

Abstract
DNA methylation-dependent epigenetic regulation plays important roles in the development and function of the mammalian nervous system. MeCP2 is a key player in recognizing methylated DNA and interpreting the epigenetic information encoded in different DNA methylation patterns. Mutations in the MECP2 gene cause Rett syndrome, a devastating neurological disease that shares many features with autism. One interesting aspect of MeCP2 function is that it can be phosphorylated in response to diverse stimuli. Insights into the regulation and function of MeCP2 phosphorylation will help improve our understanding of how MeCP2 integrates environmental stimuli in neuronal nuclei to generate adaptive responses and may eventually lead to treatments for patients.
Keywords: MeCP2, phosphorylation, Rett syndrome
Introduction
DNA methylation and histone tail modifications are the two most extensively studied epigenetic marks. Methyl-DNA binding proteins can act as a molecular linker between these two epigenetic marks. As the founding member of the methyl- DNA binding domain containing protein (MBD) gene family (Lewis et al., 1992), the methyl-CpG binding protein 2 gene (MECP2) has been linked to Rett syndrome (RTT) (Amir et al., 1999), an autism spectrum developmental disorder (Hagberg, 1985). Moreover, several recent studies have found that MECP2 is altered at both the genomic level and the expression level in many autism patients (Nagarajan et al., 2006; Xi et al., 2007; Nagarajan et al., 2008; Ramocki et al., 2009). Thus, studying the function of MeCP2 will not only advance our understanding of RTT, but may also provide insights into the mechanisms underlying a broad spectrum of neurological diseases.
The MeCP2 protein specifically binds to methylated DNA (Lewis et al., 1992; Nan et al., 1997). Earlier studies are mostly consistent with MeCP2 acting as a transcription repressor through its interaction with a core repressor complex containing mSin3A and histone deacetylases (Jones et al., 1998; Nan et al., 1998). However, recent evidence suggests MeCP2 can also activate gene transcription through its interaction with CREB and co-activators (Chahrour et al., 2008). MeCP2 protein is almost as abundant as the histone octamers in the mouse brain, and is widely distributed across the entire genome tracking the density of 5-methylcytosine (Skene et al., 2010). Similar to histones, MeCP2 is subject to posttranslational modifications, such as phosphorylation (Chen et al., 2003). Thus, MeCP2 appears to have the necessary molecular properties in serving as a master molecular switch on the chromatin to integrate diverse extracellular signals and generate adaptive transcriptional/functional outputs. To test this hypothesis, several key questions need to be addressed. First, how many of these potential sites get phosphorylated in neurons in vivo? Second, what stimulus induces phosphorylation at which site in what type of neurons? Third, what is the in vivo function of any such phosphorylation? Fourth, does any such phosphorylation change the ability of MeCP2 to bind to either methyl-CpG or MeCP2-interacting proteins? Here, we will review the recent advances in studying MeCP2 phosphorylation, focusing on the mechanisms of how MeCP2 phosphorylation is regulated and how phosphorylation fine-tunes MeCP2 function. We will also summarize the results from mouse models in understanding the in vivo roles of MeCP2 phosphorylation in the development and function of the mammalian brain.
MeCP2 phosphorylation
MeCP2 phosphorylation was initially discovered by the Greenberg group in a study aimed to identify the role of MeCP2 in neuronal activity-dependent transcription regulation (Chen et al., 2003). A previously unknown slow-migrating form of MeCP2 was observed from protein lysate of membrane-depolarized cortical neurons in SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Alkaline phosphatase treatment of the lysate led to the disappearance of this slow-migrating form of MeCP2, suggesting this MeCP2 species is a result of phosphorylation (Chen et al., 2003). This phosphorylation site was later identified as serine 421 (S421), because S421 to alanine mutation abolished this neuronal activity-induced MeCP2 mobility shift (Zhou et al., 2006). However, S421 is not the only site of MeCP2 that can be phosphorylated, as mass spectrometry analysis of immuno-precipitated MeCP2 from normal and epileptic rodent brains identified 8 potential phosphorylation sites, including S80, T148/S149, S164, S229, S399, S421 and S424 (Tao et al., 2009). Interestingly, phosphorylation of S421 and S424 is only present in the slow-migrating form of MeCP2 purified from the epileptic brain, whereas phosphorylation of other sites exists in both the basal and slow-migrating forms of MeCP2 (Tao et al., 2009). Most recently, three additional MeCP2 phosphorylation sites (S86, S274 and T308) have been identified by phosphotryptic mapping (Ebert et al., 2013). MeCP2 phosphorylation at S86, S274 and T308 is detectable under basal condition, but is greatly induced by neuronal activity in both cultured cortical neurons and intact brains. Many of the phosphorylation sites identified so far are located in important functional domains of the MeCP2 protein (Figure 1), suggesting that the precise regulation of the phosphorylation state at these sites may significantly influence the molecular function of MeCP2.
