

The guideline group was selected to be representative of UK-based medical experts. MEDLINE and EMBASE were searched systematically for publications in English from 2002 using the key word Willebrand. The writing group produced the draft guideline, which was subsequently reviewed by the A United Kingdom Haemophilia Centre Doctors Organization (UKHCDO) advisory committee, a British Committee for Standards in Haematology (BCSH) sounding board of approximately 50 UK haematologists, and the BCSH executive; comments were incorporated where appropriate. The ‘GRADE’ system was used to quote levels and grades of evidence, details of which can be found in at http://www.bcshguidelines.com/BCSH_PROCESS/EVIDENCE_LEVELS_AND_GRADES_OF_RECOMMENDATION/43_GRADE.html. The objective of this guideline is to provide healthcare professionals with clear guidance on the diagnosis and management of patients with von Willebrand disease.
Guideline update
This is a single guideline replacing two separate guidelines on diagnosis and management respectively, published in 2004 (Laffan, et al 2004, Pasi, et al 2004). Where there has been no significant change in understanding or practice, the reader is referred to the earlier guidelines.
Major changes since last guideline
The principal changes have been increased understanding of the genetic basis of von Willebrand disease, a relaxation of definition and a focus on how laboratory tests can guide management.
1. Introduction
Von Willebrand factor (VWF) is a large and complex plasma glycoprotein that is essential for normal haemostasis. It is well recognized that deficiency of VWF results in a bleeding disorder that varies in severity according to the degree of deficiency and the specific characteristics of the molecule and which may have features of both primary and secondary haemostatic defects. The complex structure of the protein and the wide range of plasma levels encountered in the population make laboratory assessment and diagnosis a challenging proposition. Since the last guidelines by this group (Laffan, et al 2004, Pasi, et al 2004), there have been considerable advances in understanding the genetics, function and clinical correlates of VWF, which have been incorporated into this revised and unified document. Here we define von Willebrand disease (VWD) as a bleeding disorder that is predominantly attributable to reduced levels of VWF activity. We recognize that this is frequently, but not always, attributable to a defect in the VWF gene (VWF). Our emphasis remains on practical guidance rather than taxonomic purity.
2. What is Von Willebrand Disease?
When patients present with mucocutaneous bleeding symptoms suggestive of a primary haemostatic disorder, a quantitative or qualitative abnormality of VWF is a possible cause or contributory factor. During the initial assessment it is important to remember that bleeding histories can be subjective and the disease characteristics can take time to evolve; there is also overlap between symptoms suffered by people with VWD and the normal population (Sadler 2003, Tosetto, et al 2013). Fig. 1 shows the bleeding symptoms in an international study of type 1 VWD patients in comparison with unaffected family members (Tosetto, et al 2006). To improve decisions regarding the significance of bleeding symptoms, attempts have been made to develop a standardized bleeding assessment tool (BAT), with the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis (ISTH/SSC) BAT being the most recent iteration (Rodeghiero, et al 2010, Rydz and James 2012). These tools can help predict the likelihood of a bleeding disorder being present and have good negative predictive value but studies evaluating their ability to predict future bleeding episodes are lacking (Tosetto, et al 2013).
Figure 1.
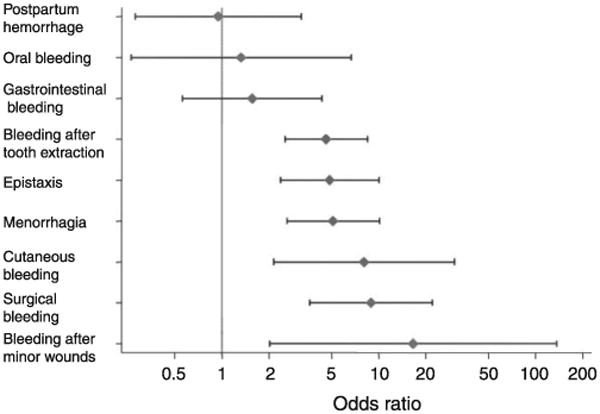
Predictive value of bleeding symptoms in diagnosis of type 1 VWD. Reproduced with permission, from Tosetto, A., Rodeghiero, F., Castaman, G., Goodeve, A., Federici, A.B., Batlle, J., Meyer, D., Fressinaud, E., Mazurier, C., Goudemand, J., Eikenboom, J., Schneppenheim, R., Budde, U., Ingerslev, J., Vorlova, Z., Habart, D., Holmberg, L., Lethagen, S., Pasi, J., Hill, F. & Peake, I. (2006) A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1 VWD). Journal of Thrombosis & Haemostasis, 4, 766-773. Copyright © 2006, John Wiley and Sons
The plasma level of VWF in normal individuals varies over a six-fold range, from 0.40–2.40 iu/ml (Abildgaard, et al 1980), and VWF levels are approximately 25% lower in blood group O individuals than in non-O (Gill, et al 1987). A VWF activity <0.30 iu/ml is usually associated with bleeding symptoms and is more likely to be associated with a mutation in VWF, however these associations are less strong for VWF levels between 0.30 and 0.50 iu/ml (Eikenboom, et al 2006, James, et al 2006). In patients recruited to the MCMDM-1 VWD study, VWF antigen (VWF:Ag) or VWF ristocetin cofactor activity (VWF:RCo) values below 0.40 iu/ml significantly increased the likelihood of type 1 VWD (Tosetto, et al 2006). Amongst 117 obligate carriers of type 3 VWD (type 3 OC) with VWF <0.5 iu/ml, only 26% had bleeding symptoms (Sadler 2003) and a more recent study (Castaman et al, 2006) did not demonstrate an independent correlation between bleeding score and VWF:Ag in 70 type 3 OC. These patients are also likely to have a normal physiological rise in VWF in response to stress. Therefore, mildly reduced VWF activity in isolation may be insufficient to result in significant bleeding. Nonetheless, some patients with only mildly reduced VWF levels do have significant bleeding symptoms: this is likely to reflect interaction with additional abnormalities in the haemostatic pathway, including mild platelet defects (Daly, et al 2009, Millar, et al 2008a).
A study of 280 patients with hereditary mucocutaneous bleeding found abnormalities of VWF and /or platelets in approximately one third, whilst the majority had no identifiable laboratory abnormality. (Quiroga, et al 2007). Therefore, while identification of laboratory abnormalities may guide management, the primary diagnosis remains ‘abnormal bleeding’; for which risk factors may or may not be identified. Because VWF levels are relatively easy to measure (in comparison to platelet function) VWD has often been diagnosed in patients with bleeding symptoms and VWF levels that are only slightly reduced (0.3-0.5 iu/ml), giving the potentially misleading impression that this is the sole responsible factor.
A Bayesian approach to diagnosis of VWD combining laboratory data, personal bleeding history and family data has been evaluated: however an area of uncertainty remained (Tosetto, et al 2008). Thus, caution should be exercised in diagnosing VWD in patients with borderline VWF levels in the range 0.3-0.5 iu/ml in order to avoid the burden of an unnecessary diagnosis and the hazard of failing to complete further necessary investigations.
Recommendations
We recommend against the use of reference ranges or blood group-specific ranges for the diagnosis of von Willebrand disease (VWD) (2C)
When investigating a patient with mucocutaneous bleeding a diagnosis of VWD can be made when von Willebrand factor (VWF) activity is <0.30 iu/ml (1B)
Patients with an appropriate bleeding history and VWF activity 0.3-0.5 iu/ml should be regarded as having primary haemostatic bleeding with reduced VWF as a risk factor rather than VWD. We suggest referring to this as ‘Low VWF’ (2C)
We recommend use of a bleeding score (e.g. Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis bleeding assessment tool) to standardize history taking (2C)
When reviewing patients and families with an historical diagnosis of VWD, we suggest confirming the accuracy of that diagnosis (2A)
The incidental finding of VWF activity <0.30 iu/ml should be taken to indicate VWD or acquired von Willebrand syndrome (AVWS).
Classification of VWD
The simplified classification of VWD proposed by the ISTH (Sadler 1994) is still in common use. Despite the potential for reclassification based on molecular defects, there has been a reluctance to move to a more complex taxonomy; only minor qualifications were introduced when last reviewed (Sadler, et al 2006). Table I summarizes how the classification is currently applied.
Table I. Classification of VWD.
| Type | Description | Comments | Inheritance |
|---|---|---|---|
| 1 | Partial quantitative deficiency of VWF | Includes VWF mutations causing rapid VWF clearance (e.g. VWF Vicenza) and requires function:antigen ratio >0.6 | Mostly autosomal dominant inheritance when VWF <0.3 iu/ml. Mutations of VWF in kindred with levels >0.3 iu/ml show variable penetrance |
| 2 | Qualitative VWF defects | ||
| 2A | Decreased VWF-dependent platelet adhesion with selective deficiency of high-molecular-weight multimers | Some controversy exists regarding classification of VWF mutations associated with subtle reductions in HMW multimers | Mostly autosomal dominant |
| 2B | Increased affinity for platelet GPIb | Should be distinguished from PT-VWD, using either platelet agglutination tests or genetic testing. Cases with normal VWF multimer and platelet count have been described | Autosomal dominant |
| 2M | Decreased VWF-dependent platelet adhesion without selective deficiency of HMW multimers | This also includes defects of VWF collagen binding. May be combined quantitative/qualitative defect | Autosomal dominant |
| 2N | Markedly decreased binding affinity for FVIII | Should be distinguished from mild haemophilia A | Reduced VWF:FVIII binding defects are more commonly identified in a compound heterozygote state with a VWF null allele rather than the classical homozygous form |
| 3 | Virtually complete deficiency of VWF | Equivalent to <0.03 iu/ml in most assays | Autosomal recessive, frequent null VWF alleles. Bleeding symptoms in 26-48% of obligate carriers |
VWD, von Willebrand disease; PT-VWD, platelet type pseudo-VWD; VWF, von Willebrand factor; VWF, VWF gene; FVII, factor VIIIGPIb, glycoprotein Ib; HMW, high molecular weight.
