

Abstract
Background & Aims
Apoptosis mediated by p53 plays a pathological role in the progression of hepatosteatosis. It is noteworthy that p53 can promote the expression of damage-regulated autophagy modulator (DRAM), an inducer of autophagy-mediated apoptosis. However, the relationship between p53-mediated apoptosis and autophagy in hepatosteatosis remains elusive. This study aimed to examine how p53 orchestrates autophagy and apoptosis to affect hepatosteatosis.
Methods
HepG2 cells were treated with oleic acid (OA) for 24 h to induce hepatosteatosis. Mice were fed a high-fat diet for 20 or 40 weeks to induce hepatosteatosis.
Results
OA induced a dose-dependent increase in steatosis severity and apoptosis. OA also induced autophagy, which was a critical inducer of apoptosis in mild steatosis induced by 400 μM OA, but not in the more severe steatosis induced by 800 and 1200 μM OA. p53 inhibition by siRNA mostly blocked OA-induced apoptosis and autophagy. Moreover, OA-induced autophagy was DRAM-dependent and primarily occurred in the mitochondria (mitophagy), where DRAM was localized. In severe steatosis induced by 1200 μM OA, apoptosis was mainly dependent on p53-induced expression of BAX, which was also localized to the mitochondria. Our in vivo study showed that p53 expression increased in both mild and severe hepatosteatosis. Increased DRAM expression and autophagy were identified in mild hepatosteatosis, whereas greater BAX expression was observed in severe hepatosteatosis.
Conclusions
p53 may induce apoptosis via different mechanisms. DRAM-mediated mitophagy is a primary apoptotic inducer in mild hepatosteatosis, whereas p53-induced BAX expression mainly induces apoptosis in severe hepatosteatosis.
Keywords: apoptosis, damage-regulated autophagy modulator, mitophagy, non-alcoholic fatty liver disease
Non-alcoholic fatty liver disease (NAFLD) is a burgeoning health problem in developed countries; it affects one-third of adults and an increasing number of children 1. Many individuals with NAFLD have a benign prognosis of simple hepatosteatosis; however, in some cases, this can progress to non-alcoholic steatohepatitis (NASH), which can induce liver fibrosis, cirrhosis and even hepatocellular carcinoma 2. NAFLD deterioration is closely associated with lipid metabolism dysfunction and lipid accumulation, which induces lipotoxicity and promotes the progression of hepatosteatosis 3,4. Previous data have shown that apoptosis plays a critical role in the progression of this disease 5. Apoptosis induces hepatocytic impairment and promotes inflammation 5. Responses to inflammation include the production of inflammatory factors that can further impair cells and induce more apoptosis 6. NAFLD is characterized by the accumulation of triglycerides in hepatocytes, which impairs the oxidative capacity of their mitochondria. Poor mitochondrial function increases the reduced state of electron transport chain complexes and stimulates the peroxisomal and microsomal pathways of fat oxidation 7. Impaired mitochondria are also sources of reactive oxygen species (ROS), which trigger apoptosis and the production of inflammatory cytokines, causing inflammation and furthering the development of NASH 7.
Autophagy also plays a critical role in NAFLD. Free fatty acids can induce autophagy in vitro 8, and autophagy impairment can aggravate insulin resistance and Endoplasmic reticulum (ER) stress in NAFLD 9. A recent finding also showed that autophagy can mediate lipid metabolism in hepatocytes (called lipophagy), further highlighting the role of autophagy in hepatosteatosis. In general, the autophagy-dependent degradation of impaired organelles—including the selective degradation of the endoplasmic reticulum (reticulophagy), mitochondria (mitophagy), and lipid droplets (lipophagy)—is a crucial cellular pathway for maintaining cell homeostasis 10,11. However, autophagy is also regarded as a pro-apoptotic factor and is the cause of ‘type II’ programmed cell death 12–14.
p53 functions primarily as a transcription factor and induces the expression of its target genes, including PUMA and BAX. PUMA, a protein in the proapoptotic BH3-only Bcl-2 family, activates BAX and induces mitochondria-dependent apoptosis 15. The relationship between the p53 signalling pathway and autophagy development was established by the discovery of DRAM and Sestrin2, which are p53-induced modulators of autophagy and are critical for apoptosis 16,17. Studies have also reported that the activation of the p53 signalling pathway is critical for hepatocytic apoptosis 18.