Figure 1.
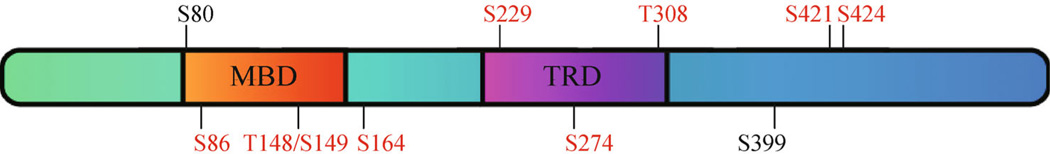
Distribution of known phosphorylation sites on the MeCP2 protein. Neuronal activity-induced phosphorylation sites are marked in red. MBD, methyl-CpG binding domain; TRD, transcriptional repression domain.
Regulation of MeCP2 phosphorylation
In cultured cortical neurons, membrane depolarization-induced MeCP2 S421 phosphorylation can be detected as early as 5 min after stimulation, and gradually reaches its maximal level in 30–60 min after depolarization (Chen et al., 2003; Zhou et al., 2006). Neurotransmission stimulation with glutamate, NMDA or GABAA-receptor antagonist bicuculline and neurotrophin treatment with BDNF, NT3, or NT4 are also sufficient to induce MeCP2 S421 phosphorylation in cortical neurons (Zhou et al., 2006). The induction of MeCP2 S421 phosphorylation requires calcium influx through the L-type voltage-gated calcium channels (L-VSCCs), as LVSCCs antagonist nimodipine blocks neuronal activity-dependent MeCP2 S421 phosphorylation (Chen et al., 2003; Zhou et al., 2006).
Consistent with the involvement of calcium signaling pathway, two calcium/calmodulin-dependent protein (CaM) kinases, CamKII and CamKIV, have been implicated in mediating MeCP2 S421 phosphorylation. Zhou et al. showed that specific inhibitor of CamKII abolishes membrane depolarization-induced MeCP2 S421 phosphorylation, and recombinant constitutively active CamKII is able to phosphorylate MeCP2 in vitro (Zhou et al., 2006). Buchthal et al. further confirmed that CamKII, generally considered as a cytosolic enzyme, is also present in the nucleus, and overexpression of a constitutively active form of CamKII in cultured hippocampal neurons is sufficient to induce MeCP2 S421 phosphorylation (Buchthal et al., 2012). Tao and colleagues, however, showed that siRNA knockdown of CamKIV greatly attenuates membrane depolarization-induced MeCP2 S421 phosphorylation (Tao et al., 2009). No direct binding has been established between MeCP2 and either kinase. Whether CamKII/IV indeed directly phosphorylates MeCP2 in vivo or actually acts as an upstream intermediate for the induction of S421 phosphorylation remains unknown.
Although MeCP2 is ubiquitously expressed in many mammalian tissues, MeCP2 S421 phosphorylation is only detected in the brain (Zhou et al., 2006). Stimulus-induced MeCP2 S421 phosphorylation has been reported in specific neuronal subtypes in various brain regions. The first reported case is light-induced S421 phosphorylation in neurons of the suprachiasmatic nucleus (SCN) (Zhou et al., 2006). In the hypothalamus, early life stress induces S421 phosphorylation specifically in neurons in the parvocellular division of the hypothalamic paraventricular nucleus (Murgatroyd et al., 2009). In the striatum, antidepressant drugs, such as amphetamine (AMPH) and imipramine, have been shown to induce S421 phosphorylation selectively in GABAergic interneurons of the ventral structure of nucleus accumbens (NAc) (Deng et al., 2010; Hutchinson et al., 2012b), while cocaine has been shown to induce S421 phosphorylation in both ventral (NAc) and dorsal (caudate putamen) structures (Mao et al., 2011). In the hippocampus, both fear conditioning and Morris water maze training induce S421 phosphorylation in CA1–3 neurons (Li et al., 2011). In the peripheral nervous system (PNS), S421 phosphorylation was first observed in the superficial dorsal horn neurons in the spinal cord after induction of peripheral joint inflammation (Geranton et al., 2007). Later work from the same group suggested S421 phosphorylation in the superficial dorsal horn neurons is regulated by the convergence of the descending serotonergic inputs and the ascending primary afferent stimulation (Géranton et al., 2008), which provides the first example of integration of synaptic inputs through MeCP2 phosphorylation. Such type of integration also appears to be responsible for the neuronal subtype specificity of AMPH-induced S421 phosphorylation in the NAc. AMPH activates multiple monoamine neurotransmitter systems, among which activation of the dopamine (DA) and serotonin (5-HT) systems, but not the norepinephrine (NE) system, are sufficient to induce S421 phosphorylation (Hutchinson et al., 2012a). A separate study by Mao et al. (2011) has implicated the NMDA receptor as the mediator of cocaine-induced S421 phosphorylation in the dorsal, but not the ventral striatum. To date, S421 phosphorylation has been observed only in postmitotic neurons.
