

Abstract
Genetic studies in patients reveal that mutations to genes that encode contractile proteins in medial smooth muscle cells can cause thoracic aortic aneurysms and dissections. Mouse models of such mutations, including Acta2−/− and Myh11 R247C/R247C, surprisingly do not present with any severe vascular phenotype under normal conditions. This observation raises the question whether these mutations nevertheless render the thoracic aorta increasingly vulnerable to aneurysms or dissections in the presence of additional, epigenetic, factors such as hypertension, a known risk factor for thoracic aortic disease. Accordingly, we compared the structure and biaxial mechanical properties of the ascending and descending thoracic aorta from male wild-type and Myh11 R247C/R247C mice under normotension and induced hypertension. On average, the mutant aortas exhibited near normal biomechanics under normotensive hemodynamics and near normal adaptations to hypertensive hemodynamics, yet the latter led to intramural delaminations or premature deaths in over 20 percent of these mice. Moreover, the delaminated vessels exhibited localized pools of mucoid material, similar to the common histopathologic characteristic observed in aortas from humans affected by thoracic aortic aneurysms and dissections. The present findings suggest, therefore, that mutations to smooth muscle cell contractile proteins may place the thoracic aorta at increased risk to epigenetic factors and that there is a need to focus on focal, not global, changes in aortic structure and properties, including the pooling of glycosaminoglycans / proteoglycans that may lead to thoracic aortic dissection.
Keywords: familial thoracic aortic aneurysms and dissections, smooth muscle myosin heavy chain, actomyosin functionality, wall stress and stiffness
1. Introduction
Advances in medical genetics have revealed diverse mutations that predispose individuals to thoracic aortic aneurysms and dissections (Lindsay and Dietz, 2011; Milewicz et al., 2008). One sub-class of these mutations directly affects actomyosin contractile activity within the medial smooth muscle cells (Guo et al., 2007, 2013; Wang et al., 2010; Zhu et al., 2006). That is, causative mutations include those that encode either the contractile proteins (e.g., ACTA2, which encodes smooth muscle specific a-actin, and MYH11, which encodes smooth muscle myosin heavy chain) or kinases involved in the actomyosin-mediated contraction and relaxation (e.g., MLCK, which encodes myosin light chain kinase, and PKG-1, which encodes a type 1 cGMP-dependent protein kinase). Notwithstanding accumulating clinical and histopathological data from patients harboring these mutations, the continuing lack of sufficient healthy and diseased tissue limits our ability to elucidate the underlying molecular and biomechanical mechanisms that lead to aneurysms or dissections. There is strong motivation, therefore, to exploit mouse models that can help increase our understanding of genetically induced aneurysms and dissections of the thoracic aorta.
Kuang et al. (2012) recently reported a knock-in mouse model (Myh11 R247C/R247U) of a recurrent, rare variant in Myh11 that results in a substitution of a cysteine for an arginine in the motor head region of the smooth muscle myosin heavy chain, which affects both ATP and actin-binding. The mutant aorta displays a decreased contractile response as expected, but there is no overall vascular phenotype under normal conditions. That is, the mice exhibit normal growth, aortic structure, systemic blood pressure, and life expectancy. Yet, smooth muscle cells explanted from the mutant aortas exhibit increased proliferation compared with wild type cells both in vitro and in vivo in a carotid artery ligation model (i.e., increased neointimal proliferation under no-flow conditions). The latter observation suggests that, despite an apparently near normal vascular development and maintenance under physiologic conditions, the elastic arteries in these mutants may nevertheless be more vulnerable to maladaptive responses to alterations in hemodynamic loading or vascular injury. Noting that hypertension is a risk factor for thoracic aortic aneurysms and dissections (Chan and Rabkin, 2014; Elefteriades, 2008), the goal of this work was to biomechanically phenotype the ascending and proximal descending thoracic aorta in Myh11 R247C/R247C mice under both normotensive and induced hypertensive conditions and to assess possible increased structural vulnerability in hypertension.
2. Methods
Mouse Models
All animal procedures conformed with NIH guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at Houston, and all transfer of tissue between Houston and New Haven was in compliance with a Material Transfer Agreement established between the two institutions. As described previously (Kuang et al., 2012), a germline Myh11 R247C/R247C mutation was achieved in mice having a mixed C57BL/6J × 129SvEv background. Male mice, both wild-type (Myh11 +/+, denoted herein as WT) and homozygous mutant ( Myh11 R247C/R247C, denoted herein as R247C) were challenged, beginning at 8 to 9 weeks of age, with a 18-week long combined treatment with NG-nitro-1-arginine methyl ester hydrochloride (3 g/L of drinking water), or L-NAME, and a high salt diet (83 NaCl chow, Harlan TD.92012). L-N AME increases blood pressure by blocking endothelial-derived nitric oxide synthase and salt loading is a common risk factor for hypertension. For completeness, data were collected for both normotensive (i.e., untreated, denoted uWT or uR247C) and hypertensive (i.e., treated, denoted tWT or tR247C) mice, the former of which allowed the first detailed assessments of the effects of genotype alone on the biomechanical properties. Blood pressures were measured in all four groups of mice using a standard tail-cuff method.
Mechanical testing
The ascending thoracic aorta (ATA) and proximal descending thoracic aorta (DTA) were obtained via a euthanize and harvest procedure (i.e., 350 mg/kg tribromoethanol IP and exsanguination upon removal of the aorta) and shipped overnight from Houston to New Haven in an iced HankaĂŹs balanced salt solution (HBSS, GIBCO by Life Science Technologies Corporation, Grand Island, NY). Consistent with prior reports (cf. Humphrey (2002)), preliminary experiments revealed that overnight storage does not affect the passive mechanical properties. Upon receipt, the vessels were cleaned of excessive perivascular tissue and mounted on glass cannulae for mechanical testing. In particular, the ATA was cannulated between the aortic root and brachiocephalic artery, while the left subclavian artery, left common carotid artery, and aortic arch (just distal to the left carotid) were ligated to allow pressurization. Similarly, the proximal DTA was cannulated approximately between the left subclavian artery and the third pair of intercostal arteries, with each included intercostal branch ligated to enable pressurization. The two extremities of the arteries were coupled securely to the glass cannulae using sutures. The mounted specimens were placed within a custom, computer-controlled biaxial device (Gleason et al., 2004), and tested in HBSS at ~ 37° C using established protocols (Ferruzzi et al., 2013). Briefly, following mechanical reacclimation (i.e., pulsatile loading from 90 to 110 mmHg at the in vivo axial length for 15 minutes) and preconditioning (i.e., 4 cycles of pressurization from 10 to 140 mmHg near the in vivo axial length), the luminal pressure, outer diameter, axial force, and axial extension were measured on-line using standard transducers, a video microscope, and custom software during two classes of tests: cyclic pressure-diameter ( P-d) tests at three different levels of axial extension (in vivo and ± 53 of in vivo) and cyclic axial force-length (f-l) tests at four different levels of luminal pressure (10, 60, 100, and 140 mmHg). To minimize possible testing-induced mechanical damage during these seven protocols, imposed transmural pressures did not exceed 140 mmHg and imposed axial loads did not exceed 5.5 grams, each consistent with prior studies of normotensive vessels (Ferruzzi et al., 2013). In this way, the associated data can also be compared directly, if desired, with those collected in similar studies of other mouse models (Ferruzzi et al., 2015; Wan et al., 2010). Immediately following biaxial testing, a ~ 2 mm long ring was cut from the proximal region of the specimen and imaged with a dissecting microscope at 20× magnification to measure the unloaded (i.e., traction-free) thickness H.
