relative rates of GTP hydrolysis.
Data information: Experiments were repeated two to three times; standard deviations are given by error bars (in some cases not visible because they are smaller than the symbol size).
The structure of the aEF1A/aRF1 complex was solved at 2.3 Å resolution and contains one copy of GTP-bound aEF1A per asymmetric unit (Kobayashi et al, 2012). This structure as well has been built with a water molecule in the pentameric coordination sphere described above. Its contact distances lie between 2.6 and 3.1 Å [2.7–3.0 Å after re-evaluation of the experimental data, downloaded from the PDB (3VMF)], which are typical for the coordination of a K+ ion (Harding, 2002). However, the electron density in this position does not correspond to K+. This observation is supported by the fact that the crystallization condition did not contain K+ but instead 200 mM of NH4+ ions (Kobayashi et al, 2012). NH4+ has a similar ion radius as K+ (1.46 and 1.38 Å, respectively) and was shown to substitute for the latter structurally and functionally in the K+-dependent GTPase MnmE (Scrima & Wittinghofer, 2006). Thus, the properties of the observed coordination point toward an NH4+ ion lying in the same position as the K+ and Na+ ions in eIF5B·GTP and the aEF1A·GTP/pelota complex (Fig 4B and Supplementary Fig S3D).
The intrinsic GTPase activity of eIF5B depends on monovalent cations
The position of the observed M+ ion in eIF5B suggests a direct involvement in the catalysis of GTP hydrolysis in analogy to MnmE and dynamin. To test this possibility, we monitored the intrinsic GTPase activity of either wild-type eIF5B(517–858) or various AspMC mutants (D533A, D533R and D533N) under different salt conditions (Fig 2C–F and Table3).
Table 3.
Intrinsic GTPase activities of wt and mutant ctelF5B(517–858) and ecEF-Tu
| Protein | Construct | KCl (mM) | k (min−1) | Fold reduction compared to wild-type at 200 mM KCl |
|---|---|---|---|---|
| ctelF5B | wt | 200 | 0.023 ± 0.0015 | |
| wt | 0 | 0.0021 ± 0.001 | 11 | |
| D533A | 200 | 0.0018 ± 0.0005 | 13 | |
| D533A | 0 | 0.0016 ± 0.0008 | 14 | |
| D533N | 200 | 0.017 ± 0.002 | 1.4 | |
| D533R | 200 | 0.0028 ± 0.0007 | 8 | |
| ecEF-Tu | wt | 200 | 0.026 ± 0.0005 | |
| wt | 0 | 0.003 ± 0.0002 | 9 | |
| D21A | 200 | 0.0035 ± 0.0008 | 7 | |
| D21A | 0 | 0.0041 ± 0.0001 | 6 |
Measurements were performed two to three times.
k, rate of GTP hydrolysis under the used experimental conditions.
Measured under multiple turnover conditions at 35°C.
Measured under single turnover conditions at 30°C.
First, we tested whether GTPase stimulation in eIF5B depends on the species of the available M+ ion (at 200 mM) (Fig 2C). The experiments revealed that the GTPase activity is stimulated most in the presence of Na+ and K+, followed by Rb+, Li+ and finally Cs+ and NH4+ with the lowest degree of activation. Importantly, this dependency was lost for the eIF5B mutant carrying Ala in lieu of AspMC. In the presence of 200 mM K+ or Na+, wild-type eIF5B catalyzed GTP hydrolysis at rates of 0.023 and 0.027 min−1, respectively. The Ala mutant of AspMC exhibited significantly reduced rates of 0.0018 min−1 in K+ or Na+, corresponding to a 13- to 15-fold reduction of the intrinsic GTPase activity. Similar results were obtained for the substitution of AspMC by Arg (D533R) (Table3). By contrast, an Asn mutant retained the ability to catalyze GTP hydrolysis nearly at wild-type rates (Fig 2D), in line with the assumption that an Asn but not an Ala or Arg residue would retain the ability to coordinate the M+ ion. This is corroborated by crystal structures of the respective mutants in complex with GTP (or GTPγS), which reveal that D533N contains the M+ ion in its active site, whereas the cation is not present in D533R and D533A and exchanged for a water molecule in the latter (Supplementary Fig S5). Moreover, fluorescence measurements with mant-labeled GTP show that all three mutants bind the nucleotide with a Kd comparable to that of the wild-type protein, speaking against the possibility that the observed effects on the GTPase activity are due to reduced affinities to the substrate (Supplementary Fig S5E–H).
Next, we studied the effect of increasing Na+/K+ ion concentrations (Fig 2E and F). In the absence of M+ ions (∼600 μM Na+ were added with the GTP), eIF5B hydrolyzed GTP with a rate of 0.002 min−1. This rate successively increased with increasing concentrations of K+ or Na+ ions, resulting in an 11- to 12-fold rate enhancement at 200 mM salt (Table3). By contrast, the rate of GTP hydrolysis in the D533A mutant was found to be insensitive to the salt concentration with invariably low rates in the absence or presence of M+ ions, whereas D533R exhibited a slightly increased GTPase activity in the absence of M+ ions (0.005 min−1), which successively decreased with increasing salt concentrations (Fig 2F).
The intrinsic GTPase activity of EF-Tu depends on monovalent cations
The GTPase center of bacterial EF-Tu is virtually identical to that of its archaeal ortholog and therefore most likely coordinates an identically positioned M+ ion in its GTP-bound form (Supplementary Fig S3B). To test the generality of our assumption of M+-dependency in trGTPases, we studied the influence of M+ ions on the intrinsic GTPase activity of E. coli EF-Tu by monitoring GTP hydrolysis at different K+ concentrations (Fig 4D). As observed for eIF5B, the rate of GTP hydrolysis successively increased with increasing concentrations of K+, corresponding to a nearly tenfold rate enhancement for the intrinsic GTPase reaction at 200 mM (Table3). This dependency is lost when Asp21 (AspMC) in EF-Tu is mutated to Ala (D21A), consistent with a role of AspMC as key ligand for the coordination of an M+ ion in EF-Tu at physiological salt concentration as observed for GTP-bound aEF1A (see above; Fig 4A and B).