In contrast to type 1 VWD, type 2 variants are usually linked to VWF and usually have a predictable laboratory and clinical phenotype. Misclassification remains an issue, typically between type 1 and type 2M (Nitu-Whalley, et al 2000). In the MCMDM-1 VWD study a third of the families recruited with an historical diagnosis of type 1 VWD had minor multimer abnormalities and a proportion of these were subsequently classified as type 2 VWD (Budde, et al 2008, Goodeve, et al 2007). However the relevance of subtle abnormalities in VWF multimer patterns remains controversial and they lack clear clinical significance (Budde, et al 2008, Castaman, et al 2008, James and Lillicrap 2012).
Classification remains an artificial exercise and although it is of great use for study and research, it does not completely define or predict response to therapy and also has a variable relationship to genetics. From a clinical perspective, any further reclassification of VWD subtypes should focus on clinical utility and, ideally, correlate with responses to therapy.
3. Tests Used for the Primary Diagnosis of VWD
VWD cannot be excluded by a normal activated partial thromboplastin time (APTT). Although the overall sensitivity of the Platelet Function Analyser (PFA) for detecting VWD is reported to be 90%, it is close to 100% in type 2 (excluding 2N) and type 3 VWD but lower in type 1 VWD and may be normal when VWF activity is 0.3-0.5 iu/ml. Factor VIII (FVIII), VWF:Ag and measurements of VWF activity are the initial laboratory tests required to make a diagnosis of VWD and must be performed if VWD is suspected. A diagnostic algorithm is shown in Fig 2. Factors that can affect VWF levels (such as anxiety and needle phobia, the combined contraceptive pill, pregnancy and strenuous exercise) were discussed fully in our previous guideline (Laffan, et al 2004). Misdiagnosis can be minimized by ensuring at least two concordant sets of results are obtained. Samples should not be put on ice (Bohm, et al 2006).
Figure 2.
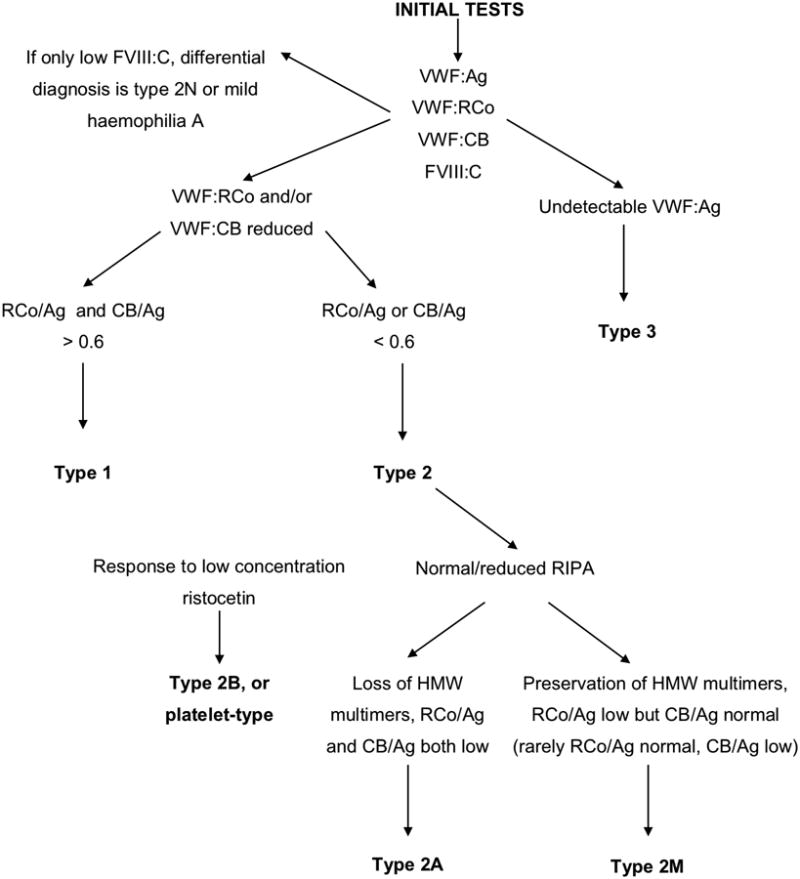
An algorithm for the investigation of suspected von Willebrand disease. VWF, von Willebrand factor; VWF:Ag, VWF antigen; VWF:RCo, ristocetin cofactor activity; VWFCB, collagen binding activity; FVIII, factor VIII; FVIII:C, Factor VIII coagulant activity; GPIb, glycoprotein Ib; HMW, high molecular weight; RIPA, ristocetin-induced platelet agglutination.
Factor VIII assay
FVIII is measured using an APTT-based one stage clotting assay or chromogenic assay. Although FVIII half-life is regulated by VWF and is frequently reduced in VWD, FVIII levels may be normal in VWD.
Von Willebrand factor antigen
Plasma VWF:Ag levels are measured by immunological methods, usually by enzyme-linked immunosorbent assay (ELISA) or by automated immunoturbidimetric methods relying on latex particle agglutination. The latter may give falsely high results in the presence of rheumatoid factor.
Von Willebrand factor activity
Assessment of the ability of VWF to bind FVIII is discussed below. Here we examine the ability of VWF to support platelet adhesion by binding to platelet glycoprotein Ib (GPIb) and collagen.
Binding of VWF to platelet GPIb
Binding of VWF to platelet GPIb has traditionally been assessed by ristocetin cofactor activity (VWF:RCo). Ristocetin dimers bind to VWF and induce a conformational change facilitating VWF binding to platelet GPIb and thus cross-linking of platelets. Putative ristocetin binding sites flank the A1 domain, which contains the GPIb binding region. In the traditional assay the agglutination of normal fixed platelets is measured in dilutions of test plasma containing an excess of ristocetin and the patient's VWF:RCo is determined by reference to a plasma standard. The agglutination is dependent on the presence of high molecular weight (HMW) multimers and an intact GPIb binding site.
The platelet agglutination method has been automated, with improvement in sensitivity and reproducibility (Lawrie, et al 2011). Other approaches to automation have used a monoclonal antibody to link recombinant GPIb to latex beads (Lawrie, et al 2011, Lawrie, et al 2013) or magnetic particles (Cabrera, et al 2013). The use of a recombinant GPIb with gain-of-function mutations can remove the requirement for ristocetin (Flood, et al 2011, Lawrie, et al 2013).
It must be recognized that using ristocetin rather than shear to induce VWF binding to GPIb is unphysiological. Thus sequence variations affecting the ristocetin binding sites on VWF can result in low VWF:RCo estimation in the absence of a physiological defect of VWF (Flood, et al 2009, Flood, et al 2010).
Assays based on monoclonal antibodies directed against the VWF GPIb-binding site, sometimes called ‘VWF activity assays’ have previously failed to detect type 2 VWD (Preston 1998) and although more recent versions have improved precision and sensitivity, they are not yet sufficiently reliable to replace VWF:RCo (Chen, et al 2011, Favaloro, et al 2012). Pending further data we recommend they are not relied on for diagnosis.
Binding of VWF to collagen (collagen binding activity, VWF:CB)
The primary collagen binding site of VWF is in the A3 domain. ELISAs are available to measure binding of patient VWF to immobilized collagen. VWF:CB is dependent on the presence of HMW multimers and an intact collagen binding site: assays using type III and type I/III mixtures of collagen have been found to give the best sensitivity to these factors (Favaloro 2000). Although VWF:RCo and VWF:CB assess different aspects of VWF function, both are sensitive to the loss of HMW multimers and both the VWF:RCo to VWF:Ag ratio and the VWF:CB to VWF:Ag ratio will be reduced in type 2A disease, though the latter ratio is better at differentiating type 2A from type 1 disease (Favaloro, et al 2000).
Recommendations
In the initial investigation for VWD, FVIII, VWF:Ag and VWF activity should be measured(1A)
VWF activity should be assessed by its ability to bind both GPIb and collagen (2B)
We recommend against using assays based on monoclonal antibodies directed against the VWF GPIb-binding site (1B)
4. Secondary Classification of VWD
Types 2A and 2M are qualitative disorders in which VWF function is significantly more reduced than VWF:Ag. Consequently, the VWF:RCo/VWF:Ag and the VWF:CB/VWF:Ag ratios are critically important in their differentiation from type 1. In the MCMDM-1 VWD study the median VWF:RCo/VWF:Ag ratio for 1166 healthy controls was 1.00 with a range of 0.59-1.62 (2.5th – 97.5th centiles) (Goodeve, et al 2007). In a comparison of collagen-binding assays in 232 healthy controls, the lower limit of normal for VWF:CB/VWF:Ag was >0.76 for all types of collagen (Flood, et al 2012) and >0.6 for VWF:RCo/VWF:Ag. Thus patients with a function:antigen ratio <0.6 should be considered to have type 2 VWD and further tests should be performed for classification.