Quantification of Mechanical Properties
Pressure-diameter and axial force-length data provide insights into the structural stiffness of an artery, including via the clinical metric of distensibility
| (1) |
where di is the current inner diameter and subscripts sys and dias refer to configurations corresponding to systolic and diastolic pressures, respectively. Nevertheless, a detailed understanding of arterial mechanics requires an analysis of the associated stress-stretch behaviors and multiple metrics that can be inferred from them (Humphrey, 2002). Mean values of the circumferential and axial stretches were calculated via
| (2) |
where a, h, and l are values of the inner radius, wall thickness, and axial length in any loaded configuration and A, H, and L are similar values in an unloaded reference configuration. Note that the inner radii were calculated from the measured outer diameters, d0 and D 0 , via a = d0 /2 − h and A = D0 /2 − H and the assumption of volume preserving (isochoric) motions during testing. Mean values of the biaxial (i.e., in-plane) components of the Cauchy stress were calculated via
| (3) |
where, pexp is the luminal pressure measured by the transducers and fexp =fT + πa2 Pexp is the axial force acting on the closed-ended vessel in the device, with fT being the axial force measured by the force transducer. Following established methods (Ferruzzi et al., 2013), we quantified the mechanical behavior using a microstructurally motivated constitutive equation that relates the biaxial stresses and stretches via a stored energy function. When evaluated using unloading data, this function represents the energy stored elastically in the artery upon pressurization that would be available to do work on the distending fluid when depressurized. Our constitutive equation contains 8 parameters that can be determined via nonlinear regression by minimizing differences between theoretically predicted and experimentally measured P-d and f-l data, the latter of which can also be used to estimate energy dissipation during loading. These best-fit model parameters, in turn, were used to compute the material stiffness and related metrics. Details can be found elsewhere (Ferruzzi et al., 2013), but the basic equations are summarized in the Appendix. Finally, note that it is often best to evaluate the stress, stiffness, and energy at in vivo values of pressure and axial stretch, the latter of which is estimated easily in vitro as the value of axial stretch, Az, at which the transducer measured axial force does not change with changes in pressure. This value of stretch appears to be energetically favorable and has proven to be a good indicator of arterial adaptations to altered hemodynamic loads as well as genetic mutations (Humphrey et al., 2009).
Statistical Analyses and Data Presentation
Values of select geometric, structural, and material metrics were organized into quartiles for each of the eight experimental groups (i.e., uWT, tWT, uR247C, and tR247C for both aortic segments, ATA and DTA) and graphically represented in the form of boxplots to highlight the median (2nd quartile), spread (interquartile range between the 1st and the 3rd quartiles), and outliers (values below the 1st quartile or above the 3rd quartile by at least 1.5 the interquartile range). Non-parametric Kruskall-Wallis analyses were performed to test differences amongst groups for each metric; post-hoc pairwise comparisons were used to assess statistical significance (p < 0.05, with adjustment for multiple comparisons). Finally, data collected during the three P-d and four f-l protocols on each specimen were averaged using methods in Ferruzzi et al. (2013) to create ‘average datasets’ for each of the four treatment/genotype pairs (uWT, tWT, uR247C, tR247C) at each of the two anatomical locations (ATA, DTA). Specimens identified as outliers by the quartile analysis were not included in these averaged data sets.
Histology
Arterial segments were fixed in formalin following mechanical testing and stored in 703 EtOH until all specimens could be embedded in paraffin, sectioned, and stained together. Standard Hematoxylin & Eosin (H&E), Verhoeff-Van Gieson (VVG), MassonaAZs Trichrome (MTC), and Movat pentachrome (Mov) stains were used to assess cell morphology and the organization of extracellular constituents such as the elastic laminae, collagen fibers, smooth muscle cells, and glycosaminoglycans (GAGs). An Olympus BX/51 microscope (Olympus Inc., Center Valley, PA), equipped with an Olympus DP70 digital camera, was used to acquire images of the stained aortic cross-sections using consistent setting. Fdisentisbility
3. Results
Table 1 lists physiological data (mean ± SD) for all surviving mice for the four basic experimental groups: untreated wild-type (uWT), L-NAME and salt treated wild-type (tWT), untreated Myh11 R247C/R247C (uR247C), and L-NAME and salt treated Myh11 R247C/R247C (tR247C). The blood pressure of untreated R247C mice was not significantly different from that of the WT mice. Treatment with L-NAME and salt significantly increased the diastolic and systolic pressures in the WT (by 40 and 41%, respectively) and especially in the R247C (by 80 and 58%) mice. Percent increase in pulse pressure due to treatment was higher in WT (41%, statistically significant) than in R247C (26%) mice. Neither genotype nor hypertension affected body mass.
Table 1.
Mean ± SD physiologic data for the four groups: untreated wild-type (uWT), treated wild-type (tWT), untreated Myh11R247C/R247C (uR247C), and treated Myh11R247C/R247C (tR247C), where treatment combined L-NAME and high salt for 18 weeks beginning at 8 to 9 weeks of age.
| uWT | tWT | uR247C | tR247C | |
|---|---|---|---|---|
| Number of Animals | 5 | 5 | 5 | 8 |
| Age (weeks) | 27.3 ± 1.0 | 26.7 ± 1.0 | 26.0 ± 2.0 | 26.9 ± 1.4 |
| Body Mass (g) | 27.4 ± 4.3 | 27.2 ± 4.0 | 29.0 ± 2.6 | 23.6 ± 2.4 |
| Blood Pressure (mmHg) | ||||
| Diastolic | 91 ± 6 | 127 ± 8* | 82 ± 2 | 141 ± 18* |
| Systolic | 124 ± 7 | 175 ± 12* | 117 ± 3 | 185 ± 16* |
p < 0.05 between treated and untreated mice with the same genotype.