Opposite to the dynamics of S421 phosphorylation, S80 is the most constitutively phosphorylated residue in resting neurons and undergoes dephosphorylation upon membrane depolarization (Tao et al., 2009). S80 dephosphorylation also requires calcium influx through L-VSCCs, as nimodipine effectively blocks membrane depolarization-induced S80 dephosphorylation (Tao et al., 2009). Different from S421 phosphorylation, S80 phosphorylation has been observed in non-neuronal cells, such as Hela cells and human fibroblasts (Bracaglia et al., 2009; Tao et al., 2009). Homeodomain-interacting protein kinase 2 (HIPK2) binds to MeCP2 and is able to phosphorylate MeCP2 at S80 in vitro (Bracaglia et al., 2009). Furthermore, overexpression of HIPK2 in human fibroblasts results in a significant increase in S80 phosphorylation, and siRNA knockdown of HIPK2 in cortical neurons strongly reduces the level of S80 phosphorylation, further supporting the involvement of HIPK2 in mediating S80 phosphorylation (Bracaglia et al., 2009). The phosphatase responsible for the neuronal activity-dependent S80 dephosphorylation is still unknown.
In cortical neurons, MeCP2 phosphorylation at S86, S274 and T308 is also induced by membrane depolarization. Interestingly, stimulation with BDNF or with forskolin to activate protein kinase A (PKA) is able to induce S86 and S274 phosphorylation, but fails to activate T308 phosphorylation (Ebert et al., 2013).
A summary of our current knowledge on the extracellular signals and the intracellular signaling pathways leading to the phosphorylation/dephosphorylation of various residues on MeCP2 protein is shown in Figure 2.
Figure 2.

Neuronal activity-induced phosphorylation and dephosphorylation modify MeCP2 function. Stimulation of NMDA receptors, DA receptors and 5-HT receptors, as well as membrane depolarization, activates L-type calcium channels on the cellular membrane and induces calcium influx. Specific kinases and phosphatases in the nucleus are subsequently activated and modify the phosphorylation status of MeCP2. Phosphoprylation of S421, together with dephosphorylation of S80, may modulate the binding of MeCP2 to the promoters of specific genes. Dephosphorylation of S80 releases DGCR8 from MeCP2 to regulate microRNA processing. Phosphorylation of T308 interrupts the association between MeCP2 and NCoR co-repressor complex. NMDA receptors, N-methyl-D-aspartate receptors; DA receptors, Dopamine receptors; 5-HT receptors, 5-hydroxytryptamine receptors or serotonin receptors.
Phosphorylation modifies the function of MeCP2
Phosphorylation is an important regulator of protein function. The addition of a phosphate group can modify the activity of the target protein by modifying protein electrostatics, by inducing protein conformational changes, and by altering protein–protein interactions. Here, we summarize recent findings in how phosphorylation at multiple residues modifies the function of MeCP2 in various ways, including DNA binding, transcription regulation, and protein–protein interactions.
The relative slow kinetics of MeCP2 S421 phosphorylation after membrane depolarization correlates with the slow induction of a subgroup of neuronal activity-regulated genes, such as Bdnf and Narp (Chen et al., 2003; Zhou et al., 2006), raising the possibility that the phosphorylation of MeCP2 S421 plays a role in the activation of this class of genes. Consistent with this hypothesis, Chen et al. showed that membrane depolarization induces the release of MeCP2 from Bdnf promoter III in rat cortical neurons, and the slow-migrating phosphorylated form of MeCP2 binds poorly to methylated Bdnf promoter in vitro (Chen et al., 2003). Zhou et al. (2006) further presented that neuronal activity-dependent Bdnf induction is attenuated in cortical neurons overexpressing the non-phosphorylatable MeCP2S421A mutant protein, compared to neurons overexpressing wild type (WT) MeCP2. However, these results were challenged by a recent study, in which Cohen et al. (2011) showed that membrane depolarization does not alter the binding of MeCP2 to the promoters of multiple neuronal activity-dependent genes, and the MeCP2 occupancy at Bdnf promoter is indistinguishable between cortical neurons cultured from WT and Mecp2S421A knockin mice. The authors reasoned that the inconsistent results among multiple studies are possibly due to the fact that the chromatin immunoprecipitation (ChIP) assays performed in the earlier work were semiquantitative and more subject to error. In addition, the extent and time course of Bdnf induction upon membrane depolarization is not significantly different between WT and Mecp2S421A cortical neurons (Cohen et al., 2011). But it remains possible that phosphorylation of MeCP2 at additional sites are involved in regulating Bdnf expression.