4.1 Multimer analysis
Plasma VWF multimer distribution can be analysed using non-reducing sodium dodecyl sulphate agarose gel electrophoresis. Following electrophoresis, a number of different methodologies have been described for VWF multimer visualization (Krizek and Rick 2000, Ott, et al 2010, Pruthi, et al 2010). The gel agarose concentration can be modified in order to examine either the presence of HMW VWF multimers (∼1%) or abnormalities of VWF satellite bands (1.5-3%)
VWF multimer analysis should not be performed until a diagnosis of VWD has been established. Multimer analysis is essential for the strict classification of type 2 VWD, but if not available, similar information can be cautiously inferred from the VWF:CB/VWF:Ag ratio (Favaloro 2007).
4.2 Ristocetin-induced platelet agglutination (RIPA)
In normal individuals, low concentrations of ristocetin are not sufficient to initiate VWF-dependent platelet agglutination. Platelet agglutination at low ristocetin concentrations (< 0.5-0.7 mg/ml) suggests a pathological enhancement of the VWF – GPIb interaction, which is characteristic of type 2B VWD and platelet-type pseudo-VWD (PT). Type 2B comprises a wide spectrum of severity overlapping with normality and in some patients the only abnormal feature present is RIPA positivity. If it is not practical to perform RIPA on all cases then it should always be performed when the VWF:RCo/VWF:Ag or VWF:CB/VWF:Ag ratio is reduced or if thrombocytopenia is present, bearing in mind that this may miss some mild forms of type 2B VWD (Federici, et al 2009).
4.3 VWF-FVIII binding assay (VWF:FVIIIB)
Mutations of the FVIII binding site within the D′ and D3 domains of VWF can impair FVIII binding. If present in the homozygous state or in trans with a null allele this gives rise to type 2N VWD in which plasma VWF:Ag, VWF:RCo and VWF:CB are often normal but FVIII coagulant (FVIII:C) levels are markedly reduced. These mutations may also complicate a type 1 phenotype causing a disproportionate reduction in FVIII level. The ability of patient VWF to bind added exogenous normal FVIII can be assessed using an immunosorbent plate-binding assay (Zhukov, et al 2009) but these assays are technically difficult and genetic analysis provides a practical alternative.
4.4 Making a diagnosis in neonates and children
The diagnosis of VWD in neonates and infants is complicated by pre-analytical variation due to the difficulty in obtaining an unactivated and uncontaminated venous or cord sample and the prolonged physiological increase of VWF following delivery. In a study of full term normal infants the mean VWF level was 1.53 iu/ml at day 1 of life, reducing to 1.07 iu/ml at day 180 (Andrew, et al 1987). Thus the diagnosis of VWD should not normally be attempted before six months of age. However when testing is clinically necessary, a severe deficiency or function-antigen discrepancy indicating type 2 disease should be apparent. Samples from children are particularly prone to the elevation by stress and if borderline results are obtained they may need to be repeated when the child is older. Modified bleeding scores for use in children are available (Marcus, et al 2011).
Recommendations
A function:antigen ratio of <0.6 should be used to identify patients with type 2 VWD (1B)
RIPA should be performed on all patients with reduced VWF:RCo/VWF:Ag or VWF:CB/VWF:Ag ratios or when thrombocytopenia is present (1B)
Multimer analysis should be used to distinguish between types 2A and 2M (1B)
If multimer analysis is not available then the ratios of VWF:RCo and VWF:CB to VWF:Ag should be used to distinguish types 2A and 2M (1B)
5. Genetic Analysis and Family Testing
5.1 Genetic testing
For many patients, phenotypic analysis yields sufficient information for VWD classification and thus appropriate treatment. However, certain circumstances, indicated below, may warrant VWF analysis to help clarify disease type or risk of disease inheritance, or to facilitate prenatal diagnosis.
Mild or moderate haemophilia A and 2N VWD can be challenging to discriminate, especially in the absence of a family history. The VWF:FVIIIB assay can discriminate between the two disorders, but is not widely available. Analysis of VWF exons 17-25 can identify missense VWF:FVIIIB mutations in patients that have type 2N VWD; individuals lacking these mutations should be investigated for sequence variation in the F8 gene.
Type 2B VWD and PT-VWD patients present with similar phenotypes and can be discriminated by plasma/platelet mixing studies or by genetic analysis. All 2B VWD mutations have been identified between amino acid residues 1266-1461 encoded by exon 28 of VWF whilst PT-VWD mutations affect either the beta hairpin or macroglycopeptide regions of GPIbα encoded by the central region of GP1BA exon 2 (Hamilton, et al 2011). Discrimination using genetic analysis is straightforward and can help guide appropriate therapy.
Prenatal diagnosis is occasionally requested by families with type 3 VWD, particularly where the parents already have one affected child. Mutation analysis of the index case can identify the familial mutation(s), which should be confirmed to be present in each parent and can subsequently be sought in a fetus using chorionic villus or amniocentesis samples. Both sequence and gene dosage analyses may be required to identify the two VWF mutations although mutations are not yet identified in all cases.(Keeney, et al 2008)
For those patients where phenotypic analysis does not clarify VWD type, genetic analysis can be used to try and identify explanatory mutation(s), e.g. some patients with D3 domain missense mutations may present with a pleiotropic phenotype with reduced VWF-FVIII binding in addition to reduced VWF-GPIbα binding (Hampshire, et al 2010). Similarly, for some affected individuals without a clear family history of bleeding, VWD inheritance risks for family members are unclear and genetic analysis may help provide clarity through identification of mutation(s) that suggest either dominant or recessive inheritance.
5.2 Family testing
When a diagnosis of VWD is made it is appropriate to test first degree relatives with or without a positive bleeding history. In this circumstance, a presumptive diagnosis of VWD may be made on the basis of laboratory findings alone. For patients with ‘low VWF’ (0.3-0.5 iu/ml), family testing may be justifiable depending on bleeding and family history.
Recommendations
Type 1 VWD should not be excluded in children before the age of 6 months (1B)
Family testing in VWD is appropriate prior to development of bleeding symptoms (2B)
Genetic analysis should be used where beneficial to clarify diagnosis and aid management (1B)
6. Management
6.1 Haemostatic therapies
Available therapies to correct haemostasis in VWD comprise the non-concentrate therapies tranexamic acid and desmopressin or concentrates containing either high purity VWF alone or intermediate purity concentrates containing FVIII-VWF.
6.1.1 Desmopressin
The pharmacology and clinical use of desmopressin to temporarily elevate FVIII and VWF levels by releasing endothelial stores have been extensively reviewed and were discussed in the previous version of this guideline (Mannucci 1997, Pasi, et al 2004). Intravenous, subcutaneous and intranasal desmopressin all have a UK product license for the treatment of VWD.
Desmopressin frequently causes flushing and sometimes hypotension, which are not harmful. Excessive fluid retention should be avoided by limiting fluid intake to 1 litre in the subsequent 24 h. Serum sodium should be monitored if desmopressin is used in children <2 years old or whenever repeated doses are given. Desmopressin use has rarely been followed by occlusive arterial events and should not be used in patients who are likely to have atherosclerosis (Federici 2008).
Because of its clinical utility, and the wide variation in response, a trial of desmopressin should be considered in all patients with type 1, type 2A, 2M and 2N VWD who do not have a contraindication to its use. It is also useful in patients with bleeding and low VWF as a risk factor (‘low VWF’). In type 2B a transient thrombocytopenia frequently follows desmopressin administration (Casonato, et al 1999). This has been regarded as a reason for contraindication and although no harmful effects have been reported, the therapeutic response is usually poor and desmopressin is not recommended for type 2B VWD (Federici 2008, Mannucci 1988).
Doses, routes of administration and responses are given in Table II. The magnitude of response will depend on the underlying defect and there are several VWF mutations (including R1205H Vicenza) that have been associated with rapid VWF clearance (Casari, et al 2013): it is therefore important to measure both the peak response at 30-60 min and the maintenance of response at 4-6 h (Castaman, et al 2013, Millar, et al 2008b). However, even when the VWF or FVIII response is shortened, most dental extractions, minor surgeries and deliveries may be successfully managed with desmopressin (Castaman, et al 2011, Trigg, et al 2012).
Table II. Desmopressin doses and response for VWD.
| Route of administration | Suggested formulation | Dose | Peak levels achieved |
|---|---|---|---|
| Intravenous infusion over 30-60 min | 4 μg/ml diluted in 100ml 0.9%NaCl for infusion | 0.3 μg/kg [maximum dose 28 μg] | 15 min after infusion completed |
| Subcutaneous | 15 μg/ml | 0.3 μg/kg [maximum dose 28 μg] | 60-90 min after injection |
| Intranasal | 150 μg per metered spray | > 50 kg: 150 μg spray to each nostril ≤ 50 kg: single 150 μg spray |
1-4 h after administration |
Desmopressin is relatively contraindicated in children <2 years old. If, after careful consideration, it is to be used in this age group then fluid restriction, avoidance of hyponatraemic solutions and close monitoring of serum electrolytes and urine output for at least 24 h after administration is advised.