Table 2 lists morphological and mechanical data for all 45 aortic specimens that were tested, namely, ascending thoracic aorta (ATA) and proximal descending thoracic aorta (DTA) from the four experimental groups (uWT, tWT, uR247C, tR247C). On average, aortas from the untreated mutants had smaller outer diameters than did the untreated wildtype mice despite wall thickness being slightly greater in the mutants. Treatment with L-NAME and salt resulted in similar increases in outer diameter (8 to 16%) independent of genotype. As expected, treatment-induced hypertension also tended to increase wall thickness and decrease the in vivo (i.e., preferred) axial stretch. Interestingly, however, the treatment-induced increase in wall thickness was significant only for the WT mice, in which pulse pressure increased the most (cf. Table 1). Note that the computed mean wall stresses at mean arterial pressure (MAP) were not significantly different as a function of genotype or blood pressure, with the only exception of the circumferential stress in the treated R247C mice. However, the large standard deviations in the treated mutants suggested a need to consider the possible existence of outliers.
Table 2.
Mean ± SD morphologic and mechanical data for the ascending thoracic aorta (ATA) and the proximal descending thoracic aorta (DTA) for the four groups: untreated wild-type (uWT), treated wild-type (tWT), untreated Myh11 R247C/R247C (uR247C), and treated Myh11R247C/R247C (tR247C). Note that one ATA was too short to test reliably. MAP denotes the group-specific mean arterial pressure.
|
| ||||
| Ascending Thoracic Aorta (ATA) | ||||
| uWT | tWT | uR247C | tR247C | |
|
| ||||
| Number of Specimens (n) | 5 | 5 | 5 | 7 |
| Unloaded Dimensions | ||||
| Outer Diameter (μm) | 1099 ± 106 | 1254 ± 128 | 1025 ± 58 | 1154 ± 121 |
| Wall Thickness (μm) | 104 ± 11 | 142 ± 13* | 117 ± 9 | 124 ± 19 |
| In-vitro Axial Length (mm) | 2.50 ± 0.29 | 2.96 ± 0.44 | 2.99 ± 0.57 | 2.56 ± 0.44 |
| Dimensions at MAP | ||||
| Outer Diameter (μm) | 1619 ± 132 | 1783 ± 190 | 1486 ± 110 | 1712 ± 275 |
| Wall Thickness (μm) | 38 ± 5 | 56 ± 6* | 41 ± 5 | 48 ± 11 |
| In-vitro Axial Stretch (−) | 1.73 ± 0.05 | 1.63 ± 0.09 | 1.81 ± 0.15 | 1.64 ± 0.09 |
| Cauchy Stresses (kPa) at MAP | ||||
| Circumferential | 276 ± 38 | 278 ± 26 | 212 ± 21 | 387 ± 182* |
| Axial | 313 ± 40 | 243 ± 31 | 272 ± 31 | 312 ± 112 |
|
| ||||
| Descending Thoracic Aorta (DTA) | ||||
| uWT | tWT | uR247C | tR247C | |
|
| ||||
| Number of Specimens (n) | 5 | 5 | 5 | 8 |
| Unloaded Dimensions | ||||
| Outer Diameter (μm) | 1001 ± 62 | 1089 ± 76 | 978 ± 19 | 1088 ± 182 |
| Wall Thickness (μm) | 98 ± 8 | 142 ± 31* | 106 ± 9 | 141 ± 42 |
| In-vitro Axial Length (mm) | 5.31 ± 0.78 | 7.49 ± 0.64 | 5.01 ± 1.38 | 5.64 ± 1.19 |
| Dimensions at MAP | ||||
| Outer Diameter (μm) | 1382 ± 93 | 1561 ± 125 | 1336 ± 85 | 1547 ± 141* |
| Wall Thickness (μm) | 43 ± 5 | 60 ± 13* | 46 ± 3 | 62 ± 25 |
| In-vitro Axial Stretch (−) | 1.54 ± 0.09 | 1.49 ± 0.05 | 1.54 ± 0.07 | 1.50 ± 0.09 |
| Cauchy Stresses (kPa) at MAP | ||||
| Circumferential | 205 ± 16 | 229 ± 30 | 164 ± 12 | 274 ± 101 |
| Axial | 225 ± 36 | 214 ± 30 | 190 ± 23 | 223 ± 75 |
|
| ||||
p < 0.05 between treated and untreated mice with the same genotype.
Arrangement of the data into quartiles indeed identified one mouse as an outlier in the treated wild-type group (i.e., 1 DTA and 0 ATA, or 1 out of 10 vessels) and three mice as outliers in the treated mutant group (i.e., 2 DTA and, albeit less marked, 1 ATA, or 3 out of 15 vessels), each consistent with visual inspections of the specimens during testing and examination of the raw mechanical data (Table 2). All specimens from these four outlier mice were thus excluded from the averaging procedure that was used to generate ‘representative’ datasets for the eight experimental groups (ATA and DTA for uWT, tWT, uR247C, and tR247C). Figure 1 (left panels) shows the averaged pressure-diameter responses for each group. Note the lack of a significant difference due to the mutation and only modest increases in structural stiffness with treatment in both the WT and R247C vessels (i.e., leftward shift in the curves), consistent with the aforementioned trend towards increased wall thickness due to hypertension. Table 3 lists the associated best-fit constitutive parameters for these eight groups based on the averaged datasets, and Figure 1 (right panels) shows the associated averaged circumferential stress-stretch responses. These results reveal similar material behaviors, again independent of genotype and treatment. Both Table 2 (mean ± SD for all data) and model predictions based on the averaged datasets (i.e., excluding outliers) suggested that the Myh11 R247C/R247C mutation modestly lowered both the values of circumferential and axial stress at mean arterial pressure and the individual preferred axial stretch in the untreated mice. Interestingly, the results suggested further that, when excluding outliers, L-NAME and salt treatment of the mutant mice seemed to have a partial compensatory effect in the axial direction whereby stress (279 kPa in the ATA and 254 kPa in the DTA) was restored toward control levels. Conversely, the circumferential stress (335 kPa in ATA and 290 kPa in DTA) increased above control values in the treated mutant mice, which suggested some remodeling interplay between the two principal directions. Overall, however, these findings collectively suggested that the mutant ascending and proximal descending aorta managed, on average, to compensate well biomechanically, even when challenged with induced hypertension, and to achieve a near normal gross mechanical behavior, albeit at a different in vivo working point.