In fact, S424 phosphorylation was observed together with S421 phosphorylation in slow-migrating form of MeCP2 induced by neuronal activity (Tao et al., 2009). We reported that Mecp2S421A;S424A knockin mice exhibit increased Bdnf transcript level in the hippocampus, and the MeCP2S421A;S424A protein binds more tightly to Bdnf promoters than the wild type MeCP2 does, suggesting S424 phosphorylation may collaborate with S421 phosphorylation in regulating gene transcription (Li et al., 2011). In addition to its direct effect on gene transcription, increased chromatin binding by the phosphor mutant MeCP2 protein may also influence the modification of DNA by further protecting the methylated CpG dinucleotides. This is possible because the loss of MeCP2 has been shown to correlate with an increase in hydroxymethylation at CpG sites (Szulwach et al., 2011), which may have been a result of loss of protection of the those methylated CpGs.
S80 is located at the N-terminal of the MBD domain of MeCP2, thus phosphorylation of S80 was proposed to affect the DNA binding affinity of MeCP2. Tao et al. demonstrated that mutating S80 to alanine leads to decreased binding of MeCP2 at the promoters of multiple genes, including Rab3d, Vamp3 and Igsf4b. They also showed that cortical neurons overexpressing MeCP2S80A exhibit increased Rab3d, Vamp3 and Igsf4b expression compared with neurons overexpressing WT MeCP2 (Tao et al., 2009). Moreover, S80 phosphorylation has been recently identified as a crucial regulator of the interaction between MeCP2 and DGCR8 in modulating nuclear microRNA processing (Cheng et al., 2014). Cheng at al. reported that the N terminus and the C terminus of MeCP2 form an intramolecular interaction. In resting neurons, phosphorylation at S80 blocks this intramolecular interaction, making the C terminus of MeCP2 accessible for its association with DGCR8. In response to neuronal activity, S80 undergoes dephosphorylation and the intramolecular interaction of MeCP2 is reestablished, leading to the release of DGCR8 for regulating neuronal activity-dependent micro-RNA processing (Cheng et al., 2014). Furthermore, Gonzales et al. suggested that phosphorylation of MeCP2 at S80 may influence the association of MeCP2 to transcription repressor Sin3A, heterochromatin protein 1 (HP1), cohesin complex member SMC3, and RNA binding protein YB1 (Gonzales et al., 2012). In addition, Rexach et al. demonstrated a reversible yin-yang relationship between S80 phosphorylation and MeCP2 glycosylation (Rexach et al., 2010), which was the first report suggesting a crosstalk between MeCP2 phosphorylation and other posttranslational modifications.
The phosphorylation site T308 is proximal to a common RTT missense mutation R306C, a mutation that disrupts the ability of MeCP2 to interact with the nuclear receptor co-repressor (NCoR) complex (Lyst et al., 2013). Ebert et al. demonstrated that phosphorylation of T308 abolishes the interaction between MeCP2 and NCoR complex, and suppresses MeCP2-NCoR-mediated transcription repression (Ebert et al., 2013). In cortical neurons isolated from T308A knockin mice, the induction of a subset of neuronal activity-regulated genes, including Npas4 and Bdnf, is reduced compared to the WT counterpart (Ebert et al., 2013).
In an early report, Miyake at al. suggested that phosphorylation of MeCP2 regulates the intracellular localization of MeCP2 during neuronal cell differentiation (Miyake and Nagai, 2007). However, multiple studies using phospho-specific antibodies of MeCP2 showed that the subcellular distribution of MeCP2 with phosphorylation at S80, S229, or S421 is indistinguishable from that of total MeCP2, and mutating serine to alanine or aspartic acid at these residues does not affect the subcellular localization of MeCP2 (Zhou et al., 2006; Tao et al., 2009; Gonzales et al., 2012).
A summary of our current knowledge on how phosphorylation at several sites may regulate the molecular function of MeCP2 is shown in Figure 2.