In order to identify the preferences of EF-Tu for GTPase stimulation, we analyzed the effect of different alkali salts (Li+, Na+, K+, Rb+, Cs+) or NH4+ (each at 200 mM) on the intrinsic hydrolysis rates (Fig 4E). The strongest stimulating effect was observed for NH4+ and K+. Na+, Li+ and Rb+ ions showed only a slight activation; virtually no effect was observed for the large Cs+ ion. Importantly, none of the tested M+ ion species show a significant stimulating effect on the D21A mutant of EF-Tu, which, moreover, shows no preference for NH4+ or K+ as observed for the wild-type protein (Supplementary Fig S6).
Discussion
M+-dependent conformational switching of eIF5B
Elucidation of the structural dynamics—that is the conformational switch—of the G domain in response to GDP/GTP exchange and GTP hydrolysis is one of the central problems to understand the functional cycle of GTPases. Here, we identified a monovalent cation (Na+/K+) as structural cofactor in the active site of GTP-bound eIF5B, which stabilizes the GTP-dependent reorganization of its G domain (Figs1 and 5). The M+ ion is bound in a highly conserved coordination shell formed by two nonbridging α- and γ-phosphate oxygens and the β-γ-bridging oxygen of the GTP molecule, AspMC in the P-loop and GlyMC in the MC-loop of switch 1. P-loop and GTP molecule thus form a stable socket for the M+ ion, which in turn constitutes an anchor point for the reorganized switch 1 in the “on” state. Structural and thermodynamic data demonstrate that GTPγS is a faithful replacement for GTP that coordinates the M+ ion and is thus able to stabilize the GTP-bound “on” state of eIF5B (Fig 1 and Supplementary Fig S4A). In contrast, replacement of GTP with GDPNP disrupts the coordination shell, resulting in the inability of the analog to stabilize the GTP-conformation in eIF5B through the M+ ion (Figs2A and B and 5A). As a consequence, the equilibrium between “on” state and GDP-like “off” state is shifted toward the latter in eIF5B·GDPNP allowing the GDPNP-bound factor to crystallize in the GDP-conformation, as observed for aIF5B·GDPNP from M. thermoautotrophicum (Roll-Mecak et al, 2000) (Fig 5A and Supplementary Fig S4A).
Figure 5. The mechanism of M+-dependent conformational switching in trGTPases.

- The nucleotide-dependent conformational switch in the G domain of a/eIF5B. In eIF5B·GDP, switch 1 and 2 are oriented away from the nucleotide binding pocket. Upon exchange of GDP for GTP, the switch regions undergo a large conformational rearrangement that results in direct contacts with the γ-phosphate. Here, the M+ ion (blue sphere) provides a direct contribution to the stabilization of switch 1 (inset). This contribution is missing in aIF5B·GDPNP (PDB: 1G7T), allowing it to crystallize in the GDP-like “off” state conformation.
- Schematic presentation of the M+-dependent conformational switch mechanism in trGTPases.
This interpretation is supported by the parallels between our observations and previous reports on M+-dependent GTPases, distantly related to trGTPases. In MnmE, an identically positioned K+ ion (Fig 1D) is required for the rearrangement of switch 1 and subsequent dimerization (Scrima & Wittinghofer, 2006). K+-dependent dimerization in MnmE was found to be induced by GTP and GTPγS, while GDPNP and GDPCP failed to support the stable switching of the G domain in biochemical as well as structural experiments (Scrima & Wittinghofer, 2006; Meyer et al, 2009). Similarly, GDPCP was found to be unable to induce stable G domain dimerization in dynamin or the conformational switch in EngA GD2, with structural changes limited to a shift of switch 2 (Chappie et al, 2010, 2011; Foucher et al, 2012). Moreover, FeoB was crystallized in the presence of GDP-AlFx with switch 1 stabilized in the “on” state by a K+ ion (Ash et al, 2011), whereas the GDPNP-bound structures either remained in the “off” state (2WIC) or did switch to the “on” state containing either a water molecule or nothing at all in place of the M+ ion (3B1X; 3LX5). These findings support the idea that GDPNP and GDPCP are incompatible with the coordination of an M+ ion and thereby destabilize the GTP-conformation of M+-dependent GTPases as observed for a/eIF5B.
Based on the presented data, we propose an M+-dependent conformational switch mechanism for eIF5B in solution (Fig 5) similar to that in MnmE (Scrima & Wittinghofer, 2006), where the M+ ion in tandem with Mg2+ acts as structural cofactor that supports binding of the GTP molecule and helps to reorganize and close the active site around the substrate. The M+ ion would thus form a constitutive component in the preorganized active site, required to stabilize the “on” state conformation of the trGTPase for productive interactions with the ribosome.
Universality of M+-dependent conformational switching among trGTPases
Like for eIF5B, the mechanism of nucleotide-dependent conformational switching has remained obscure for most other trGTPases. Crystal structures of isolated EF-G and eRF3 reveal essentially identical conformations for their respective apo, GDP- and GDPNP-bound forms (Kong et al, 2004; Hansson et al, 2005) (Supplementary Fig S4). In SelB, the canonical GDPNP-induced structural rearrangements are limited to switch 2, while switch 1 is only partially reorganized and remains mainly flexible (Leibundgut et al, 2005) (Supplementary Fig S4C). In the crystal structure, GDPNP is bound by SelB in the canonical way and provides the Mg2+ ion as well as its coordinating water molecules that usually contribute to the association of switch 1. However, although Thr46 (Thr557 in cteIF5B) interacts with Mg2+ in the canonical way, the preceding residues starting with Ile45 and Gly44 (GlyMC in cteIF5B) remain flexible together with the rest of switch 1 (Leibundgut et al, 2005). This observation cannot be explained by the loss of the hydrogen bond between the β-γ-bridging atom of the nucleotide and the backbone NH of the P-loop, but can be readily explained by the loss of the M+ ion as structural cofactor, required to stabilize Gly44 (GlyMC) and thereby the rest of switch 1 in the “on” state. This is corroborated by ITC experiments with SelB, showing that GDP and GDPNP binding result in similar structures for SelB·GDP and SelB·GDPNP, whereas GTP and GTPγS induce substantial structural rearrangements (Paleskava et al, 2012). Similarly, ITC experiments with EF-G indicate large differences between the GTP-bound and apo conformation (Hauryliuk et al, 2008b). In both cases, these results were interpreted as the GTP-induced burial of surface areas by switch 1 and 2 that, however, do not become apparent from the crystal structures.