6.1.2 Tranexamic acid
Tranexamic acid administered topically, as a mouthwash, orally or parenterally remains a useful therapy for minor bleeding or surgery (beginning prior to the procedure) either on its own or as an adjunctive therapy to desmopressin or concentrates. (Pasi, et al 2004)
Recommendations
A trial of desmopressin should be carried out in patients with type 1, type 2A, 2M and 2N VWD with VWF antigen, activity and FVIII measured at baseline, 30-60 min and 4-6 h (1B)
When shown to be effective, desmopressin should be used in preference to blood-derived products where possible (1B)
Adults should be warned to limit fluid intake to 1 litre in the 24 h after desmopressin (2C)
Desmopressin should be avoided in those with known atherosclerosis.(1C)
6.1.3 VWF-containing concentrates
A number of plasma-derived concentrates containing VWF are available for replacement therapy in patients whose desmopressin response is inadequate for the relevant bleeding episode or surgical procedure (Batlle, et al 2009). In choosing which concentrate to use, the plasma source, purification and viral inactivation measures should first be considered although all currently available concentrates have an excellent safety record. For haemostatic activity, the relevant characteristics of these concentrates are the multimeric composition of the VWF, reflected in the VWF:RCo/VWF:Ag ratio, and the amount of FVIII contained per unit of VWF. A VWF:RCo/VWF:Ag ratio close to 1 is desirable because it indicates the VWF has normal multimeric structure and adhesive function, but the appropriate amount of FVIII is debatable and will vary according to the circumstance. Some high purity concentrates contain virtually no FVIII and if given alone to a patient with type 3 VWD it will be >12 hours before the FVIII level has risen to normal (Borel-Derlon, et al 2007, Goudemand, et al 1998).
Comparison of different concentrates yields a uniform VWF:RCo recovery of 0.021 – 0.024 (iu/ml)/(u/kg) (Mannucci, et al 1992a) but reveals significant differences in specific activity (function:antigen) and in their content of HMW multimers.(Batlle, et al 2009, Budde, et al 2006).
Initial (primary) haemostasis is dependent upon elevation of the plasma VWF:RCo activity to normal, but longer term secure haemostasis is dependent on a normal level of Factor VIII (Mannucci, et al 1987, Sakurai, et al 2006). Because endogenous FVIII production is normal, continued administration of large amounts of FVIII may result in an undesirably high plasma FVIII level.
Recommendations
For replacement therapy a VWF-containing concentrate manufactured from a safe plasma source with adequate viral testing and inactivation procedures should be chosen (1A)
For treatment of acute bleeding or emergency surgery, a VWF-FVIII concentrate or a combination of high purity FVIII and high purity VWF concentrates should be used (1A)
6.2 Treatment of bleeding episodes
6.2.1 Treatment of acute bleeding episodes
Typical bleeding problems in VWD are epistaxes, gum bleeding and menorrhagia. Post-traumatic bleeding can also occur and type 3 patients can develop spontaneous joint or muscle bleeding. When any of these are frequent, self-administration of desmopressin and tranexamic acid can be helpful (Leissinger, et al 2001). If the patient is non-desmopressin responsive, then acute treatment and secondary prophylaxis using concentrates should be considered. Hormonal management of menorrhagia should also be considered, including use of the Mirena coil (Laffan, et al 2004).
6.2.2 Surgery including dentistry
Management of surgery in patients with VWD may be straightforward in those with mild forms of the disease. However, type 3 and type 2 variants may be extremely difficult to manage and there is no guarantee that haemostasis will be achieved even when plasma concentrations have apparently been corrected into the normal range. In these patients, surgery should be carried out in a centre where experienced haematology and laboratory support is available and after careful consultation between responsible teams.
Minor surgery including dental work using an inferior dental block in patients with type 1 disease or ‘low VWF’ (and a minority of 2A and 2M) can often be carried out using desmopressin with or without tranexamic acid. The level of VWF:RCo and FVIII required will vary with the particular procedure but is likely to be satisfactory if >0.5 iu/ml. The response to desmopressin should be measured unless it is well known and also when repeated doses are given. The response to a second dose of desmopressin is, on average, 30% lower than the first but tends not to fall further thereafter (Mannucci, et al 1992b). Particular care is required when the endogenous VWF has a shortened half-life and in patients with type 2N VWD. When desmopressin is contraindicated or the response is inadequate, a VWF-FVIII concentrate should be used. The recovery of both VWF and FVIII is approximately 0.02 iu/ml per iu/kg given.
For major surgery in all types of VWD the VWF:RCo and FVIII should be corrected to ≥1.0 iu/ml preoperatively with either desmopressin or concentrate(s). In the postoperative period, recent studies have shown efficacy by measuring and maintaining VWF:RCo >0.5 iu/ml for 6 days (Mannucci, et al 2013). However, maintaining FVIII >0.5 iu/ml for 7-10 days using a suitable VWF-FVIII concentrate has also proved effective (Windyga, et al 2011) and was common practice before rapid VWF:RCo measurement became available (Lusher 1998, Mannucci 2001). It does not appear necessary to correct the bleeding time or PFA100 time and these need not be measured. Surgery should therefore be carried out only in centres with the ability to perform these measurements with the required frequency.
Tranexamic acid remains a useful adjunctive therapy.
When bleeding persists despite apparently normal plasma levels of VWF activity, platelet transfusion may be helpful.
Platelet transfusion is also the treatment of choice in platelet-type VWD pseudo (PT-VWD) and may be supplemented by FVIII-VWF concentrate (O'Connor, et al 2011, Othman 2011).
Recommendations
Factor VIII levels should be monitored regularly in all major and most minor surgical procedures (1B). The FVIII plasma concentration should be ≥ 1.0 iu/ml to cover major surgery and sustained above 0.5 iu/ml in the postoperative period (1B).
The VWF:RCo should be monitored in major surgical procedures, particularly in the perioperative period (grade C, level IV). The VWF:RCo should be maintained above 0.5 iu/ml in the perioperative period (1B).
6.3 Prophylactic therapy
Because most cases of VWD are relatively mild and patients do not suffer from serious spontaneous bleeding, prophylaxis is rarely indicated. Exceptions include patients with type 3 disease plus haemarthroses, severe epistaxis, women with menorrhagia, and those with VWD in conjunction with an on-going risk factor for bleeding, such as angiodysplasia. In 2006 an international survey reported that 74.5% type 3, 17.6% type 2 and 7.8% type 1 patients were receiving prophylaxis (Berntorp, et al 2006). The most frequent indications were epistaxis/oral bleeding (23.6%), gastrointestinal (GI) bleeding (23.6%), joint bleeding (21.8%), and menorrhagia (7.3%). (Berntorp, et al 2006) Prophylaxis was able to almost abolish joint bleeding but was less effective against mucosal bleeding (50-60% reduction). A German cohort study also reported that prophylaxis was almost completely effective in abolishing bleeding (Halimeh, et al 2011). Typical doses were 20-50 iu/kg VWF:RCo given 2-3 times per week. In these studies a variety of VWF concentrates were used, but notably, for prophylaxis, a FVIII-containing concentrate may not be necessary. There are no data to indicate the appropriate trough level of FVIII or VWF but analogy with experience in haemophilia seems appropriate. Prophylaxis beginning at age <5 years is reported to prevent arthropathy (Berntorp, et al 2010).
Recommendations
Prophylaxis should be considered for recurrent bleeding in all types of VWD (1A)
In children with type 3 VWD, consider prophylaxis 2-3 times per week at 30-50 iu VWF:RCo/kg when joint bleeding develops (as for haemophilia). (1A)
Intermediate purity FVIII-VWF or high purity VWF concentrates are both appropriate for prophylaxis (1B)
7. Pregnancy
Levels of VWF and FVIII rise from early in the first trimester of pregnancy and increase with gestational stage, reaching two to three times those of the non-pregnant state at term. Levels start to fall soon after delivery, returning to non-pregnant values within a few weeks (Mahieu, et al 2007, Sanchez-Luceros, et al 2003, Stirling, et al 1984).
Women with VWD whose VWF activity does not rise to normal levels during pregnancy should be delivered in an obstetric unit that can easily and quickly access Haemophilia Centre and comprehensive neonatal care facilities. A plan for management of delivery and the puerperium should be drawn up jointly between the obstetrician and Haemophilia Centre and agreed with the patient. The increase in VWF is often sufficient to correct the VWF deficiency in women with type 1 VWD, but not in qualitative or severe VWF deficiency (Lee, et al 2006, Walker, et al 1994). Pregnancy may exacerbate the thrombocytopenia and bleeding tendency in women with type 2B VWD (Ranger, et al 2012).