In vivo function of MeCP2 phosphorylation
Among all the phosphorylation sites of MeCP2, S421 has been the most extensively studied. In vitro results have suggested that neuronal activity-induced S421 phosphorylation may regulate MeCP2 binding to the Bdnf promoter (Chen et al., 2003), Bdnf transcription, dendritic growth and spine maturation (Zhou et al., 2006). Several correlative in vivo studies have implicated S421 phosphorylation in regulating pain sensitivity (Géranton et al., 2007, 2008), encoding early life stress (Murgatroyd et al., 2009), and modulating behavioral responses to psychostimulants (Deng et al., 2010). To directly study the in vivo function of S421 phosphorylation, two knockin mice were independently generated. The Greenberg group made a Mecp2S421A allele. Our group made a Mecp2S421A;S424A allele. We decided to make the S421A;S424A double knockin allele because our mass spectrometry study identified both serines potentially phosphorylated in response to neuronal activity (Tao et al., 2009).
Neural plasticity and cognitive function
Rett syndrome features mental retardation and loss of acquired language and motor skills (Chahrour and Zoghbi, 2007). Mecp2 deficient mice display impaired learning and memory (Moretti et al., 2006), attenuated LTP and LTD (Asaka et al., 2006; Moretti et al., 2006). Furthermore, neurons from Mecp2 deficient mice present decreased dendritic complexity and reduced excitatory synapse formation (Chao et al., 2007). Elucidating the role of MeCP2 S421 phosphorylation in brain development and plasticity is a key step to understand how S421 phosphorylation modifies MeCP2 function and how S421 phosphorylation might contribute to Rett syndrome.
Because both increased and decreased expression of MeCP2 leads to deficits in neuronal function, we first confirmed that the level of MeCP2S421A;S424A is comparable to that of the wild type MeCP2, and that S421 phosphorylation is abolished in the Mecp2S421A;S424A/y mice (Li et al., 2011). While we made multiple attempts to generate an antibody that specifically recognizes phosphorylated S424, we have yet to succeed. The lack of a phosphor-S424 antibody prevents us from directly assess the status S424 phosphorylation in vivo. Nonetheless, we performed detailed analysis of the Mecp2S421A;S424A/y mice, and found that, comparing to WT mice, they perform better in two hippocampus-dependent spatial learning/memory assays (fear conditioning and Morris water maze), have enhanced long-term potentiation at two hippocampal synapses (the Schaffer collateral-CA1 synapse and the dentate-CA3 synapse), have increased number of excitatory synapses on hippocampal and cortical neurons, and show related gene expression changes in the adult hippocampus (Li et al., 2011). It is worth noting that these phenotypes of Mecp2S421A;S424A/y mice are remarkably similar to what was observed in mice with MeCP2 overexpression, and opposite to that of Mecp2 deficient mice (Collins et al., 2004; Chao et al., 2007; Chahrour et al., 2008). In addition, the MeCP2S421A;S424A protein appears to bind more tightly to gene promoters than the wild type MeCP2 does (Li et al., 2011). These results indicate that loss of S421/S424 phosphorylation might turn MeCP2 to a hypermorph.
MeCP2 has also been implicated in homeostatic synaptic scaling, a non-Hebbian form of synaptic plasticity (Qiu et al., 2012). MeCP2 S421 phosphorylation is dynamically regulated during the process of synaptic scaling (Zhou et al., 2006; Tao et al., 2009; Qiu et al., 2012). Our group tested whether S421 and/or S424 phosphorylation is required for homeostatic synaptic scaling. We demonstrated that hippocampal neurons cultured from Mecp2S421A;S424A/y mice present normal tetrodotoxin (TTX)-induced synaptic scaling up, but exhibit impaired bicuculline-induced synaptic scaling down (Zhong et al., 2012). In addition, neurons from Mecp2S421A;S424A/y mice showed reduced metabotropic glutamate receptor 5 (mGluR5) at both mRNA and protein levels than neurons from WT mice. Pretreatment with group I mGluR agonist or overexpression of GluR5 in Mecp2S421A;S424A/y neurons is sufficient to restore the bicuculline-induced synaptic scaling down to normal level (Zhong et al., 2012).
In a separate study, the Greenberg group reported their characterization of the Mecp2S421A/y mice (Cohen et al., 2011). Surprisingly, the Mecp2S421A/y mice only showed mild phenotypes, including increased dendritic complexity, increased inhibitory neurotransmission, and a deficit in hippocampal learning/memory (novel object recognition behavior). Moreover, they reported no change in MeCP2 binding to DNA across the genome after neuronal activity-induced S421 phosphorylation (Cohen et al., 2011). It should be cautioned that it is difficult to achieve the necessary sequencing depth in ChIP-seq analysis to accurately quantify MeCP2 binding changes across the genome, because MeCP2 binds to so many places across the genome. Despite of this caveat, the Mecp2S421A/y mice appear to be phenotypically different from the Mecp2S421A;S424A/y mice. The obvious genetic difference (S424 vs. A424) between these two mice may help explain the phenotypic difference. First, S424 may be either phosphorylated or modified in some unknown ways in response to neuronal activity, which work together with S421 phosphorylation to ensure the correct function of MeCP2. Indeed, the MeCP2 sequence from S421 to S424, when S421 is phosphorylated, matches the consensus phosphorylation site of Casein kinase 1 (pS/T-X-X-S/T). The possible crosstalk between S421 and S424 phosphorylation requires further investigation. Second, the A424 mutation may have affected MeCP2 function independent of neuronal activity-induced S421 phosphorylation. Generation of the Mecp2S424A/y mouse and a phospho-S424 specific antibody will undoubtedly help distinguish the two possibilities.