The parallels to the observations discussed above for a/eIF5B are obvious. It is moreover evident from sequence comparison that nearly all trGTPases, including EF-Tu, EF-G, SelB, and eRF3, contain the highly conserved AspMC and GlyMC and would thus be able to provide the coordination shell for the M+ ion (Fig 3). The loss of the M+ ion as structural cofactor between nucleotide and switch 1 due to the use of GDPNP or GDPCP therefore provides a general explanation for the observations for EF-G, SelB and eRF3, as well as other previously reported discrepancies in the behavior of trGTPases toward GTP or its nonhydrolyzable analogs (Delaria et al, 1991; Hauryliuk et al, 2006; Wilden et al, 2006; Paleskava et al, 2010, 2012; Burnett et al, 2013). The common evolutionary origin of trGTPases and their functional homology during the translation process argue in favor of a systematic origin rather than individual reasons why GDPNP and GDPCP but not GTPγS are generally unable to substitute for GTP. In light of the above presented observations, we therefore propose that with few exceptions (eIF2γ) trGTPases utilize M+ ions as structural cofactor and that the mechanism of M+-dependent conformational switching as suggested for eIF5B (Fig 5B) is universal among canonical trGTPases.
This proposed commonality of M+-dependency is corroborated by the example of elongation factor Tu (EF-Tu). In line with our prediction on the basis of sequence and structural homology to eIF5B, we found that the active site of the archaeal EF-Tu ortholog aEF1A bound to GTP as well contains an M+ ion (Na+/NH4+), bound by an identical coordination shell as observed for eIF5B·GTP (Fig 4 and Supplementary Fig S3). Likewise, M+ ion binding was demonstrated for free EF-Tu by the finding that its intrinsic GTPase activity is accelerated by M+ ions in a manner dependent on Asp21 (AspMC), in line with its role as key ligand for M+ ion coordination (Fig 4D and E). The usage of M+ ions as structural cofactor in EF-Tu provides a simple explanation why ternary complexes (TC) of EF-Tu with aa-tRNA formed in the presence of GDPNP exhibit decreased stability compared to those formed with GTP (Delaria et al, 1991; Burnett et al, 2013) due to the loss of the M+-dependent allosteric stabilization of the GTP-conformation required for aa-tRNA binding. This is highlighted by the comparison of EF-Tu·GDPNP with the structures of EF-Tu·GDPNP·aa-tRNA complexes and aEF1A·GTP, which indicates that the M+ ion specifically stabilizes a conformation of switch 1 in which helix A″ is drawn toward the GTP molecule, which seems necessary for stable TC formation (Fig 4C). Hence, in the absence of the M+ ion, the aa-tRNA itself has to overcome the entropic penalty to arrange switch 1 in the correct conformation that would otherwise be paid by the M+ ion in the correctly assembled active site with GTP.
The M+ ion as catalytic element in the GTPase reaction of trGTPases
Apart from the GTP-induced conformational switch, the mechanism of GTP hydrolysis is another unresolved problem in the universal functional cycle of trGTPases. Although it has been established that the precise ribosome-induced positioning of the invariant Hiscat from the inactive ground state to the catalytically active conformation is critical for GTP hydrolysis (Voorhees et al, 2010), it remained obscure how the ribosome-bound trGTPase stabilizes the transition state (TS) of the hydrolysis reaction in the absence of an arginine-finger.
As demonstrated above, trGTPases coordinate an M+ ion next to the GTP-γ-phosphate in a conserved coordination shell, where it forms a structurally relevant component of the catalytic center (Figs1 and 4). The M+ ion thus adds another positive charge to the preorganized active site of trGTPases that together with the invariant lysine of the P-loop and the Mg2+ ion forms a triangle of positively charged moieties around the β-γ-bridging oxygen of the GTP molecule (Fig 6A). Following the ribosome-induced activation of Hiscat [which leaves the coordination shell for the M+ ion intact (Fig 3B) (Voorhees et al, 2010)], the M+ ion would thus be in a suitable position to neutralize negative charges of the TS in the γ-phosphate as well as the designated leaving group (GDP) (Fig 6A and B). This suggests that the M+ ion might function as the so far elusive catalytic element in trGTPases that acts in the second step of the ribosome-dependent GTPase reaction and contributes to rapid GTP hydrolysis by providing electrostatic stabilization for the TS, in analogy to the arginine-finger in the Ras-RasGAP system (Fig 6) or the M+ ion in MnmE (Fig 1D).
Figure 6. The M+ ion as catalytic element in the GTP hydrolysis reaction.

A Model of aEF1A·GTP·M+ on the ribosome [the sarcin-ricin loop (SRL) is shown as green sticks], based on a superposition with ribosome-bound EF-Tu (gray; PDB: 2XQD). Upon productive interactions with the SRL, the imidazole moiety of Hiscat is reoriented from its inactive ground state (purple) to the active position (yellow) in which it forms a hydrogen bond to Wcat (Voorhees et al, 2010). The invariant P-loop lysine, the Mg2+ ion (light brown sphere) and the M+ ion (blue sphere) form a triangle of positively charged moieties around the β-γ-bridging oxygen. The M+ ion is thus suitably positioned to stabilize negative charges that develop in the TS of the hydrolysis reaction.