Figure 1.

Mean ± SEM pressure - diameter responses (left panels) and circumferential Cauchy stress - stretch responses (right panels) for the ascending thoracic aorta (ATA, top panels) and the proximal descending thoracic aorta (DTA, bottom panels), excluding data from the 4 outliers. Datasets for the untreated (u) and treated (t, L-NAME + NaCl) wild-type (WT) as wells as for untreated (u) and treated (t) Myh11 R247C/R247C (R247C) mice reveal only modest differences due to either the mutation or the induced hypertension (i.e., L-NAME plus high salt treatment). Mean P-d curves were obtained by averaging 5 ATAs/DTAs for the uWT, the uR247C, and the tR247C groups, and 4 ATAs/DTAs for the tWT group.
Table 3.
Material parameters estimated by fitting the microstructurally motivated constitutive relation (Appendix 1) to average datasets (excluding outliers) for the ascending thoracic aorta (ATA) and proximal descending thoracic aorta (DTA) from untreated wild-type (uWT), treated wild-type (tWT), untreated R247C mutant (uR247C), and treated R247C mutant (tR247C) mice. RMSE denotes the root mean sum-of-the-squares of the error for the regression. Although individual parameters can be compared across groups, better interpretations come from resulting values of the linearized stiffness and stored energy (cf. Figures 3 and 5).
| c (kPa) |
(kPa) |
(−) |
(kPa) |
(−) |
(kPa) |
(−) |
α0
(deg) |
RSME (−) |
|
|---|---|---|---|---|---|---|---|---|---|
| ATA | |||||||||
| uWT | 32.38 | 12.67 | 0.04 | 15.00 | 0.24 | 4.57 | 0.50 | 42.08 | 0.060 |
| tWT | 29.06 | 9.54 | 0.10 | 14.40 | 0.47 | 3.50 | 0.82 | 41.76 | 0.053 |
| uR247C | 20.01 | 6.62 | 0.12 | 16.02 | 0.16 | 3.70 | 0.40 | 41.31 | 0.064 |
| tR247C | 26.10 | 6.89 | 0.19 | 20.94 | 0.26 | 5.69 | 0.63 | 41.25 | 0.058 |
| DTA | |||||||||
| uWT | 45.17 | 0.65 | 1.60 | 13.14 | 0.64 | 0.20 | 2.50 | 30.75 | 0.061 |
| tWT | 33.73 | 2.96 | 1.26 | 14.04 | 0.48 | 0.27 | 2.53 | 28.57 | 0.054 |
| uR247C | 36.19 | 4.66 | 0.34 | 23.21 | 0.25 | 0.27 | 2.26 | 26.85 | 0.087 |
| tR247C | 28.25 | 2.11 | 0.99 | 18.18 | 0.36 | 1.12 | 1.46 | 25.52 | 0.078 |
Although stress-stretch plots (Figure 1, right panels) and associated modeling (Table 3) provide considerable insight into the material behavior, the primary function of an elastic artery is to store energy when distended during systole and to use that energy to work on the blood during diastole to augment blood flow and smooth the pulse pressure. Plots of average energy storage as a function of biaxial stretch (i.e., circumferential distension and axial extension) thus provide useful functional information. For example, Figure 2 reveals that the ATA was more compliant and isotropic in its biaxial behavior than the DTA, independent of genotype and treatment, and that the average energy stored at the group specific mean arterial pressure and in vivo axial stretch (solid circle) was greater in the ATA than in the DTA in the untreated vessels, independent of genotype. These two observations are consistent with in vivo differences in loading of the ATA and DTA, with the ATA alone experiencing both cyclic distension and extension. Figure 2 reveals further, however, that treatment-induced hypertension reduced energy storage in all cases, due in part to the decreased in vivo axial stretch (cf. Table 2). Finally, stored energy was less for untreated mutants compared with untreated wild-type for both the ATA and DTA, yet the hypertension, on average, restored the mutant stored energy contours closer to control. Overall, therefore, consistent with the initial report of no vascular phenotype due to the Myh11 R247C/R247C mutation (Kuang et al., 2012), differences in biaxial mechanical behaviors between average wild-type and average mutant aortas were subtle, not dramatic, in both normotension and induced hypertension. In other words, with the exception of the few outliers, the thoracic aorta appeared both to develop and to adapt to increased blood pressures reasonably well despite the mutation.
Figure 2.
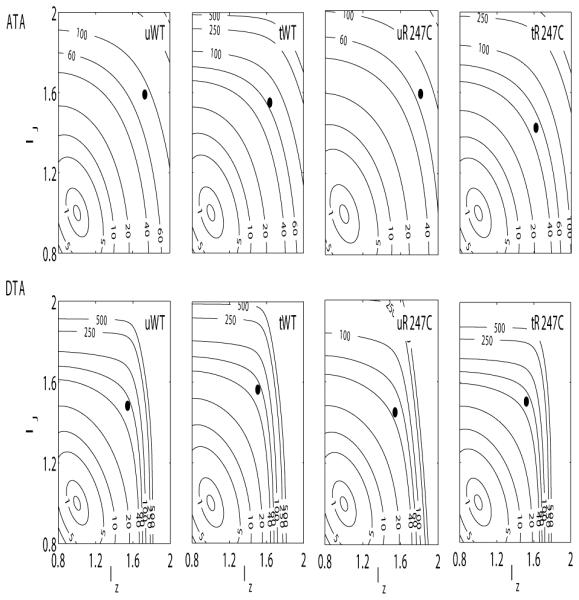
Contour plots of constant strain energy (W) as a function of biaxial stretch λθ - λz for the untreated wild-type (first column), treated wild-type (second column), untreated Myh11 R247C/R247C (third column) and treated Myh11 R247C/R247C (fourth column) mice. The strain energy functions were estimated from average datasets for the ascending thoracic aorta (ATA, top panels) and proximal descending thoracic aorta (DTA, bottom panels). The solid black circle in each plot indicates the in vivo conditions (mean arterial pressure and preferred axial stretch). The superimposed numerical values on the contours denote the values of strain energy in kPa. The mean strain energy functions (W) representative for each combination of genotype, treatment, and location were obtained by averaging experimental data from 5 ATAs/DTAs for the uWT, the uR247C, and the tR247C groups, and 4 ATAs/DTAs for the tWT group.