Monoamine neurotransmission related mood regulation and drug addiction
Monoamine neurotransmission is essential in emotion regulation and has been implicated in mood disorders, such as depression and anxiety. RTT patients develop anxiety, aggression, and mood alterations (Chahrour and Zoghbi, 2007), accompanied with reduced levels of dopamine and serotonin (5-HT) metabolite (Fyffe et al., 2008). The activation of dopamine or 5-HT receptor is sufficient to induce MeCP2 S421 phosphorylation in NAc (Hutchinson et al., 2012a). Antidepressants, such as imipramine, increase the extracellular levels of monoamine neurotransmitters and induce S421 phosphorylation selectively in GABAergic interneurons of the ventral structure of nucleus accumbens (NAc) (Hutchinson et al., 2012b). Hunchinson et al. (2012b) showed that Mecp2S421A/y mice exhibit increased sensitivity to environmental stress in tail-suspension test and forced-swim test and do not generate the normal response to chronic imiparmine treatment in a chronic social defeat stress paradigm, suggesting a role of MeCP2 S421 phosphorylation in depression-like behavior and the behavioral response to antidepressant treatment.
Similar to antidepressants, psychostimulants, including cocaine and AMPH, activate monoamine receptors and are able to induce MeCP2 S421 phosphorylation in NAc (Deng et al., 2010; Mao et al., 2011). Deng at al. (2014) reported that Mecp2S421A/y mice display a reduced threshold for behavioral sensitization to experimenter-administered AMPH and increased sensitivity to chronically self-administered cocaine. At the cellular level, the authors observed a decrease in intrinsic excitability of the medium spiny neurons (MSNs) in the NAc shell of Mecp2S421A/y mice exposed to repetitive AMPH. At the molecular level, repetitive cocaine administration- induced CREB expression is impaired in the NAc of Mecp2S421A/y mice (Deng et al., 2014). These results indicated that S421 phosphorylation is required to induce both behavioral and cellular adaptations to psychostimulants.
The in vivo function of MeCP2 phosphorylation at other sites
Phosphorylation of MeCP2 at S80 and S421/S424 are dynamically regulated in opposite manners by neuronal activity (Tao et al., 2009). More interestingly, Mecp2S80A/y mice showed decreased locomotor activity, whereas Mecp2S421A;S424A/y mice presented increased locomotor activity (Tao et al., 2009), suggesting that S80 and S421/S424 phosphorylation might indeed play opposing roles in the brain. A more comprehensive behavioral analysis of Mecp2S80A/y will be needed to gain better understanding in this area. In vitro results also suggest that S80 phosphorylation play roles in regulating apoptosis (Bracaglia et al., 2009) and microRNA processing (Cheng et al., 2014), and it is necessary to examine whether loss of S80 phosphorylation in Mecp2S80A/y mice will lead to the corresponding alterations in such processes in vivo.
Besides S421 and S80, the other phosphorylation site that draws a lot of recent attention is T308, whose phosphorylation leads to the disruption of the interaction between MeCP2 and NCoR complex (Ebert et al., 2013). Mecp2T308A/y mice have a normal bodyweight, but show a mild decrease in brain weight. Mecp2T308A/y mice display hindlimb clasping and show motor coordination deficit in accelerating rotarod test. Mecp2T308A/y mice also have a reduced seizure threshold compared to WT mice (Ebert et al., 2013). Although more detailed behavioral characterization needs to be performed on Mecp2T308A/y mice, these initial results indicate that Mecp2T308A/y mice present some types of RTT-like phenotype, suggesting disruption of T308 phosphorylation and disregulation of MeCP2/NcoR interaction may contribute to RTT pathogenesis caused by mutations in the vicinity of T308.