B Schematic presentation of the active site of a ribosome-bound trGTPase with GTP in the transition state of the hydrolysis reaction stabilized by the M+ ion (blue). TrGTPase and ribosome are colored in purple and green, respectively; negative charges, the P-loop lysine and the Mg2+ ion are omitted for clarity. For simplicity, Hiscat is shown in its neutral form, although it might be double protonated in its activated state (Adamczyk & Warshel, 2011; Liljas et al, 2011; Aleksandrov & Field, 2013; Wallin et al, 2013) (modified from Bos et al, 2007 and Rodnina, 2009).
C, D The M+ ion in eIF5B and aEF1A (purple) is coordinated in a position analogous to the arginine-finger in the complex of Ras (gray) and RasGAP (green) (modified from Bos et al, 2007 and Rodnina, 2009).
Structurally, this role for the M+ ion in trGTPases is supported by the superposition of GTP-bound eIF5B and aEF1A with MnmE, dynamin or the Ras-RasGAP complex where the catalytic M+ ions and the guanidino group are located in virtually identical positions (Figs1D and 6C, and Supplementary Fig S1E) (Scheffzek et al, 1997; Scrima & Wittinghofer, 2006; Chappie et al, 2010). For AspMC, this indicates a role of vital importance in the universal functional cycle of trGTPases as key ligand for the M+ ion that provides an explanation for its invariant conservation (Fig 3A). Consistently, we found that the loss of AspMC in the D533A mutant results in M+-independency of the intrinsic GTPase activity in eIF5B, whereas the M+-dependency of the wild-type protein is retained in the D533N mutant. The marked effect of AspMC mutations in eIF5B, which directly correlates with the ability of the substituted residue to coordinate M+ ions, clearly argues in favor of a direct contribution by the M+ ion to the GTPase reaction.
Further evidence is provided by experiments with bacterial EF-Tu. In agreement with earlier observations from Parmeggiani and co-workers (Fasano et al, 1982), we found the intrinsic GTPase activity of EF-Tu to depend on the concentration as well as the species of M+ ions, with a preference for K+ and NH4+ (Fig 4D and E, and Supplementary Fig S6). As for eIF5B, the reduced activity in the absence of M+ ions and the loss of M+-dependency in the D21A (AspMC) mutant argues for a direct effect on GTP hydrolysis. Moreover, kinetic experiments indicated that mutations of Asp21 in E. coli EF-Tu result in a significant reduction of the GTPase activity in the EF-Tu·GTP·aa-tRNA complex in the presence of the correct codon on the ribosome (C. Maracci and M.V. Rodnina, personal communication). This observation as well is consistent with a role of AspMC as key ligand for a catalytic M+ ion in EF-Tu, involved in ribosome-dependent GTP hydrolysis. Moreover, the ribosome-dependent GTPase activity of EF-Tu, EF-G as well as IF2 was reported to depend on M+ ions (Dubnoff et al, 1972; Parmeggiani & Sander, 1981; Fasano et al, 1982). However, in all these cases, the GTPase activity reaches maximum values between 20 and 100 mM M+ ions, most likely reflecting compensating effects of the salt concentration, for example, on the stability and/or conformation of the ribosomal complexes and their interactions with trGTPases (Nagel & Voigt, 1993) that necessarily obscure a direct effect of M+ ions on the GTPase reaction. Importantly, similar observations are known from dynamins, where the stimulating effect of M+ ions on GTP hydrolysis is compensated at increasing salt concentrations due to an inhibition of oligomerization and consequently dimerization-dependent GTPase activation (Warnock et al, 1996; Praefcke & McMahon, 2004; Chappie et al, 2011).
The question whether trGTPases are specific for K+ ions under physiological conditions cannot be unambiguously answered at present. The structural and biochemical analyses indicate that eIF5B and EF-Tu/aEF1A have slightly different specificities in their usage of M+ ions in vitro. However, given the general preference of the translation apparatus for K+ or ions with similar ionic radii (Conway, 1964; Conway & Lipmann, 1964; Dubnoff et al, 1972; Parmeggiani & Sander, 1981; Fasano et al, 1982), the similarly strong stimulation of the GTPase activity in eIF5B and EF-Tu by K+, and the usually high cellular K+/Na+ ratio, we assume that K+ is preferred by trGTPases under physiological conditions.
An interesting case that provides further indirect evidence for M+-dependency among trGTPases is the γ-subunit of eukaryal initiation factor 2 (eIF2γ). Notably, eIF2γ is the only trGTPase for which a specific GAP (eIF5) has been identified that was reported to provide an arginine-finger as catalytic element to promote GTP hydrolysis in eIF2 (Das & Maitra, 2001; Paulin et al, 2001). The coordination sphere for the M+ ion would therefore not be required in eIF2γ. Consistently, eIF2γ is the only trGTPase that contains neither an AspMC nor GlyMC (Fig 3A). Instead, AspMC is replaced by Ala, which might be necessary to allow the introduction of the bulky guanidino group into the active site instead of the M+ ion. Importantly, the archaeal ortholog, aIF2γ, contains AspMC and GlyMC and no GAP is known indicating that aIF2γ in contrast to eIF2γ may be M+ dependent.
Implications for the evolution of trGTPases
At the center of this work stands the conclusion that trGTPases belong to the group of M+-dependent G proteins. This establishes trGTPases as a functionally distinct subfamily among GTPases in which the ribosome, as an RGS-like GAP, stabilizes the active conformation of the catalytic machinery of the GTPase (Voorhees et al, 2010) which includes an M+ ion as an additional trans-acting catalytic element, constitutively bound in the active site. Conceptually, this places trGTPases between the classical GAP- or RGS-activated GTPases and GTPases activated by homodimerization (GADs) (e.g. MnmE and dynamin), which, like trGTPases, directly couple the GTPase reaction to their biological function in the cell (Gasper et al, 2009).