VWF parameters should be checked at booking and at around 34 weeks gestation unless levels have already been shown to have risen to normal. Neuraxial anaesthesia, vaginal delivery and Caesarean section can all be regarded as safe in type 1 when VWF:RCo > 0.5 iu/ml. In type 2 and 3 VWD, restoration of normal haemostasis is not reliably achievable even following replacement therapy. Therefore, neuraxial anaesthesia is not recommended for use in types 2 or 3 VWD irrespective of whether VWF activity has been restored to apparently normal levels. Patients may require several days' treatment, especially after Caesarean section. Even if VWF parameters are satisfactory at the time of delivery, abnormal bleeding may ensue following discharge from hospital. It is important that women are made aware of this and advised to seek appropriate medical help should they develop persistent heavy postpartum bleeding. Thromboprophylaxis may be given to patients with adequate correction of VWF parameters.
Desmopressin is safe both in pregnancy and at delivery (Trigg, et al 2012) (Ray 1998) but should be avoided in pre-eclampsia. Repeated administration should be avoided in view of the sensitivity of the fetus to the effects of hyponatraemia.
Tranexamic acid is not contraindicated during pregnancy or the puerperium; a limited quantity (∼1%) may be secreted in breast milk http://www.medicines.org.uk/emc/medicine/27753, although is unlikely to produce an antifibrinolytic effect in the infant.
Where a fetus is at risk of having significantly reduced plasma VWF activity levels, invasive monitoring procedures, mid-cavity rotational forceps and Ventouse delivery should be avoided with early recourse to Caesarean section. Administration of intramuscular vitamin K therapy can be considered safe, unless the infant is at risk of type 3 VWD.
Recommendations
All women with type 2 and type 3 VWD, and women with type 1 VWD in whom VWF levels are unlikely to normalize should be delivered in a obstetric unit with close collaboration between haematology, obstetric, anaesthetic and neonatal teams and access to 24-h monitoring of FVIII-VWF (2C)
The delivery of women with type 1 VWD can be managed as normal when VWF:RCo activity is > 0.5 iu/ml by 34-36 weeks gestation (1C)
Vaginal delivery or Caesarean section can be performed when VWF:RCo activity is maintained > 0.5 iu/ml and platelet count maintained > 50 × 109/l (2C)
Neuraxial anaesthesia is not recommended in women with type 2 and type 3 VWD, or in women with type 1 VWD in whom plasma VWF levels have failed to normalize (1B)
Intermediate or high purity VWF concentrates, desmopressin and tranexamic acid can be used to support haemostasis during pregnancy, delivery and the puerperium (1B)
8. VWF Inhibitors
VWF inhibitors have been described in multi-transfused patients with type 3 VWD at a frequency of 5-10% (James, et al 2013) but not in other VWD subgroups. Patients who develop inhibitors may present with loss of response to VWF concentrate, sometimes with an associated anaphylactic reaction (Mannucci, et al 1987).
Inhibitors to VWF cannot be reliably detected by either conventional mixing tests or ELISA-based assays (James, et al 2013). Poor VWF recovery and/or rapid clearance after infusion of VWF concentrate may therefore be the only indication of an inhibitor.
Treatment options for type 3 VWD patients with inhibitors include recombinant FVIII administered by continuous intravenous infusion at very large doses sufficient to maintain FVIII levels > 0.50 iu/ml (Mannucci, et al 1987), recombinant activated FVII (rVIIa) (Boadas, et al 2011, Ciavarella, et al 1996, Mannucci 2001), platelet transfusions (James, et al 2013) and tranexamic acid. Patients with low-level inhibitors may still respond to infusions of VWF-containing concentrate but anamnestic responses may be seen. A case of successful immune tolerance in a 9-year-old boy with type 3 VWD and inhibitors has been described (Pergantou, et al 2012).
Recommendations
Traditional mixing studies are an unreliable method for inhibitor screening in type 3 VWD and measurement of in vivo recovery and survival should be considered if an inhibitor is suspected (2A)
If there is no response to VWF concentrate or when anaphylaxis occurs, high dose rVIII infusion, rVIIa, platelet transfusion and tranexamic acid should be considered (2A)
9. Acquired Von Willebrand Syndrome (AVWS)
Acquired von Willebrand syndrome is the term used to describe an acquired loss of VWF function. It can arise via a wide range of mechanisms and should be suspected when a patient's current symptoms or laboratory results do not match their clinical history. The AVWS will respond to removal of the cause where this is possible, such as aortic valve replacement, treatment of Wilms tumour or correction of hypothyroidism. Paraproteins causing AVWS are not easily removed but AVWS associated with IgG paraproteins frequently responds to intravenous immunoglobulin (Federici, et al 1998). Other treatment options include VWF concentrates, desmopressin, plasma exchange and immunoadsorption (Tiede, et al 2011).
Topics for Audit.
Registration for those patients who fulfil diagnostic criteria for VWD.
Patient registrations with appropriate classification of type entered.
Proportion of eligible patients who have had test dose of desmopressin administered
Proportion of desmopressin test doses that include a measurement of fall-off at 4-6 h
Proportion of patients registered with mild haemophilia who have had possible 2N VWD excluded.
Acknowledgments
J.S.O'D is supported by a Science Foundation Ireland Principal Investigator Award (11/PI/1066). ACG is funded through the NIH Program Project Grant, Zimmerman Program for the Molecular and Clinical Biology of VWD study (HL-081588). ML and CM acknowledge support from the Imperial College BRC.
Footnotes
Conflict-of-Interest Disclosure: ML has received speaker fees from Bayer, Octapharma and Pfizer, advisory board fees from CSL-Behring, Pfizer, Bayer and Grifols and research support from Bayer and CSL Behring. WL has received speaker fees and travel support from CSL and advisory board fees from Octapharma. JSOD has served on the speakers bureau for Baxter, Bayer, Novo Nordisk, Leo Pharma and Octapharma; served on the advisory boards of Baxter, Bayer, Octapharma and Pfizer and received research grant funding awards from Baxter, Bayer and Novo Nordisk. AW has no declarations of interest relating to this guideline. AG has received speaker fees from Octapharma and VWF mutation database support from CSL Behring. CMM has received research grant funding award and speaker fees from CSL Behring and Baxter and served on advisory boards for CSL and NovoNordisk. DMK has served on advisory boards for Baxter, CSL, Bayer, Pfizer and NovoNordisk. RCT has received speaker and/or consultancy fees from Baxter, Pfizer & Bayer
Author Contributions: All authors took an active role in drafting, reviewing and approving the guideline in a series of round table meetings and subsequent correspondence. ML assembled the final draft.
Publisher's Disclaimer: Disclaimer: While the advice and information in these guidelines is believed to be true and accurate at the time of going to press, neither the authors, the UKHCDO, the British Society for Haematology nor the publishers accept any legal responsibility for the content of these guidelines
References
- Abildgaard CF, Suzuki Z, Harrison J, Jefcoat K, Zimmerman TS. Serial studies in von Willebrand's disease: variability versus “variants”. Blood. 1980;56:712–716. [PubMed] [Google Scholar]
- Andrew M, Paes B, Milner R, Johnston M, Mitchell L, Tollefsen DM, Powers P. Development of the human coagulation system in the full-term infant. Blood. 1987;70:165–172. [PubMed] [Google Scholar]
- Batlle J, Lopez-Fernandez MF, Fraga EL, Trillo AR, Perez-Rodriguez MA. Von Willebrand factor/factor VIII concentrates in the treatment of von Willebrand disease. Blood Coagulation & Fibrinolysis. 2009;20:89–100. doi: 10.1097/MBC.0b013e3283254570. [DOI] [PubMed] [Google Scholar]
- Berntorp E, Abshire T von Willebrand Disease Prophylaxis Network Steering, C. The von Willebrand disease prophylaxis network: exploring a treatment concept. Journal of Thrombosis & Haemostasis. 2006;4:2511–2512. doi: 10.1111/j.1538-7836.2006.02179.x. [DOI] [PubMed] [Google Scholar]