Figure 3 supports this conclusion, but shows additional measures of stiffness and energy dissipation for all specimens in terms of boxplots to highlight the overall median and spread of these metrics as well as the associated outliers (circled cross). As it can be seen, most of the metrics followed similar patterns in the ATA and DTA. Circumferential (material) stiffness increased with hypertension, but only significantly so in the mutant, whereas axial stiffness was not different statistically. Consistent with this finding, plus that of increased wall thickness due to treatment (cf. Table 2), distensibility D (Equation 1) decreased dramatically with hypertension in all cases. Due to similarities between the wild-type and the ‘adapted mutants’ in both normotension and hypertension, however, this common clinical metric would likely not be revealing of disease or dissection potential.
Figure 3.

Mechanical metrics of material stiffness (left panels), and structural stiffness and energy dissipation (rigth panels) for the ascending thoracic aorta (ATA, top panels) and proximal descending thoracic aorta (DTA, bottom panels) of untreated (u) and treated (L-Name + NaCl, t) wild-type (WT) as well as for untreated (u) and treated (t) Myh11R247C/R247C (R247C) mice, showed at in vivo conditions (mean arterial pressure and preferred axial stretch). Cvvvv and Czzzz are values of linearized stiffness in circumferential and axial direction, respectively. D is the distensibility computed at in vivo conditions (mean arterial pressure and preferred axial stretch). Wdis is the percentage of energy dissipated during the mechanical test, relative to the energy stored after loading. Note: circled cross symbols indicate outliers. Data from all collected vessels were included in the statistical analysis, i.e. 5 ATAs/DTAs for the uWT, tWT, and the uR247C groups, 7 ATAs and 8 DTAs for the tR247C group.
Consistent with observed phenotypic diversity in patients harboring various mutations, we hypothesized that the mutant outliers identified by statistical analysis of the gross mechanical data may have actually manifested thoracic disease, likely driven in part by the epigenetic factor of induced hypertension. Figure 4 thus contrasts averaged pressure-diameter (left panels) and stress-stretch (right panels) results versus those for the 3 mutant outliers identified by analysis of stiffness. These outliers clearly represent maladaptations that did not compensate for the combined effects of mutation and treatment. In particular, outlier 3 exhibited a much stiffer behavior compared with the average, mainly in the DTA, whereas the other two outliers were globally more compliant, mostly in the circumferential direction. Most strikingly, however, Figure 5 shows gross images, histological sections, and energy storage from four representative R247C descending thoracic aortas: one untreated ‘average compensated’ DTA (first column), one treated ‘average adapted’ DTA (second column), and two treated outlier DTAs (third and fourth columns). Similar to prior tests on thoracic aorta from wild-types (Ferruzzi et al., 2013), the untreated mutant vessels tended to have a smooth, nearly translucent appearance during in vitro testing. Induced hypertension reduced this translucency, consistent with a thickening of the wall that appeared to include changes in both the media (e.g., increased inter-lamellar spacing) and adventitia. Whereas cross-sections from the average mutant appeared contiguous whether treated or not, note the marked intramural delaminations within the two outliers (white arrows). Note, too, the dramatic thickening of the wall in the two outliers, perhaps due to an attempted reparative response (i.e., these delaminations appeared to have occurred in vivo, not during testing, which would likely have elicited a healing response). Although there was some increase in GAGs (blue in Movat stained sections) due to the induced hypertension (compare sections in the first and second columns), there was a much greater accumulation in the two outliers (black arrows), including a marked pooling of GAGs in outlier 2 in the outer media that appeared to correlate with the location of the delamination. Unfortunately, it was not possible to correlate the precise regions of delaminations and mucoid staining because these segments were used for different measurements. Finally, note the marked differences in energy storage in the vessels exhibiting a delamination, which revealed decreased ranges of distension and extension as well as energy storage, all indicators of a compromised mechanical functionality.
Figure 4.

Pressure - diameter (left panels) and circumferential stress - stretch (rigth panels) data for the ascending thoracic aorta (ATA, top panels) and the proximal descending thoracic aorta (DTA, bottom panels). The datasets corresponding to the 3 treated Myh11 R247C/R247C mutant outliers (ol, o2, o3) are compared with the average dataset for the treated mutant group in each plot, for both ATA and DTAs. The standard error of the mean is shown for selected points of the average dataset. Mean P-d curves for the tR247C group were obtained by averaging the data from the 5 ATAs and the 5 DTAs (5 treated mutant mice) that were not classified as outliers.
Figure 5.

Gross appearance, microstructure and mechanical behavior of descending thoracic aortas from one representative ‘average’ untreated Myh11 R247C/R247C mouse (first column, uR247C), one representative ‘average’ treated Myh11 R247C/R247C mouse (second column, tR247C), and two of the three treated Myh11 R247C/R247C 'outlier’ mice (third and fourth columns, tR247C-o2 and tR247C-o3 ). The panels in the first row show the gross appearance during biaxial testing, the panels in the second row show cross-sections of rings cut from the vessel after mechanical testing, the panels in the third row show Movat-stained histological sections (60x magnification), and the panels in the forth row show the contour plots of constant strain energy (W). The thin white arrows highlight intramural delaminations that were evident by eye. The thick white arrows highlight accumulated mucoid material, which tended to increase diffusely with induced hypertension but to accumulate more focally in the outliers having evidence of intramural delaminations. Note that the histological sections were necessarily taken from different regions than the isolated rings. Similar to Figure 3, the solid black circle in each contour plot indicates the specimen-specific in vivo condition (mean arterial pressure and preferred axial stretch). The superimposed numerical values on the contours denote the values of strain energy in kPa.
Figure 6 shows an unusual feature in the DTA of the second mutant outlier. On gross imaging, there was a significant perivascular accumulation of collagen near one of the intercostal branches. Movat and VVG-stained sections suggested that there was an underlying focal intramural delamination near the inlet to the intercostal artery as well as frank interlamellar delaminations at sites adjacent to this branch, again corresponding to an apparent pooling of GAGs in the outer medial lamellae (Figure 5, Movat-stained cut in the third column). It thus appeared that this marked increase in perivascular tissue may have been stimulated as a healing response by an increased density of cells (H&E staining), likely fibroblasts, given the nearby delamination.
Figure 6.
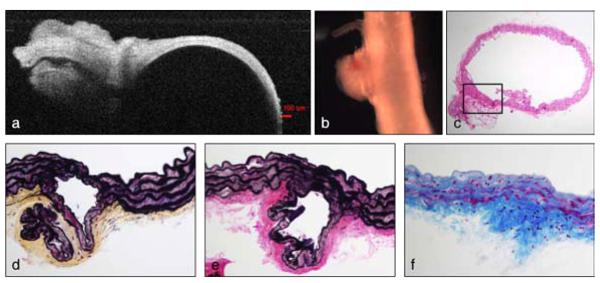
Full vessel and stained cross-sectional views of the proximal descending thoracic aorta from the second outlier in the Myh11 R247C/R247C treated group. A dense mesh of fibrotic tissue originated from the adventitial layer of the aorta (b, naked eye), in the proximity of one of the intercostal branches (a, Optical Coherence Tomographic image during mechanical testing). Cross-sectional cuts stained with H&E (c, lOx magnification), Movat (d, 40x magnification), VVG (e, 40× magnification), and MTC (f, 40× magnification) selectively show cells and/or ECM components and reveal a separation between the elastic laminae within the fibrotic region (magnified).