The proposed universality of M+-dependency among canonical trGTPases is particularly interesting from the evolutionary perspective, as it points toward a common origin in an ancestral M+-dependent trGTPase. At the same time, usage of M+ ions as structural and catalytic cofactor constitutes a functional link to other known M+-dependent GTPases that—like trGTPases—usually belong to particularly ancient lineages of the TRAFAC class, associated with basic cellular functions such as tRNA modification or ribosome assembly (Leipe et al, 2002; Ash et al, 2012). It is therefore conceivable that M+-dependency might represent the primordial form of catalysis of GTP hydrolysis in GTPases of the TRAFAC class that antedates the convergent occurrence of arginine-finger-dependent catalysis.
Materials and Methods
Protein purification, crystallization and structure determination
C. thermophilum eIF5B constructs containing residues 517–858 [cteIF5B(517–858)] were purified as previously described (Kuhle & Ficner, 2014). Escherichia coli EF-Tu was prepared essentially as described in Perla-Kajan et al (2009). Escherichia coli EF-Ts was prepared using standard procedures (see Supplementary Materials and Methods for details).
Crystallization trials were performed by sitting-drop vapor diffusion method using either self-made optimization screens for the original crystallization condition for cteIF5B·GTP [0.1 M HEPES/NaOH (pH 7); 13% PEG 4000; 0.1 M NaOAc] (Kuhle & Ficner, 2014) or standard screens in the presence of Mg2+ and varying concentrations of guanine nucleotides. Crystals of cteIF5B(517–858)·GTP used for structure determination grew overnight at 20°C in 100 mM HEPES (pH 7.25), 12% PEG 4000 and 100 mM NaOAc with 10 mg ml−1 of protein in the presence of 2 mM GTP. Crystals of cteIF5B(517–858) bound to GTPγS grew overnight at 20°C under similar conditions [100 mM HEPES (pH 7), 13% PEG 4000 and 125 mM NaOAc] with 12 mg ml−1 protein and 2 mM GTPγS. The crystals for both proteins grew in space group P21.
Crystals of cteIF5B(517–858)·GTPγS with a potassium ion bound in the active site were obtained after 2 weeks at 20°C in 11% PEG 8000, 6% glycerol and 50 mM KCl. The best diffracting crystals grew when using 10 mg ml−1 protein in the presence of 2 mM GTPγS. The crystals grew in space group P41212.
Crystals of the AspMC mutant cteIF5B(517–858)D533N were obtained with 8 mg ml−1 protein and 3 mM GTP at 20°C in a condition containing 100 mM HEPES (pH 7), 13% PEG 4000 and 125 mM NaOAc. As for the wild-type protein crystals that were obtained under similar conditions, the crystals of cteIF5B(517–858)D533N belonged to space group P21. Initially, no crystals were obtained for the mutants D533A and D533R. However, high-quality crystals for both mutants could finally be obtained by repeated microseeding experiments (see Supplementary Materials and Methods for details). Crystals of cteIF5B(517–858)D533A (with 15 mg ml−1 protein and 6 mM GTPγS) grew in space group P21 in a condition containing 0.1 M HEPES/NaOH (pH 7.3), 15% PEG 4000 and 150 mM NaOAc. Crystals of cteIF5B(517–858)D533R (with 15 mg ml−1 protein and 4 mM GTPγS) grew in space group P41212 in a condition containing 100 mM MES (pH 6.7), 13% PEG 8000 and 225 mM NaOAc.
X-ray diffraction data were collected using synchrotron radiation. For all structures, the phase problem was solved by molecular replacement using the program PHASER (McCoy et al, 2007). Structures were refined to reasonable R-values and stereochemistry using the program PHENIX (Adams et al, 2010). Data collection and refinement statistics for the structures of wild-type eIF5B(517–858) and for the three AspMC mutants are summarized in Table1 and Supplementary Table S2, respectively. See Supplementary Materials and Methods for details.
Isothermal titration calorimetry
The thermodynamic parameters of eIF5B binding to GDP or GDPNP were measured using a MicroCal VP-ITC instrument (GE Healthcare). Experiments were carried out as previously described (Kuhle & Ficner, 2014) in ITC buffer [30 mM HEPES/KOH (pH 7.5), 100 mM KCl, 10% glycerol, 4 mM β-mercaptoethanol, 0.01% Tween-20, 2.5 mM MgCl2] at different temperatures (10, 15, 20, 25 or 30°C). 14 μl aliquots of 200–400 μM ligand were injected into the 1.42-ml cell containing 10–30 μM cteIF5B(517–858). The heat of dilution was measured by injecting the ligand into the buffer solution without protein; the values were then subtracted from the heat of the individual binding reactions to obtain the effective heat of binding. The final titration curves were fitted using the “Origin” based MicroCal software, assuming one binding site per protein molecule. For each isotherm, the binding stoichiometry (N), enthalpy changes (ΔH) and the association constants (Ka) were obtained by a nonlinear regression fitting procedure. These directly measured values were used to estimate the Gibbs energy (ΔG) from the relation ΔG = −R·T·lnKa and the entropy changes (ΔS) through ΔG = ΔH – T·ΔS.
In order to estimate the change in heat capacity (ΔCp) upon complex formation between eIF5B and guanine nucleotides, the measured ΔH values were plotted against the temperature, where the slope of the fitted line directly represents the ΔCp of the binding reaction (Jelesarov & Bosshard, 1999; Prabhu & Sharp, 2005). See Supplementary Materials and Methods for details about the correlation between ΔCp and the change in solvent accessible surface area (ΔASA).