- Berntorp E, de Moerloose P, Ljung RCR. The role of prophylaxis in bleeding disorders. Haemophilia. 2010;16 Suppl 5:189–193. doi: 10.1111/j.1365-2516.2010.02319.x. [DOI] [PubMed] [Google Scholar]
- Boadas A, Fernandez-Palazzi F, De Bosch NB, Cedeno M, Ruiz-Saez A. Elective surgery in patients with congenital coagulopathies and inhibitors: experience of the National Haemophilia Centre of Venezuela. Haemophilia. 2011;17:422–427. doi: 10.1111/j.1365-2516.2010.02427.x. [DOI] [PubMed] [Google Scholar]
- Bohm M, Taschner S, Kretzschmar E, Gerlach R, Favaloro EJ, Scharrer I. Cold storage of citrated whole blood induces drastic time-dependent losses in factor VIII and von Willebrand factor: potential for misdiagnosis of haemophilia and von Willebrand disease. Blood Coagul Fibrinolysis. 2006;17:39–45. doi: 10.1097/01.mbc.0000198990.16598.85. [DOI] [PubMed] [Google Scholar]
- Borel-Derlon A, Federici AB, Roussel-Robert V, Goudemand J, Lee CA, Scharrer I, Rothschild C, Berntorp E, Henriet C, Tellier Z, Bridey F, Mannucci PM. Treatment of severe von Willebrand disease with a high-purity von Willebrand factor concentrate (Wilfactin®): a prospective study of 50 patients. Journal of Thrombosis and Haemostasis. 2007;5:1115–1124. doi: 10.1111/j.1538-7836.2007.02562.x. [DOI] [PubMed] [Google Scholar]
- Budde U, Metzner HJ, Muller HG. Comparative analysis and classification of von Willebrand factor/factor VIII concentrates: impact on treatment of patients with von Willebrand disease. Seminars in Thrombosis & Hemostasis. 2006;32:626–635. doi: 10.1055/s-2006-949668. [DOI] [PubMed] [Google Scholar]
- Budde U, Schneppenheim R, Eikenboom J, Goodeve A, Will K, Drewke E, Castaman G, Rodeghiero F, Federici AB, Batlle J, Perez A, Meyer D, Mazurier C, Goudemand J, Ingerslev J, Habart D, Vorlova Z, Holmberg L, Lethagen S, Pasi J, Hill F, Peake I. Detailed von Willebrand factor multimer analysis in patients with von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 von Willebrand disease (MCMDM-1VWD) Journal of Thrombosis & Haemostasis. 2008;6:762–771. doi: 10.1111/j.1538-7836.2008.02945.x. [DOI] [PubMed] [Google Scholar]
- Cabrera N, Moret A, Caunedo P, Cid AR, Vila V, Espana F, Aznar JA. Comparison of a new chemiluminescent immunoassay for von Willebrand factor activity with the ristocetin cofactor-induced platelet agglutination method. Haemophilia. 2013 doi: 10.1111/hae.12203. [DOI] [PubMed] [Google Scholar]
- Casari C, Lenting PJ, Wohner N, Christophe OD, Denis CV. Clearance of von Willebrand factor. Journal of Thrombosis & Haemostasis. 2013;11 Suppl 1:202–211. doi: 10.1111/jth.12226. [DOI] [PubMed] [Google Scholar]
- Casonato A, Steffan A, Pontara E, Zucchetto A, Rossi C, De Marco L, Girolami A. Post-DDAVP Thrombocytopenia in Type 2B von Willebrand Disease Is not Associated with Platelet Consumption: Failure to Demonstrate Glycocalicin Increase or Platelet Activation. Thrombosis and Haemostasis. 1999;81:224–228. [PubMed] [Google Scholar]
- Castaman G, Rodeghiero F, Tosetto A, Cappelletti A, Baudo F, Eikenboom JCJ, Federici AB, Lethagen S, Linari S, Lusher J, Nishino M, Petrini P, Srivastava A, Ungerstedt JS. Hemorrhagic symptoms and bleeding risk in obligatory carriers of type 3 von Willebrand disease: an international, multicenter study. Journal of Thrombosis & Haemostasis. 2006;4:2164–2169. doi: 10.1111/j.1538-7836.2006.02070.x. [DOI] [PubMed] [Google Scholar]
- Castaman G, Lethagen S, Federici AB, Tosetto A, Goodeve A, Budde U, Batlle J, Meyer D, Mazurier C, Fressinaud E, Goudemand J, Eikenboom J, Schneppenheim R, Ingerslev J, Vorlova Z, Habart D, Holmberg L, Pasi J, Hill F, Peake I, Rodeghiero F. Response to desmopressin is influenced by the genotype and phenotype in type 1 von Willebrand disease (VWD): results from the European Study MCMDM-1VWD. Blood. 2008;111:3531–3539. doi: 10.1182/blood-2007-08-109231. [DOI] [PubMed] [Google Scholar]
- Castaman G, Tosetto A, Federici AB, Rodeghiero F. Bleeding tendency and efficacy of anti-haemorrhagic treatments in patients with type 1 von Willebrand disease and increased von Willebrand factor clearance. Thrombosis & Haemostasis. 2011;105:647–654. doi: 10.1160/TH10-11-0697. [DOI] [PubMed] [Google Scholar]
- Castaman G, Goodeve A, Eikenboom J European Group on von Willebrand, D. Principles of care for the diagnosis and treatment of von Willebrand disease. Haematologica. 2013;98:667–674. doi: 10.3324/haematol.2012.077263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Tange JI, Meyers BJ, Pruthi RK, Nichols WL, Heit JA. Validation of an automated latex particle-enhanced immunoturbidimetric von Willebrand factor activity assay. Journal of Thrombosis & Haemostasis. 2011;9:1993–2002. doi: 10.1111/j.1538-7836.2011.04460.x. [DOI] [PubMed] [Google Scholar]
- Ciavarella N, Schiavoni M, Valenzano E, Mangini F, Inchingolo F. Use of recombinant factor VIIa (NovoSeven) in the treatment of two patients with type III von Willebrand's disease and an inhibitor against von Willebrand factor. Haemostasis. 1996;26 Suppl 1:150–154. doi: 10.1159/000217258. [DOI] [PubMed] [Google Scholar]
- Daly ME, Dawood BB, Lester WA, Peake IR, Rodeghiero F, Goodeve AC, Makris M, Wilde JT, Mumford AD, Watson SP, Mundell SJ. Identification and characterization of a novel P2Y 12 variant in a patient diagnosed with type 1 von Willebrand disease in the European MCMDM-1VWD study. Blood. 2009;113:4110–4113. doi: 10.1182/blood-2008-11-190850. [DOI] [PubMed] [Google Scholar]
- Eikenboom J, Van Marion V, Putter H, Goodeve A, Rodeghiero F, Castaman G, Federici AB, Batlle J, Meyer D, Mazurier C, Goudemand J, Schneppenheim R, Budde U, Ingerslev J, Vorlova Z, Habart D, Holmberg L, Lethagen S, Pasi J, Hill F, Peake I. Linkage analysis in families diagnosed with type 1 von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 VWD. Journal of Thrombosis & Haemostasis. 2006;4:774–782. doi: 10.1111/j.1538-7836.2006.01823.x. [DOI] [PubMed] [Google Scholar]
- Favaloro EJ. Collagen binding assay for von Willebrand factor (VWF:CBA): detection of von Willebrands Disease (VWD), and discrimination of VWD subtypes, depends on collagen source. Thrombosis & Haemostasis. 2000;83:127–135. [PubMed] [Google Scholar]
- Favaloro EJ. An update on the von Willebrand factor collagen binding assay: 21 years of age and beyond adolescence but not yet a mature adult. Seminars in Thrombosis & Hemostasis. 2007;33:727–744. doi: 10.1055/s-2007-1000364. [DOI] [PubMed] [Google Scholar]
- Favaloro EJ, Henniker A, Facey D, Hertzberg M. Discrimination of von Willebrands disease (VWD) subtypes: direct comparison of von Willebrand factor:collagen binding assay (VWF:CBA) with monoclonal antibody (MAB) based VWF-capture systems. Thrombosis & Haemostasis. 2000;84:541–547. [PubMed] [Google Scholar]
- Favaloro EJ, Bonar R, Chapman K, Meiring M, Funk Adcock D. Differential sensitivity of von Willebrand factor (VWF) ‘activity’ assays to large and small VWF molecular weight forms: a cross-laboratory study comparing ristocetin cofactor, collagen-binding and mAb-based assays. Journal of Thrombosis & Haemostasis. 2012;10:1043–1054. doi: 10.1111/j.1538-7836.2012.04729.x. [DOI] [PubMed] [Google Scholar]
- Federici AB. The use of desmopressin in von Willebrand disease: the experience of the first 30 years (1977-2007) Haemophilia. 2008;14 Suppl 1:5–14. doi: 10.1111/j.1365-2516.2007.01610.x. [DOI] [PubMed] [Google Scholar]
- Federici AB, Stabile F, Castaman G, Canciani MT, Mannucci PM. Treatment of acquired von Willebrand syndrome in patients with monoclonal gammopathy of uncertain significance: comparison of three different therapeutic approaches. Blood. 1998;92:2707–2711. [PubMed] [Google Scholar]
- Federici AB, Mannucci PM, Castaman G, Baronciani L, Bucciarelli P, Canciani MT, Pecci A, Lenting PJ, De Groot PG. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood. 2009;113:526–534. doi: 10.1182/blood-2008-04-152280. [DOI] [PubMed] [Google Scholar]