Looking into the future
As discussed above, we have learned a great deal about MeCP2 S421 phosphorylation. Going forward, it will be important for the field to reconcile the difference between the Mecp2S421A/y mice and the Mecp2S421A;S424A/y mice, and confirm the identity of S421 kinase by direct binding. To fully understand how phosphorylation states are tightly controlled at S421, it will be important to identify the phosphatase that de-phosphorylate S421 and study the molecular connect between the phosphatase and the responsible extracellular signals. To unequivocally determine the in vivo function of S421 phosphorylation, Mecp2S421E/y and/or Mecp2S421D/y mice (serine to glutamic acid or aspartic acid to mimic phosphorylation) should be generated and analyzed. More importantly, the field needs to move downstream and investigate how S421 phosphorylation affects physical interaction between MeCP2 and its interacting partners, so that we can begin to understand how stimulus-induced S421 phosphorylation may change chromatin state and gene transcription across the neuronal genome. While most of the MeCP2 phosphorylation studies have focused on neurons, it has become increasingly clear that other cell types in the brain also express MeCP2 and play significant roles RTT pathogenesis (Ballas et al., 2009; Lioy et al., 2011; Derecki et al., 2012; Nguyen et al., 2013). Thus, it will be interesting to determine whether S421 phosphorylation is present in these other cell types, how it is regulated and what functions it may have. The same set of questions applies to S80, T308 and all other potential phosphorylation sites on the MeCP2 protein.
If phosphorylation/dephosphorylation can indeed be induced at multiple sites by distinct stimuli in the same neuron, it would suggest signal integration on MeCP2 might produce a combinatorial phosphorylation code, which can then be translated into adaptive transcriptional/functional outputs. MeCP2 is already expressed at the level of histones, and widely distributed across the genome. Why not acquire another key feature of the histone?
Acknowledgements
H.L. was supported by a pre-doctoral fellowship from the Stem Cell and Regenerative Medicine Center at the University of Wisconsin-Madison and a graduate student fellowship from the Friends of the Waisman Center. Q.C. was supported by a Young Investigator Award from NARSAD. This work was partially supported by grants from NICHD (R01 HD064743 and R21 HD066560 to Q.C. and P30 HD03352 to the Waisman Center).
Footnotes
Hongda Li and Qiang Chang declare that they have no conflict of interest.
Compliance with ethics guidelines
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21(1):217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12(3):311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracaglia G, Conca B, Bergo A, Rusconi L, Zhou Z, Greenberg ME, Landsberger N, Soddu S, Kilstrup-Nielsen C. Methyl-CpG-binding protein 2 is phosphorylated by homeodomain-interacting protein kinase 2 and contributes to apoptosis. EMBO Rep. 2009;10(12):1327–1333. doi: 10.1038/embor.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchthal B, Lau D, Weiss U, Weislogel JM, Bading H. Nuclear calcium signaling controls methyl-CpG-binding protein 2 (MeCP2) phosphorylation on serine 421 following synaptic activity. J Biol Chem. 2012;287(37):30967–30974. doi: 10.1074/jbc.M112.382507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56(3):422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56(1):58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302(5646):885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Cheng TL, Wang Z, Liao Q, Zhu Y, Zhou WH, Xu W, Qiu Z. MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell. 2014;28(5):547–560. doi: 10.1016/j.devcel.2014.01.032. [DOI] [PubMed] [Google Scholar]
- Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, Wetsel WC, West AE, Greenberg ME. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72(1):72–85. doi: 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13(21):2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- Deng JV, Rodriguiz RM, Hutchinson AN, Kim IH, Wetsel WC, West AE. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat Neurosci. 2010;13(9):1128–1136. doi: 10.1038/nn.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng JV, Wan Y, Wang X, Cohen S, Wetsel WC, Greenberg ME, Kenny PJ, Calakos N, West AE. MeCP2 phosphorylation limits psychostimulant-induced behavioral and neuronal plasticity. J Neurosci. 2014;34:4519–4527. doi: 10.1523/JNEUROSCI.2821-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484(7392):105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Gabel HW, Robinson ND, Kastan NR, Hu LS, Cohen S, Navarro AJ, Lyst MJ, Ekiert R, Bird AP, Greenberg ME. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature. 2013;499(7458):341–345. doi: 10.1038/nature12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, Zoghbi HY. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. 2008;59(6):947–958. doi: 10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géranton SM, Fratto V, Tochiki KK, Hunt SP. Descending serotonergic controls regulate inflammation-induced mechanical sensitivity and methyl-CpG-binding protein 2 phosphorylation in the rat superficial dorsal horn. Mol Pain. 2008;4(1):35. doi: 10.1186/1744-8069-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géranton SM, Morenilla-Palao C, Hunt SP. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J Neurosci. 2007;27:6163–6173. doi: 10.1523/JNEUROSCI.1306-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales ML, Adams S, Dunaway KW, LaSalle JM. Phosphorylation of distinct sites in MeCP2 modifies cofactor associations and the dynamics of transcriptional regulation. Mol Cell Biol. 2012;32(14):2894–2903. doi: 10.1128/MCB.06728-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg B. Rett’s syndrome: prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr Scand. 1985;74(3):405–408. doi: 10.1111/j.1651-2227.1985.tb10993.x. [DOI] [PubMed] [Google Scholar]