Analysis of the GTPase reaction of eIF5B and EF-Tu by HPLC
The intrinsic GTP hydrolysis of C. thermophilum eIF5B(517–858) and E. coli EF-Tu was analyzed by HPLC (GE Healthcare). Nucleotides were separated on a NUCLEOSIL 4,000 PEI (Macherey Nagel) in 10 mM Tris–HCl (pH 8.0) with a linear gradient from 0 to 1 M NaCl. For EF-Tu, the reactions were followed under single turnover conditions, for which 25 μM EF-Tu·GTP was incubated at 30°C in 25 mM Tris–HCl (pH 7.5), 7 mM MgCl2, 2 mM DTT and alkali salts at different concentrations. At various time points, 50 μl aliquots were taken and incubated at 96°C for 2 min to stop the reaction. Denatured protein was removed by centrifugation and the supernatant applied to the HPLC.
For eIF5B, the reactions were followed under multiple-turnover conditions, for which 25 μM nucleotide-free eIF5B was incubated with 300 μM GTP at 35°C in 25 mM Tris–HCl (pH 7.5), 3 mM MgCl2, 2 mM DTT and alkali salts at different concentrations. 50 μl aliquots were taken at various time points and treated as described above.
Multiple sequence alignments
Multiple sequence alignments were done using the iterative alignment program MUSCLE (Edgar, 2004).
Coordinates
Coordinates and structure factors have been deposited in the PDB: 4TMW (ct5B(517–858)·GTP·Na+); 4TMV (ct5B(517–858)·GTPγS·Na+); 4TMZ (ct5B(517–858)·GTPγS·K+); 4TMT (ct5B(517–858)D533A·GTPγS); 4TMX (ct5B(517–858)D533N·GTP·Na+); 4TN1 (ct5B(517–858)D533R·GTPγS).
Acknowledgments
We thank the beam line scientists at EMBL/DESY (Hamburg) and ESRF (Grenoble) as well as P. Neumann for support during X-ray diffraction data collection and L. Hsu for help with figures. We are also grateful to L. K. Dörfel and A. Dickmanns for critical reading of the manuscript as well as C. Maracci and M.V. Rodnina for comments on the manuscript and for sharing data before publication.
Author contributions
BK designed the experiments, prepared and crystallized proteins, collected X-ray data, solved structures, performed GTPase and ITC experiments and created figures. BK and RF analyzed the data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Adamczyk AJ, Warshel A. Converting structural information into an allosteric-energy-based picture for elongation factor Tu activation by the ribosome. Proc Natl Acad Sci USA. 2011;108:9827–9832. doi: 10.1073/pnas.1105714108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(2):213–221. doi: 10.1107/S0907444909052925. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrov A, Field M. Mechanism of activation of elongation factor Tu by ribosome: catalytic histidine activates GTP by protonation. RNA. 2013;19:1218–1225. doi: 10.1261/rna.040097.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash MR, Maher MJ, Guss JM, Jormakka M. The initiation of GTP hydrolysis by the G-domain of FeoB: insights from a transition-state complex structure. PLoS ONE. 2011;6:e23355. doi: 10.1371/journal.pone.0023355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash MR, Maher MJ, Mitchell Guss J, Jormakka M. The cation-dependent G-proteins: in a class of their own. FEBS Lett. 2012;586:2218–2224. doi: 10.1016/j.febslet.2012.06.030. [DOI] [PubMed] [Google Scholar]
- Berchtold H, Reshetnikova L, Reiser CO, Schirmer NK, Sprinzl M, Hilgenfeld R. Crystal structure of active elongation factor Tu reveals major domain rearrangements. Nature. 1993;365:126–132. doi: 10.1038/365126a0. [DOI] [PubMed] [Google Scholar]
- Bondi A. van der Waals volumes and Radii. J Phys Chem. 1964;68:441–451. [Google Scholar]
- Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- Burnett BJ, Altman RB, Ferrao R, Alejo JL, Kaur N, Kanji J, Blanchard SC. Elongation factor Ts directly facilitates the formation and disassembly of the Escherichia coli elongation factor Tu.GTP.aminoacyl-tRNA ternary complex. J Biol Chem. 2013;288:13917–13928. doi: 10.1074/jbc.M113.460014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappie JS, Acharya S, Leonard M, Schmid SL, Dyda F. G domain dimerization controls dynamin's assembly-stimulated GTPase activity. Nature. 2010;465:435–440. doi: 10.1038/nature09032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappie JS, Mears JA, Fang S, Leonard M, Schmid SL, Milligan RA, Hinshaw JE, Dyda F. A pseudoatomic model of the dynamin polymer identifies a hydrolysis-dependent powerstroke. Cell. 2011;147:209–222. doi: 10.1016/j.cell.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway TW. On the role of ammonium or potassium ion in amino acid polymerization. Proc Natl Acad Sci USA. 1964;51:1216–1220. doi: 10.1073/pnas.51.6.1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway TW, Lipmann F. Characterization of a ribosome-linked guanosine triphosphatase in Escherichia coli extracts. Proc Natl Acad Sci USA. 1964;52:1462–1469. doi: 10.1073/pnas.52.6.1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Maitra U. Functional significance and mechanism of eIF5-promoted GTP hydrolysis in eukaryotic translation initiation. Prog Nucleic Acid Res Mol Biol. 2001;70:207–231. doi: 10.1016/s0079-6603(01)70018-9. [DOI] [PubMed] [Google Scholar]
- Delaria K, Guillen M, Louie A, Jurnak F. Stabilization of the Escherichia coli elongation factor Tu-GTP-aminoacyl-tRNA complex. Arch Biochem Biophys. 1991;286:207–211. doi: 10.1016/0003-9861(91)90029-i. [DOI] [PubMed] [Google Scholar]