4. Discussion
Genetic mutations affecting either smooth muscle α-actin (encoded by the gene ACTA2) or smooth muscle myosin heavy chain (encoded by the gene MYH11) are causative for human thoracic aortic aneurysms and dissections (Guo et al., 2007; Zhu et al., 2006). Histopathological characteristics in the media include disruption or loss of elastic fibers, disarray or loss of smooth muscle cells, and significant accumulations of mucoid material (i.e., glycosaminoglycans / proteoglycans). Many of these patients present with aortic dissection, with or without aneurysm, at ages ranging from teenagers to older adults. Zhu et al. (2006) reported further that the MYH11 mutation correlated with a ~ 3-fold increased aortic structural stiffness and an associated ~ 1.7-fold increase in pulse wave velocity, not unlike that seen in Marfan syndrome patients (de Wit et al., 2013) who are similarly susceptible to thoracic aneurysms and dissections (El-Hamamsy and Yacoub, 2009; Lindsay and Dietz, 2011). It is now generally accepted, therefore, that the smooth muscle contractile apparatus is important in establishing, maintaining, or restoring the structural integrity of the thoracic aorta (Humphrey et al., 2014; Milewicz et al., 2008; Prakash et al., 2010; Renard et al., 2013).
It has been somewhat surprising, therefore, that mouse models of altered smooth muscle α-actin (Acta2−/−, see Schildmeyer et al. 2000) and smooth muscle myosin heavy chain (Myh11 R247C/R247C, see Kuang et al. 2012) do not exhibit a severe vascular phenotype under normal conditions. Yet, a full understanding of the biomechanical phenotype requires a detailed assessment of the biaxial mechanical properties under normal and hemodynamically stressed conditions, which has not been attempted heretofore for these two mouse models. Consistent with prior histological findings by Kuang et al. (2012), including lack of marked elastic fiber disruption, we found that, on average, the biaxial mechanical properties of the ascending and proximal descending thoracic aorta from mutants exhibited modest, not dramatic, differences from those of age-matched wild-type controls. The mutant aortas tended to be slightly less stiff materially (Figure 3), but somewhat stiffer structurally as revealed by pressure-diameter data (Figure 1) and calculated values of distensibility (Figure 3, right panels). This increased structural stiffness was likely due in part to the slight, though not statistically significant, increase in wall thickness (Table 2), but it was not nearly as dramatic as that reported for humans (Zhu et al., 2006). The mutant vessels also tended to store less elastic energy upon pressurization and to dissipate more energy than the controls (Figures 2 and 3), but the latter was marked only in the ascending aorta. Diminished energy storage decreases overall aortic function, but does not necessarily imply a compromised structural integrity (Ferruzzi et al., 2015). We conclude, therefore, that the thoracic aorta in Myh11 R247C/R247C mice compensated during development so as to exhibit sufficient mechanical functionality and structural integrity and not develop aneurysms or dissections under normotensive conditions. Indeed, it appears that many viable mutant mouse arteries similarly compensate under normal conditions to achieve sufficient biomechanical properties and functionality (Louis et al., 2007; Bersi et al., 2014; Ferruzzi et al., 2015; Sather et al., 2012).
A similar comparison of the biomechanical properties of wild-type and mutant aortas subjected to an 18-week period of induced hypertension revealed that, on average, the aortas from the surviving mutants were also able to adapt well. In particular, similar to wild-types, the two primary changes with hypertension were a further increase in wall thickness and decrease in the in vivo value of axial stretch, both of which helped to decrease the pressureinduced increases in intramural stress toward normal values. A similar adaptation to induced hypertension has been reported for an elastin haploinsufficient mouse model (Wagenseil et al., 2007). Yet, there was an increased vulnerability to intramural delaminations by the R247C aortas in hypertension (Figure 5): only 1of10 wild-type specimens (103), but 3of15 mutant specimens (203) showed signs of intramural delamination, that is, separation of layers within the wall without frank dissection. In each of these cases, however, the adventitia appeared robust, which likely prevented any progression to rupture (Li et al., 2014). To these numbers, we add that the overall mortality of the R247C mice with hypertension was also greater than WT (263 vs. 73 after 8 weeks of treatment), with deaths in the R247C ascribed to acute aortic dissections based on necropsy (unpublished data). It should be noted, therefore, that complementary studies in wild-type mice have not identified thoracic aortic failures in either the two-kidney, 1-clip (2K1C) model or the aortic arch banding model of hypertension (Chen et al., 2011; Kuang et al., 2013; Wagenseil et al., 2007), though chronic infusion of a high concentration of angiotensin-II can lead to dissection (Ju et al., 2013).
It is well known that GAGs accumulate within the aorta in both aging and hypertension (Sawabe, 2010; Wight, 2008), yet these accumulations tend to be diffuse, not focal, and not to result in aneurysms or dissections. In contrast, one of the most distinguishing histopathological features of thoracic aortic aneurysms and dissections is a focal accumulation, often pooling, of GAGs (Humphrey, 2013; Jain et al., 2011). We observed herein that the hypertensive aortas appearing mechanically as outliers (i.e., not adaptive) tended both to have focal accumulations of GAGs within the media and evidence of intralamellar delaminations (cf. Figure 5), though not frank dissections. Although we cannot establish causality based on the present results (e.g., GAGs could accumulate in response to prior injury), a recent computational model supports the hypothesis that pooled GAGs can precede spontaneous intramural delaminations since they represent sites of geometric and material discontinuity as well as possibly increased osmotic (Donnan) swelling pressures (Roccabianca et al., 2014). This hypothesis thus merits further testing. Finally, althought it is not clear what leads to a local production and pooling of GAGs in the case of diminished actomyosin activity, we suggest that compromised mechancosensing may play an important role (Humphrey et al., 2014). This hypothesis also merits testing.