- Hutchinson AN, Deng JV, Aryal DK, Wetsel WC, West AE. Differential regulation of MeCP2 phosphorylation in the CNS by dopamine and serotonin. Neuropsychopharmacology. 2012a;37:321–337. doi: 10.1038/npp.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson AN, Deng JV, Cohen S, West AE. Phosphorylation of MeCP2 at Ser421 contributes to chronic antidepressant action. J Neurosci. 2012b;32:14355–14363. doi: 10.1523/JNEUROSCI.2156-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19(2):187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69(6):905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- Li H, Zhong X, Chau KF, Williams EC, Chang Q. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nat Neurosci. 2011;14(8):1001–1008. doi: 10.1038/nn.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, Mandel G. A role for glia in the progression of Rett’s syndrome. Nature. 2011;475(7357):497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima Alves F, Rappsilber J, Greenberg ME, Bird A. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci. 2013;16(7):898–902. doi: 10.1038/nn.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao LM, Horton E, Guo ML, Xue B, Jin DZ, Fibuch EE, Wang JQ. Cocaine increases phosphorylation of MeCP2 in the rat striatum in vivo: a differential role of NMDA receptors. Neurochem Int. 2011;59(5):610–617. doi: 10.1016/j.neuint.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K, Nagai K. Phosphorylation of methyl-CpG binding protein 2 (MeCP2) regulates the intracellular localization during neuronal cell differentiation. Neurochem Int. 2007;50(1):264–270. doi: 10.1016/j.neuint.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmühl Y, Fischer D, Holsboer F, Wotjak CT, Almeida OF, Spengler D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 2009;12(12):1559–1566. doi: 10.1038/nn.2436. [DOI] [PubMed] [Google Scholar]
- Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics: official journal of the DNA Methylation Society. 2006;1:e1–e11. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Patzel KA, Martin M, Yasui DH, Swanberg SE, Hertz-Picciotto I, Hansen RL, Van de Water J, Pessah IN, Jiang R, Robinson WP, LaSalle JM. MECP2 promoter methylation and X chromosome inactivation in autism. Autism Res. 2008;1:169–178. doi: 10.1002/aur.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88(4):471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393(6683):386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Nguyen MV, Felice CA, Du F, Covey MV, Robinson JK, Mandel G, Ballas N. Oligodendrocyte lineage cells contribute unique features to Rett syndrome neuropathology. J Neurosci. 2013;33:18764–18774. doi: 10.1523/JNEUROSCI.2657-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Sylwestrak EL, Lieberman DN, Zhang Y, Liu XY, Ghosh A. The Rett syndrome protein MeCP2 regulates synaptic scaling. J Neurosci. 2012;32:989–994. doi: 10.1523/JNEUROSCI.0175-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Peters SU, Tavyev YJ, Zhang F, Carvalho CM, Schaaf CP, Richman R, Fang P, Glaze DG, Lupski JR, Zoghbi HY. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol. 2009;66(6):771–782. doi: 10.1002/ana.21715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach JE, Rogers CJ, Yu SH, Tao J, Sun YE, Hsieh-Wilson LC. Quantification of O-glycosylation stoichiometry and dynamics using resolvable mass tags. Nat Chem Biol. 2010;6(9):645–651. doi: 10.1038/nchembio.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37(4):457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, Irier H, Upadhyay AK, Gearing M, Levey AI, Vasanthakumar A, Godley LA, Chang Q, Cheng X, He C, Jin P. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14(12):1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Hu K, Chang Q, Wu H, Sherman NE, Martinowich K, Klose RJ, Schanen C, Jaenisch R, Wang W, Sun YE. Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proc Natl Acad Sci USA. 2009;106(12):4882–4887. doi: 10.1073/pnas.0811648106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi CY, Ma HW, Lu Y, Zhao YJ, Hua TY, Zhao Y, Ji YH. MeCP2 gene mutation analysis in autistic boys with developmental regression. Psychiatr Genet. 2007;17(2):113–116. doi: 10.1097/YPG.0b013e3280114a5c. [DOI] [PubMed] [Google Scholar]
- Zhong X, Li H, Chang Q. MeCP2 phosphorylation is required for modulating synaptic scaling through mGluR5. J Neurosci. 2012;32:12841–12847. doi: 10.1523/JNEUROSCI.2784-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52(2):255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]