- Dubnoff JS, Lockwood AH, Maitra U. Studies on the role of guanosine triphosphate in polypeptide chain initiation in Escherichia coli. J Biol Chem. 1972;247:2884–2894. [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano O, De Vendittis E, Parmeggiani A. Hydrolysis of GTP by elongation factor Tu can be induced by monovalent cations in the absence of other effectors. J Biol Chem. 1982;257:3145–3150. [PubMed] [Google Scholar]
- Foucher AE, Reiser JB, Ebel C, Housset D, Jault JM. Potassium acts as a GTPase-activating element on each nucleotide-binding domain of the essential Bacillus subtilis EngA. PLoS ONE. 2012;7:e46795. doi: 10.1371/journal.pone.0046795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasper R, Meyer S, Gotthardt K, Sirajuddin M, Wittinghofer A. It takes two to tango: regulation of G proteins by dimerization. Nat Rev Mol Cell Biol. 2009;10:423–429. doi: 10.1038/nrm2689. [DOI] [PubMed] [Google Scholar]
- Gomez J, Hilser VJ, Xie D, Freire E. The heat capacity of proteins. Proteins. 1995;22:404–412. doi: 10.1002/prot.340220410. [DOI] [PubMed] [Google Scholar]
- Hansson S, Singh R, Gudkov AT, Liljas A, Logan DT. Crystal structure of a mutant elongation factor G trapped with a GTP analogue. FEBS Lett. 2005;579:4492–4497. doi: 10.1016/j.febslet.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Harding MM. Metal-ligand geometry relevant to proteins and in proteins: sodium and potassium. Acta Crystallogr D Biol Crystallogr. 2002;58(5):872–874. doi: 10.1107/s0907444902003712. Pt. [DOI] [PubMed] [Google Scholar]
- Hauryliuk V, Zavialov A, Kisselev L, Ehrenberg M. Class-1 release factor eRF1 promotes GTP binding by class-2 release factor eRF3. Biochimie. 2006;88:747–757. doi: 10.1016/j.biochi.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Hauryliuk V, Hansson S, Ehrenberg M. Cofactor dependent conformational switching of GTPases. Biophys J. 2008a;95:1704–1715. doi: 10.1529/biophysj.107.127290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauryliuk V, Mitkevich VA, Eliseeva NA, Petrushanko IY, Ehrenberg M, Makarov AA. The pretranslocation ribosome is targeted by GTP-bound EF-G in partially activated form. Proc Natl Acad Sci USA. 2008b;105:15678–15683. doi: 10.1073/pnas.0807912105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelesarov I, Bosshard HR. Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics of biomolecular recognition. J Mol Recognit. 1999;12:3–18. doi: 10.1002/(SICI)1099-1352(199901/02)12:1<3::AID-JMR441>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Kikuno I, Kuroha K, Saito K, Ito K, Ishitani R, Inada T, Nureki O. Structural basis for mRNA surveillance by archaeal Pelota and GTP-bound EF1alpha complex. Proc Natl Acad Sci USA. 2010;107:17575–17579. doi: 10.1073/pnas.1009598107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Saito K, Ishitani R, Ito K, Nureki O. Structural basis for translation termination by archaeal RF1 and GTP-bound EF1alpha complex. Nucleic Acids Res. 2012;40:9319–9328. doi: 10.1093/nar/gks660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong C, Ito K, Walsh MA, Wada M, Liu Y, Kumar S, Barford D, Nakamura Y, Song H. Crystal structure and functional analysis of the eukaryotic class II release factor eRF3 from S. pombe. Mol Cell. 2004;14:233–245. doi: 10.1016/s1097-2765(04)00206-0. [DOI] [PubMed] [Google Scholar]
- Kubarenko AV, Sergiev PV, Rodnina MV. GTPases of translational apparatus. Mol Biol. 2005;39:746–761. [PubMed] [Google Scholar]
- Kuhle B, Ficner R. eIF5B employs a novel domain release mechanism to catalyze ribosomal subunit joining. EMBO J. 2014;33:1177–1191. doi: 10.1002/embj.201387344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibundgut M, Frick C, Thanbichler M, Bock A, Ban N. Selenocysteine tRNA-specific elongation factor SelB is a structural chimaera of elongation and initiation factors. EMBO J. 2005;24:11–22. doi: 10.1038/sj.emboj.7600505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipe DD, Wolf YI, Koonin EV, Aravind L. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 2002;317:41–72. doi: 10.1006/jmbi.2001.5378. [DOI] [PubMed] [Google Scholar]
- Liljas A, Ehrenberg M, Aqvist J. Comment on “The mechanism for activation of GTP hydrolysis on the ribosome”. Science. 2011;333:37. doi: 10.1126/science.1202532. author reply 37. [DOI] [PubMed] [Google Scholar]
- Marintchev A, Wagner G. Translation initiation: structures, mechanisms and evolution. Q Rev Biophys. 2004;37:197–284. doi: 10.1017/S0033583505004026. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(4):658–674. doi: 10.1107/S0021889807021206. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesters JR, Potapov AP, de Graaf JM, Kraal B. Synergism between the GTPase activities of EF-Tu.GTP and EF-G.GTP on empty ribosomes. Elongation factors as stimulators of the ribosomal oscillation between two conformations. J Mol Biol. 1994;242:644–654. doi: 10.1006/jmbi.1994.1614. [DOI] [PubMed] [Google Scholar]
- Meyer S, Bohme S, Kruger A, Steinhoff HJ, Klare JP, Wittinghofer A. Kissing G domains of MnmE monitored by X-ray crystallography and pulse electron paramagnetic resonance spectroscopy. PLoS Biol. 2009;7:e1000212. doi: 10.1371/journal.pbio.1000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr D, Wintermeyer W, Rodnina MV. GTPase activation of elongation factors Tu and G on the ribosome. Biochemistry. 2002;41:12520–12528. doi: 10.1021/bi026301y. [DOI] [PubMed] [Google Scholar]