In summary, it is widely recognized that the primary function of the thoracic aorta is mechanical and consequently that there is a pressing need for more analyses of the mechanics (de Wit et al., 2013; Elefteriades, 2008; Wu et al., 2013). The present results represent the first assessment of the biaxial mechanical properties of the thoracic aorta in any mouse model of altered smooth muscle functionality. Somewhat surprisingly, we found that, on average, the thoracic aorta inMyh11 R247C/R247C mice appears both to develop under physiologic loading and to adapt under induced hypertensive loading so as to maintain sufficient mechanical functionality and structural integrity. Nevertheless, however, phenotypic diversity manifested as a 263 early mortality of the hypertensive mutants and evidence of intramural delaminations in 203 percent of the surviving hypertensive mutants. The latter associated with focal, not diffuse, accumulations of GAGs. Because of the apparent highly focal nature of these intramural defects, one must be careful not to over interpret any results that are necessarily averaged, including those from Western blots, PCR, or measures of structural stiffness such as distensibility (D). Rather, there is a need for increased spatially-specific immunohistochemical data and computational models to test further the hypothesis that highly focal defects may increase overall vulnerability to intramural delaminations, which in turn may lead to lethal dissections or ruptures.
Supplementary Material
Acknowledgments
This work was supported, in part, by grants from the National Institutes of Health [ROl HL062594 and P50 HL083794 to D.M.M., R03 EB016810 and UOl HL116323 to J.D.H.] and the National Marfan Foundation (to J .D.H.).
Appendix 1
The structurally motivated ‘four-fiber family’ model for the stored energy W can be written as (Ferruzzi et al., 2013)
| (4) |
where c and are material parameters having the dimension of stress, are dimensionless material parameters, C = FT F is the left Cauchy-Green deformation tensor, with F = diag(λr, λv, λz) the deformation gradient tensor, and is a unit vector defining the orientation of the ith family of fibers (i = 1, 2, 3, 4) in the reference configuration, with the reference angle between the ith family of fibers and the axial direction. Note, too, that IC = trC and are coordinate-invariant measures of deformation that can be expressed in terms of the principal stretches as
| (5) |
where IV Ci has the physical interpretation of the square of the stretch in the direction of the ith family of fibers. Assuming incompressibility, detF = 1, reduces the number of independent principal stretches and allows calculation of the radial stretch as λr = 1/( λvλz), where λr= h/H.
It can be shown (Humphrey, 2002) that the Cauchy stress tensor is determined by
| (6) |
where p is a Lagrange multiplier that enforces incompressibility and is the spatial second order identity tensor, with i= σij the Kronecker delta. Hence, the two theoretically determined components of Cauchy stress are
| (7) |
| (8) |
where the Lagrange multiplier p = c/ (λv2λz2) results from the assumption of plane stress, namely trr < tvv, tzz.
These equations for stress, in turn, were used to determine theoretical values of luminal pressure (Pth) and axial force (fth) as a function of the 8 unknown model parameters. A non-linear least-square regression, based on the Levenberg-Marquardt algorithm, was then used to estimate the unknown parameters by minimization of the objective function e proposed by Wan et al. 2010
| (9) |
where N is the total number of data points, the superscripts exp and th denote experimentally measured and theoretically predicted, respectively, with the overbars denoting to average values.
Finally, circumferential and axial components of the linearized stiffness, Cvvvv and Czzzz, were determined from the stored energy function, viz.
| (10) |
where lower case indices can be r, v, or z and upper case indices can be R, Θ, or Z, and the components of the deformation gradient tensor (F) as well as of the deformation-dependent part of the Cauchy stress tensor (t̄) are referred to the configuration about which the linearization is performed. Note that the values of linearized stiffness can be interpreted as the local slope of the corresponding stress-stretch curve about the configuration of interest provided that experiments are performed appropriately.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
None to declare.
References
- Bersi MR, Ferruzzi J, Eberth JF, Gleason RL, Jr, Humphrey JD. Consistent biomechanical phenotyping of common carotid arteries from seven genetic, pharmacological, and surgical mouse models. Ann Biomed Eng. 2014 Jun;42(6):1207–23. doi: 10.1007/s10439-014-0988-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KK, Rabkin SW. Increasing prevalence of hypertension among patients with thoracic aorta dissection: Trends over eight decades-a structured meta-analysis. Am J Hypertens. 2014 Jul;27(7):907–17. doi: 10.1093/ajh/hpt293. [DOI] [PubMed] [Google Scholar]
- Chen J, Wu J, Li L, Zou Y-Z, Zhu D-L, Gao P-J. Effect of an acute mechanical stimulus on aortic structure in the transverse aortic constriction mouse model. Clin Exp Pharmacol Physiol. 2011 Sep;38(9):570–6. doi: 10.1111/j.1440-1681.2011.05544.x. [DOI] [PubMed] [Google Scholar]
- de Wit A, Vis K, Jeremy RW. Aortic stiffness in heritable aortopathies: relationship to aneurysm growth rate. Heart Lung Circ. 2013 Jan;22(1):3–11. doi: 10.1016/j.hlc.2012.08.049. [DOI] [PubMed] [Google Scholar]
- El-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol. 2009 Dec;6(12):771–86. doi: 10.1038/nrcardio.2009.191. [DOI] [PubMed] [Google Scholar]
- Elefteriades JA. Thoracic aortic aneurysm: reading the enemy's playbook. Curr Probl Cardiol. 2008 May;33(5):203–77. doi: 10.1016/j.cpcardiol.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Ferruzzi J, Bersi MR, Humphrey JD. Biomechanical phenotyping of central arteries in health and disease: advantages of and methods for murine models. Ann Biomed Eng. 2013 Jul;41(7):1311–30. doi: 10.1007/s10439-013-0799-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferruzzi J, Bersi MR, Yanagisawa H, Humphrey JD. Fibulin-5 deficiency increases structural stiffness and decreases elastic energy storage consistently within central arteries. J Biomech Eng. 2015 doi: 10.1115/1.4029431. (accepted, pending minor revisions) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason RL, Gray SP, Wilson E, Humphrey JD. A multiaxial computer-controlled organ culture and biomechanical device for mouse carotid arteries. J Biomech Eng. 2004 Dec;126(6):787–95. doi: 10.1115/1.1824130. [DOI] [PubMed] [Google Scholar]