- Flood VH, Friedman KD, Gill JC, Morateck PA, Wren JS, Scott JP, Montgomery RR. Limitations of the ristocetin cofactor assay in measurement of von Willebrand factor function. J Thromb Haemost. 2009;7:1832–1839. doi: 10.1111/j.1538-7836.2009.03594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood VH, Gill JC, Morateck PA, Christopherson PA, Friedman KD, Haberichter SL, Branchford BR, Hoffmann RG, Abshire TC, Di Paola JA, Hoots WK, Leissinger C, Lusher JM, Ragni MV, Shapiro AD, Montgomery RR. Common VWF exon 28 polymorphisms in African Americans affecting the VWF activity assay by ristocetin cofactor. Blood. 2010;116:280–286. doi: 10.1182/blood-2009-10-249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood VH, Gill JC, Morateck PA, Christopherson PA, Friedman KD, Haberichter SL, Hoffmann RG, Montgomery RR. Gain-of-function GPIb ELISA assay for VWF activity in the Zimmerman Program for the Molecular and Clinical Biology of VWD. Blood. 2011;117:e67–74. doi: 10.1182/blood-2010-08-299016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood VH, Gill JC, Christopherson PA, Wren JS, Friedman KD, Haberichter SL, Hoffmann RG, Montgomery RR. Comparison of type I, type III and type VI collagen binding assays in diagnosis of von Willebrand disease. Journal of Thrombosis and Haemostasis. 2012;10:1425–1432. doi: 10.1111/j.1538-7836.2012.04747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Jr, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood. 1987;69:1691–1695. [PubMed] [Google Scholar]
- Goodeve A, Eikenboom J, Castaman G, Rodeghiero F, Federici AB, Batlle J, Meyer D, Mazurier C, Goudemand J, Schneppenheim R, Budde U, Ingerslev J, Habart D, Vorlova Z, Holmberg L, Lethagen S, Pasi J, Hill F, Hashemi Soteh M, Baronciani L, Hallden C, Guilliatt A, Lester W, Peake I. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM-1VWD) Blood. 2007;109:112–121. doi: 10.1182/blood-2006-05-020784. Erratum appears in Blood. 2008 Mar 15;111(6):3299-300. [DOI] [PubMed] [Google Scholar]
- Goudemand J, Negrier C, Ounnoughene N, Sultan Y. Clinical management of patients with von Willebrand's disease with a VHP vWF concentrate: the French experience. Haemophilia. 1998;4 Suppl 3:48–52. doi: 10.1046/j.1365-2516.1998.0040s3048.x. [DOI] [PubMed] [Google Scholar]
- Halimeh S, Krumpel A, Rott H, Bogdanova N, Budde U, Manner D, Faeser B, Mesters R, Nowak-Gottl U. Long-term secondary prophylaxis in children, adolescents and young adults with von Willebrand disease. Results of a cohort study. Thrombosis & Haemostasis. 2011;105:597–604. doi: 10.1160/TH10-09-0616. [DOI] [PubMed] [Google Scholar]
- Hamilton A, Ozelo M, Leggo J, Notley C, Brown H, Frontroth JP, Angelillo-Scherrer A, Baghaei F, Enayat SM, Favaloro E, Lillicrap D, Othman M. Frequency of platelet type versus type 2B von Willebrand disease. An international registry-based study. Thrombosis and haemostasis. 2011;105:501–508. doi: 10.1160/TH10-08-0523. [DOI] [PubMed] [Google Scholar]
- Hampshire DJ, Burghel GJ, Goudemand J, Bouvet LC, Eikenboom JC, Schneppenheim R, Budde U, Peake IR, Goodeve AC. Polymorphic variation within the VWF gene contributes to the failure to detect mutations in patients historically diagnosed with type 1 von Willebrand disease from the MCMDM-1VWD cohort. Haematologica. 2010;95:2163–2165. doi: 10.3324/haematol.2010.027177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James PD, Lillicrap D. von Willebrand disease: clinical and laboratory lessons learned from the large von Willebrand disease studies. American Journal of Hematology. 2012;87 Suppl 1:S4–11. doi: 10.1002/ajh.23142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James PD, Paterson AD, Notley C, Cameron C, Hegadorn C, Tinlin S, Brown C, O'Brien L, Leggo J, Lillicrap D Association Of Hemophilia Clinic Directors Of, C. Genetic linkage and association analysis in type 1 von Willebrand disease: results from the Canadian type 1 VWD study. Journal of Thrombosis & Haemostasis. 2006;4:783–792. doi: 10.1111/j.1538-7836.2006.01860.x. [DOI] [PubMed] [Google Scholar]
- James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood. 2013;122:636–640. doi: 10.1182/blood-2012-10-462085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Bowen D, Cumming A, Enayat S, Goodeve A, Hill M. The molecular analysis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors' Organisation Haemophilia Genetics Laboratory Network. Haemophilia. 2008;14:1099–1111. doi: 10.1111/j.1365-2516.2008.01813.x. [DOI] [PubMed] [Google Scholar]
- Krizek DR, Rick ME. A rapid method to visualize von willebrand factor multimers by using agarose gel electrophoresis, immunolocalization and luminographic detection. Thrombosis Research. 2000;97:457–462. doi: 10.1016/s0049-3848(99)00196-6. [DOI] [PubMed] [Google Scholar]
- Laffan M, Brown SA, Collins PW, Cumming AM, Hill FGH, Keeling D, Peake IR, Pasi KJ. The diagnosis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors' Organization. Haemophilia. 2004;10:199–217. doi: 10.1111/j.1365-2516.2004.00894.x. [DOI] [PubMed] [Google Scholar]
- Lawrie AS, Mackie IJ, Machin SJ, Peyvandi F. Evaluation of an automated platelet-based assay of ristocetin cofactor activity. Haemophilia. 2011;17:252–256. doi: 10.1111/j.1365-2516.2010.02419.x. [DOI] [PubMed] [Google Scholar]
- Lawrie AS, Stufano F, Canciani MT, Mackie IJ, Machin SJ, Peyvandi F. A comparative evaluation of a new automated assay for von Willebrand factor activity. Haemophilia. 2013;19:338–342. doi: 10.1111/hae.12064. [DOI] [PubMed] [Google Scholar]
- Lee CA, Chi C, Pavord SR, Bolton-Maggs PH, Pollard D, Hinchcliffe-Wood A, Kadir RA. The obstetric and gynaecological management of women with inherited bleeding disorders--review with guidelines produced by a taskforce of UK Haemophilia Centre Doctors' Organization. Haemophilia. 2006;12:301–336. doi: 10.1111/j.1365-2516.2006.01314.x. [DOI] [PubMed] [Google Scholar]
- Leissinger C, Becton D, Cornell C, Jr, Cox Gill J. High-dose DDAVP intranasal spray (Stimate) for the prevention and treatment of bleeding in patients with mild haemophilia A, mild or moderate type 1 von Willebrand disease and symptomatic carriers of haemophilia A. Haemophilia. 2001;7:258–266. doi: 10.1046/j.1365-2516.2001.00500.x. [DOI] [PubMed] [Google Scholar]
- Lusher JM. Clinical guidelines for treating von Willebrand disease patients who are not candidates for DDAVP--a survey of European physicians. Haemophilia. 1998;4 Suppl 3:11–14. doi: 10.1046/j.1365-2516.1998.0040s3011.x. [DOI] [PubMed] [Google Scholar]
- Mahieu B, Jacobs N, Mahieu S, Naelaerts K, Vertessen F, Weyler J, Jacquemyn Y, Van der Planken M. Haemostatic changes and acquired activated protein C resistance in normal pregnancy. Blood Coagulation and Fibrinolysis. 2007;18:685–688. doi: 10.1097/MBC.0b013e3282f09835. [DOI] [PubMed] [Google Scholar]
- Mannucci P. Desmopressin: a nontransfusional form of treatment for congenital and acquired bleeding disorders. Blood. 1988;72:1449–1455. see comments. [PubMed] [Google Scholar]
- Mannucci PM. Desmopressin (DDAVP) in the treatment of bleeding disorders: the first 20 years. Blood. 1997;90:2515–2521. [PubMed] [Google Scholar]
- Mannucci PM. How I treat patients with von Willebrand disease. Blood. 2001;97:1915–1919. doi: 10.1182/blood.v97.7.1915. [DOI] [PubMed] [Google Scholar]
- Mannucci PM, Tamaro G, Narchi G, Candotti G, Federici A, Altieri D, Tedesco F. Life-threatening reaction to factor VIII concentrate in a patient with severe von Willebrand disease and alloantibodies to von Willebrand factor. European Journal of Haematology. 1987;39:467–470. doi: 10.1111/j.1600-0609.1987.tb01458.x. [DOI] [PubMed] [Google Scholar]
- Mannucci PM, Tenconi PM, Castaman G, Rodeghiero F. Comparison of four virus-inactivated plasma concentrates for treatment of severe von Willebrand disease: a cross-over randomized trial. Blood. 1992a;79:3130–3137. [PubMed] [Google Scholar]
- Mannucci PM, And DB, Cattaneo M. Patterns of development of tachyphylaxis in patients with haemophilia and von Willebrand disease after repeated doses of desmopressin (DDAVP) British Journal of Haematology. 1992b;82:87–93. doi: 10.1111/j.1365-2141.1992.tb04598.x. [DOI] [PubMed] [Google Scholar]
- Mannucci P, Kyrle P, Schulman S, Di Paola J, Schneppenheim R, G J. Prophylactic efficacy and pharmacokinetically guided dosing of a von Willebrand factor/factor VIII concentrate in adults and children with von Willebrand's disease undergoing elective surgery: a pooled and comparative analysis of data from USA and European Union clinical trials. Blood Transfusion. 2013;11:533–540. doi: 10.2450/2013.0254-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus PD, Nire KG, Grooms L, Klima J, O'Brien SH. The power of a standardized bleeding score in diagnosing paediatric type 1 von Willebrand's disease and platelet function defects. Haemophilia. 2011;17:223–227. doi: 10.1111/j.1365-2516.2010.02390.x. [DOI] [PubMed] [Google Scholar]