- Nagel K, Voigt J. Regulation of the uncoupled GTPase activity of elongation factor G (EF-G) by the conformations of the ribosomal subunits. Biochim Biophys Acta. 1993;1174:153–161. doi: 10.1016/0167-4781(93)90109-q. [DOI] [PubMed] [Google Scholar]
- Paleskava A, Konevega AL, Rodnina MV. Thermodynamic and kinetic framework of selenocysteyl-tRNASec recognition by elongation factor SelB. J Biol Chem. 2010;285:3014–3020. doi: 10.1074/jbc.M109.081380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paleskava A, Konevega AL, Rodnina MV. Thermodynamics of the GTP-GDP-operated conformational switch of selenocysteine-specific translation factor SelB. J Biol Chem. 2012;287:27906–27912. doi: 10.1074/jbc.M112.366120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmeggiani A, Sander G. Properties and regulation of the GTPase activities of elongation factors Tu and G, and of initiation factor 2. Mol Cell Biochem. 1981;35:129–158. doi: 10.1007/BF02357085. [DOI] [PubMed] [Google Scholar]
- Paulin FE, Campbell LE, O'Brien K, Loughlin J, Proud CG. Eukaryotic translation initiation factor 5 (eIF5) acts as a classical GTPase-activator protein. Curr Biol. 2001;11:55–59. doi: 10.1016/s0960-9822(00)00025-7. [DOI] [PubMed] [Google Scholar]
- Perla-Kajan J, Lin X, Cooperman BS, Goldman E, Jakubowski H, Knudsen CR, Mandecki W. Properties of Escherichia coli EF-Tu mutants designed for fluorescence resonance energy transfer from tRNA molecules. Protein Eng Des Sel. 2009;23:129–136. doi: 10.1093/protein/gzp079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozzo R, Folkers G, Scapozza L. Thermodynamics of protein-ligand interactions: history, presence, and future aspects. J Recept Signal Transduct Res. 2004;24:1–52. doi: 10.1081/rrs-120037896. [DOI] [PubMed] [Google Scholar]
- Pisareva VP, Hellen CU, Pestova TV. Kinetic analysis of the interaction of guanine nucleotides with eukaryotic translation initiation factor eIF5B. Biochemistry. 2007;46:2622–2629. doi: 10.1021/bi062134g. [DOI] [PubMed] [Google Scholar]
- Polekhina G, Thirup S, Kjeldgaard M, Nissen P, Lippmann C, Nyborg J. Helix unwinding in the effector region of elongation factor EF-Tu-GDP. Structure. 1996;4:1141–1151. doi: 10.1016/s0969-2126(96)00122-0. [DOI] [PubMed] [Google Scholar]
- Prabhu NV, Sharp KA. Heat capacity in proteins. Annu Rev Phys Chem. 2005;56:521–548. doi: 10.1146/annurev.physchem.56.092503.141202. [DOI] [PubMed] [Google Scholar]
- Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5:133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Stark H, Savelsbergh A, Wieden HJ, Mohr D, Matassova NB, Peske F, Daviter T, Gualerzi CO, Wintermeyer W. GTPases mechanisms and functions of translation factors on the ribosome. Biol Chem. 2000;381:377–387. doi: 10.1515/BC.2000.050. [DOI] [PubMed] [Google Scholar]
- Rodnina MV. Visualizing the protein synthesis machinery: new focus on the translational GTPase elongation factor Tu. Proc Natl Acad Sci USA. 2009;106:969–970. doi: 10.1073/pnas.0812576106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll-Mecak A, Cao C, Dever TE, Burley SK. X-Ray structures of the universal translation initiation factor IF2/eIF5B: conformational changes on GDP and GTP binding. Cell. 2000;103:781–792. doi: 10.1016/s0092-8674(00)00181-1. [DOI] [PubMed] [Google Scholar]
- Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, Wittinghofer A. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- Scrima A, Wittinghofer A. Dimerisation-dependent GTPase reaction of MnmE: how potassium acts as GTPase-activating element. EMBO J. 2006;25:2940–2951. doi: 10.1038/sj.emboj.7601171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon RD. Revised effective ionic radii and systematic studies of interatomie distances in halides and chaleogenides. Acta Crystallogr A. 1976;32(5):751–767. Pt. [Google Scholar]
- Spolar RS, Record MT., Jr Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- Tourigny DS, Fernandez IS, Kelley AC, Ramakrishnan V. Elongation factor G bound to the ribosome in an intermediate state of translocation. Science. 2013;340:1235490. doi: 10.1126/science.1235490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- Voorhees RM, Schmeing TM, Kelley AC, Ramakrishnan V. The mechanism for activation of GTP hydrolysis on the ribosome. Science. 2010;330:835–838. doi: 10.1126/science.1194460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees RM, Ramakrishnan V. Structural basis of the translational elongation cycle. Annu Rev Biochem. 2013;82:203–236. doi: 10.1146/annurev-biochem-113009-092313. [DOI] [PubMed] [Google Scholar]
- Wallin G, Kamerlin SC, Aqvist J. Energetics of activation of GTP hydrolysis on the ribosome. Nat Commun. 2013;4:1733. doi: 10.1038/ncomms2741. [DOI] [PubMed] [Google Scholar]
- Warnock DE, Hinshaw JE, Schmid SL. Dynamin self-assembly stimulates its GTPase activity. J Biol Chem. 1996;271:22310–22314. doi: 10.1074/jbc.271.37.22310. [DOI] [PubMed] [Google Scholar]
- Wieden HJ, Wintermeyer W, Rodnina MV. A common structural motif in elongation factor Ts and ribosomal protein L7/12 may be involved in the interaction with elongation factor Tu. J Mol Evol. 2001;52:129–136. doi: 10.1007/s002390010141. [DOI] [PubMed] [Google Scholar]
- Wilden B, Savelsbergh A, Rodnina MV, Wintermeyer W. Role and timing of GTP binding and hydrolysis during EF-G-dependent tRNA translocation on the ribosome. Proc Natl Acad Sci USA. 2006;103:13670–13675. doi: 10.1073/pnas.0606099103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.