- Guo D-C, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (acta2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007 Dec;39(12):1488–93. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- Guo D.-c., Regalado E, Casteel DE, Santos-Cortez RL, Gong L, Kim JJ, Dyack S, Horne SG, Chang G, Jondeau G, Boileau C, Coselli JS, Li Z, Leal SM, Shendure J, Rieder MJ, Bamshad MJ, Nickerson DA, GenTAC Registry Consortium, National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project. Kim C, Milewicz DM. Recurrent gain-of-function mutation in prkgl causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. 2013 Aug;93(2):398–404. doi: 10.1016/j.ajhg.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey JD. Springer-Verlag; 2002. Cardiovascular Solid Mechanics: Cells, Tissues, and Organs. [Google Scholar]
- Humphrey JD. Possible mechanical roles of glycosaminoglycans in thoracic aortic dissection and associations with dysregulated transforming growth factor-Is. J Vase Res. 2013;50(1):1–10. doi: 10.1159/000342436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey JD, Eberth JF, Dye WW, Gleason RL. Fundamental role of axial stress in compensatory adaptations by arteries. J Biomech. 2009 Jan;42(1):1–8. doi: 10.1016/j.jbiomech.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey JD, Milewicz DM, Tellides G, Schwartz MA. Cell biology. dysfunctional mechanosensing in aneurysms. Science. 2014 May;344(6183):477–9. doi: 10.1126/science.1253026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain D, Dietz HC, Oswald GL, Maleszewski JJ, Halushka MK. Causes and histopathology of ascending aortic disease in children and young adults. Cardiovasc Pathol. 2011;20(1):15–25. doi: 10.1016/j.carpath.2009.09.008. 2011/2// [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju X, Ijaz T, Sun H, Ray S, Lejeune W, Lee C, Recinos A, 3rd, Guo D-C, Milewicz DM, Tilton RG, Brasier AR. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin ii via the t-helper lymphocyte 17-interleukin 17 axis in c57bl/6 mice. Arterioscler Thromb Vase Biol. 2013 Jul;33(7):1612–21. doi: 10.1161/ATVBAHA.112.301049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang S-Q, Geng L, Prakash SK, Cao J-M, Guo S, Villamizar C, Kwartler CS, Peters AM, Brasier AR, Milewicz DM. Aortic remodeling after transverse aortic constriction in mice is attenuated with atl receptor blockade. Arterioscler Thromb Vase Biol. 2013 Sep;33(9):2172–9. doi: 10.1161/ATVBAHA.113.301624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang S-Q, Kwartler CS, Byanova KL, Pham J, Gong L, Prakash SK, Huang J, Kamm KE, Stull JT, Sweeney HL, Milewicz DM. Rare, nonsynonymous variant in the smooth muscle-specific isoform of myosin heavy chain, myhll, r247c, alters force generation in the aorta and phenotype of smooth muscle cells. Circ Res. 2012 May;110(11):1411–22. doi: 10.1161/CIRCRESAHA.111.261743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014 Feb;124(2):755–67. doi: 10.1172/JCI69942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011 May;473(7347):308–16. doi: 10.1038/nature10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis H, Kakou A, Regnault V, Labat C, Bressenot A, Gao-Li J, Gardner H, Thornton SN, Challande P, Li Z, Lacolley P. Role of alpl-integrin in arterial stiffness and angiotensin-induced arterial wall hypertrophy in mice. Am J Physiol Heart Circ Physiol. 2007;293(4):H2597–H2604. doi: 10.1152/ajpheart.00299.2007. [DOI] [PubMed] [Google Scholar]
- Milewicz DM, Guo D-C, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- Prakash SK, LeMaire SA, Guo D-C, Russell L, Regalado ES, Golabbakhsh H, Johnson RJ, Safi HJ, Estrera AL, Coselli JS, Bray MS, Leal SM, Milewicz DM, Belmont JW. Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am J Hum Genet. 2010 Dec;87(6):743–56. doi: 10.1016/j.ajhg.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard M, Callewaert B, Baetens M, Campens L, MacDermot K, Fryns J-P, Bonduelle M, Dietz HC, Gaspar IM, Cavaco D, Stattin E-L, Schrander-Stumpel C, Coucke P, Loeys B, De Paepe A, De Backer J. Novel myhll and acta2 mutations reveal a role for enhanced tgfls signaling in ftaad. Int J Cardiol. 2013 May;165(2):314–21. doi: 10.1016/j.ijcard.2011.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roccabianca S, Ateshian GA, Humphrey JD. Biomechanical roles of medial pooling of glycosaminoglycans in thoracic aortic dissection. Biomech Model Mechanobiol. 2014 Jan;13(1):13–25. doi: 10.1007/s10237-013-0482-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather BA, Hageman D, Wagenseil JE. Murray's law in elastin haploinsufficient (eln+/aLS) and wild-type (wt) mice. Journal ofBiomechanical Engineering. 2012;134(12):124504–124504. doi: 10.1115/1.4023093. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawabe M. Vascular aging: from molecular mechanism to clinical significance. Geriatr Gerontol Int. 2010 Jul;10(Suppl 1):S213–20. doi: 10.1111/j.1447-0594.2010.00603.x. [DOI] [PubMed] [Google Scholar]
- Schildmeyer LA, Braun R, Taffet G, Debiasi M, Burns AE, Bradley A, Schwartz RJ. Impaired vascular contractility and blood pressure homeostasis in the smooth muscle alpha-actin null mouse. FASEB J. 2000 Nov;14(14):2213–20. doi: 10.1096/fj.99-0927com. [DOI] [PubMed] [Google Scholar]
- Wagenseil JE, Knutsen RH, Li DY, Mecham RP. Elastin-insufficient mice show normal cardiovascular remodeling in 2klc hypertension despite higher baseline pressure and unique cardiovascular architecture. Am J Physiol Heart Circ Physiol. 2007 Jul;293(1):H574–82. doi: 10.1152/ajpheart.00205.2007. [DOI] [PubMed] [Google Scholar]
- Wan W, Yanagisawa H, Gleason RL., Jr Biomechanical and microstructural properties of common carotid arteries from fibulin-5 null mice. Ann Biomed Eng. 2010 Dec;38(12):3605–17. doi: 10.1007/s10439-010-0114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Guo D.-c., Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He W-Q, Zhu M-S, Offermanns S, Gilchrist D, Elefteriades J, Stull JT, Milewicz DM. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010 Nov;87(5):701–7. doi: 10.1016/j.ajhg.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wight TN. Arterial remodeling in vascular disease: a key role for hyaluronan and versican. Front Biosci. 2008;13:4933–7. doi: 10.2741/3052. [DOI] [PubMed] [Google Scholar]
- Wu D, Shen YH, Russell L, Coselli JS, LeMaire SA. Molecular mechanisms of thoracic aortic dissection. J Surg Res. 2013 Oct;184(2):907–24. doi: 10.1016/j.jss.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gase J-M, Brunotte F, Bruneval P, Wolf J-E, Michel J-B, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006 Mar;38(3):343–9. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.