- Millar CM, Riddell AF, Tuddenham EG. Consideration of platelet function disorders in patients with reduced VWF levels. Haemophilia. 2008a;14:1131–1132. doi: 10.1111/j.1365-2516.2008.01818.x. [DOI] [PubMed] [Google Scholar]
- Millar CM, Riddell AF, Brown SA, Starke R, Mackie I, Bowen DJ, Jenkins PV, van Mourik JA. Survival of von Willebrand factor released following DDAVP in a type 1 von Willebrand disease cohort: influence of glycosylation, proteolysis and gene mutations. Thrombosis & Haemostasis. 2008b;99:916–924. doi: 10.1160/TH07-09-0565. [DOI] [PubMed] [Google Scholar]
- Nitu-Whalley IC, Riddell A, Lee CA, Pasi KJ, Owens D, Enayat MS, Perkins SJ, Jenkins PV. Identification of type 2 von Willebrand disease in previously diagnosed type 1 patients: a reappraisal using phenotypes, genotypes and molecular modelling. Thrombosis & Haemostasis. 2000;84:998–1004. [PubMed] [Google Scholar]
- O'Connor D, Lester W, Willoughby S, Wilde JT. Pregnancy in platelet-type VWD: a case series. Thrombosis & Haemostasis. 2011;106:386–387. doi: 10.1160/TH11-02-0108. [DOI] [PubMed] [Google Scholar]
- Othman M. Platelet-type Von Willebrand disease: three decades in the life of a rare bleeding disorder. Blood Reviews. 2011;25:147–153. doi: 10.1016/j.blre.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Ott HW, Griesmacher A, Schnapka-Koepf M, Golderer G, Sieberer A, Spannagl M, Scheibe B, Perkhofer S, Will K, Budde U. Analysis of von Willebrand factor multimers by simultaneous high- and low-resolution vertical SDS-agarose gel electrophoresis and Cy5-labeled antibody high-sensitivity fluorescence detection. American Journal of Clinical Pathology. 2010;133:322–330. doi: 10.1309/AJCPZSBTD2BWOMVL. [DOI] [PubMed] [Google Scholar]
- Pasi KJ, Collins PW, Keeling DM, Brown SA, Cumming AM, Dolan GC, Hay CRM, Hill FGH, Laffan M, Peake IR. Management of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors' Organization. Haemophilia. 2004;10:218–231. doi: 10.1111/j.1365-2516.2004.00886.x. [DOI] [PubMed] [Google Scholar]
- Pergantou H, Xafaki P, Adamtziki E, Koletsi P, Komitopoulou A, Platokouki H. The challenging management of a child with type 3 von Willebrand disease and antibodies to von Willebrand factor. Haemophilia. 2012;18:e66–67. doi: 10.1111/j.1365-2516.2012.02799.x. [DOI] [PubMed] [Google Scholar]
- Preston FE. Assays for von Willebrand Factor Functional Activity: A UK NEQAS Survey. Thrombosis and Haemostasis. 1998;80:863–863. [PubMed] [Google Scholar]
- Pruthi RK, Daniels TM, Heit JA, Chen D, Owen WG, Nichols WL. Plasma von Willebrand factor multimer quantitative analysis by in-gel immunostaining and infrared fluorescent imaging. Thrombosis Research. 2010;126:543–549. doi: 10.1016/j.thromres.2010.09.015. [DOI] [PubMed] [Google Scholar]
- Quiroga T, Goycoolea M, Panes O, Aranda E, Martinez C, Belmont S, Munoz B, Zuniga P, Pereira J, Mezzano D. High prevalence of bleeders of unknown cause among patients with inherited mucocutaneous bleeding. A prospective study of 280 patients and 299 controls. Haematologica. 2007;92:357–365. doi: 10.3324/haematol.10816. [DOI] [PubMed] [Google Scholar]
- Ranger A, Manning RA, Lyall H, Laffan MA, Millar CM. Pregnancy in type 2B VWD: a case series. Haemophilia. 2012;18:406–412. doi: 10.1111/j.1365-2516.2011.02691.x. [DOI] [PubMed] [Google Scholar]
- Ray JG. DDAVP use during pregnancy: an analysis of its safety for mother and child. Obstetrical and Gynecological Survey. 1998;53:450–455. doi: 10.1097/00006254-199807000-00025. [DOI] [PubMed] [Google Scholar]
- Rodeghiero F, Tosetto A, Abshire T, Arnold DM, Coller B, James P, Neunert C, Lillicrap D On Behalf Of The Isth/Ssc Joint, V.W.F. & Perinatal/Pediatric Hemostasis Subcommittees Working, G. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. Journal of Thrombosis and Haemostasis. 2010;8:2063–2065. doi: 10.1111/j.1538-7836.2010.03975.x. [DOI] [PubMed] [Google Scholar]
- Rydz N, James PD. The evolution and value of bleeding assessment tools. Journal of Thrombosis and Haemostasis. 2012;10:2223–2229. doi: 10.1111/j.1538-7836.2012.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler JE. A revised classification of von Willebrand disease. For the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thrombosis & Haemostasis. 1994;71:520–525. [PubMed] [Google Scholar]
- Sadler JE. Von Willebrand disease type 1: a diagnosis in search of a disease. Blood. 2003;101:2089–2093. doi: 10.1182/blood-2002-09-2892. [DOI] [PubMed] [Google Scholar]
- Sadler JE, Budde U, Eikenboom JCJ, Favaloro EJ, Hill FGH, Holmberg L, Ingerslev J, Lee CA, Lillicrap D, Mannucci PM, Mazurier C, Meyer D, Nichols WL, Nishino M, Peake IR, Rodeghiero F, Schneppenheim R, Ruggeri ZM, Srivastava A, Montgomery RR, Federici AB Working Party on von Willebrand Disease, C. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. Journal of Thrombosis & Haemostasis. 2006;4:2103–2114. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- Sakurai Y, Shima M, Imai Y, Omura S, Kirita T, Yoshioka A. Successful use of recombinant factor VIII devoid of von Willebrand factor during multiple teeth extractions in a patient with type 3 von Willebrand disease. Blood Coagulation & Fibrinolysis. 2006;17:151–154. doi: 10.1097/01.mbc.0000214711.19116.09. [DOI] [PubMed] [Google Scholar]
- Sanchez-Luceros A, Meschengieser SS, Marchese C, Votta R, Casais P, Woods AI, Nadal MV, Salviu MJ, Lazzari MA. Factor VIII and von Willebrand factor changes during normal pregnancy and puerperium. Blood Coagulation and Fibrinolysis. 2003;14:647–651. doi: 10.1097/00001721-200310000-00005. [DOI] [PubMed] [Google Scholar]
- Stirling Y, Woolf L, North WR, Seghatchian MJ, Meade TW. Haemostasis in normal pregnancy. Thromb Haemost. 1984;52:176–182. [PubMed] [Google Scholar]
- Tiede A, Rand JH, Budde U, Ganser A, Federici AB. How I treat the acquired von Willebrand syndrome. Blood. 2011;117:6777–6785. doi: 10.1182/blood-2010-11-297580. [DOI] [PubMed] [Google Scholar]
- Tosetto A, Rodeghiero F, Castaman G, Goodeve A, Federici AB, Batlle J, Meyer D, Fressinaud E, Mazurier C, Goudemand J, Eikenboom J, Schneppenheim R, Budde U, Ingerslev J, Vorlova Z, Habart D, Holmberg L, Lethagen S, Pasi J, Hill F, Peake I. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1 VWD) Journal of Thrombosis & Haemostasis. 2006;4:766–773. doi: 10.1111/j.1538-7836.2006.01847.x. [DOI] [PubMed] [Google Scholar]
- Tosetto A, Castaman G, Rodeghiero F. Evidence-based diagnosis of type 1 von Willebrand disease: a Bayes theorem approach. Blood. 2008;111:3998–4003. doi: 10.1182/blood-2007-08-105940. [DOI] [PubMed] [Google Scholar]
- Tosetto A, Castaman G, Rodeghiero F. Bleeders, bleeding rates, and bleeding score. Journal of Thrombosis and Haemostasis. 2013;11 S1:142–150. doi: 10.1111/jth.12248. [DOI] [PubMed] [Google Scholar]
- Trigg DE, Stergiotou I, Peitsidis P, Kadir RA. A systematic review: The use of desmopressin for treatment and prophylaxis of bleeding disorders in pregnancy. Haemophilia. 2012;18:25–33. doi: 10.1111/j.1365-2516.2011.02573.x. [DOI] [PubMed] [Google Scholar]
- Walker ID, Walker JJ, Colvin BT, Letsky EA, Rivers R, Stevens R. Investigation and management of haemorrhagic disorders in pregnancy. Haemostasis and Thrombosis Task Force. Journal of Clinical Pathology. 1994;47:100–108. doi: 10.1136/jcp.47.2.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windyga J, von Depka-Prondzinski M on behalf of the European Wilate® Study Group. Efficacy and safety of a new generation von Willebrand factor/factor VIII concentrate (Wilate®) in the management of perioperative haemostasis in von Willebrand disease patients undergoing surgery. Thrombosis and Haemostasis. 2011;105:1072–1079. doi: 10.1160/TH10-10-0631. [DOI] [PubMed] [Google Scholar]
- Zhukov O, Popov J, Ramos R, Vause C, Ruden S, Sferruzza A, Dlott J, Sahud M. Measurement of von Willebrand factor-FVIII binding activity in patients with suspected von Willebrand disease type 2N: application of an ELISA-based assay in a reference laboratory. Haemophilia. 2009;15:788–796. doi: 10.1111/j.1365-2516.2009.01995.x. [DOI] [PubMed] [Google Scholar]