This study was designed to elucidate whether p53 induces apoptosis by promoting autophagy and to clarify the relationship between autophagy-induced apoptosis and other types of induced apoptosis in NAFLD. Our results suggest that p53 could induce apoptosis through different methods in different stages of NAFLD. In mild steatosis, p53 promoted DRAM expression; the DRAM was subcellularly localized to the mitochondria and induced autophagy development, which was the main inducer of apoptosis. In severe hepatosteatosis, p53 primarily promoted BAX expression, and the BAX then travelled to the mitochondria to induce apoptosis.
Materials and methods
Cell culture and treatment
The human hepatoblastoma cell line HepG2 was grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum. HepG2 cells were treated with 400 μM OA for 24 h. HepG2 cells were transfected with Fugene HD transfection reagent (Promega, Madison, WI, USA) for the transfection of plasmids encoding GFP-LC3 or with Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) for the transfection of DRAM siRNA and VPS34 siRNA. LY294002 (50 μM) and Bafilomycin A1 (BafA 1) (50 nM) were used to inhibit autophagy. Cells grown on glass cover slips were used for immunofluorescence detection.
Detection of autophagy and apoptosis
The level of autophagy was detected by the transfection of GFP-LC3 plasmids or western blotting. Apoptosis was detected by immunofluorescence (based on calcein AM/PI and M30 immunoreactivity) or a western blot assay of cleaved PARP (p85 fragment). The detailed immunofluorescence protocol is provided in the Supplementary Materials and Methods.
Subcellular fractionation, protein isolation and western blotting
Mitochondria- and endoplasmic reticulum-enriched fractions of HepG2 cells were isolated to execute the western blotting assay. The standard western blotting method is provided in the Supplementary Materials and Methods.
Real-time PCR
SYBR Green was used to detect the mRNA level during real-time PCR. The mRNA content was normalized to the housekeeping gene β-actin. The detailed protocol for real-time PCR is provided in the Supplementary Materials and Methods.
Lipotoxicity detection
The levels of lactate dehydrogenase (LDH) and intracellular triglycerides were determined using commercial kits. Oil Red O was used to determine the level of lipid droplets (for a detailed protocol, see the Supplementary Materials and Methods).
Animal experiment
All protocols for animal care and experimentation were approved by the Ethical Committee of YouAn Hospital, Capital Medical University, Beijing. Male C57BL mice were fed a high-fat diet (HFD) to induce NAFLD development. Detailed protocols for the animal experiments are provided in the Supplementary Materials and Methods.
Statistical analysis
All data represent at least three independent experiments and are expressed as the mean ± SEM. Differences between the groups were compared using Student's t-test. Differences were considered significant if the P-value was less than 0.05. Additional notations are included for differences associated with P-values that are less than 0.01 or 0.001.
Please refer to the Supplementary Materials and Methods section for more detailed descriptions.
Results
OA treatment induces a dose-dependent increase in lipotoxicity and apoptosis
We used three concentrations of OA (400, 800 and 1200 μM) to stimulate HepG2 cells. The greatest concentration of OA induced the highest levels of lipid droplets, intracellular triglycerides and LDH release, followed by the 800 μM and then 400 μM treatments (Fig.1A and Fig. S1 A and B). This result suggests that OA treatment induced a dose-dependent increase in lipotoxicity. Lipotoxicity-induced apoptosis plays a critical role in exacerbating the progression of NAFLD. The Calcein AM/PI assay is a method used to detect whether late apoptosis is induced. Calcein AM stains viable cells (green), whereas PI stains late apoptotic cells (red). M30 stains keratin 18, a protein that is cleaved by caspase 3, and hence identifies early apoptotic cells 19. We found that 1200 μM OA induced the highest levels of both late and early apoptosis, followed by the 800 and 400 μM treatments (Figs.1A–C). During apoptosis, PARP is cleaved by caspase-3 and caspase-7 to yield p85, which is a marker of apoptosis 20. Immunoblot assays identified that the 1200 μM treatment induced the highest levels of p85, followed by the 800 and 400 μM treatments, confirming the results of the Calcein AM/PI and M30 assays (Fig.1E). Thus, OA induced a dose-dependent increase in apoptosis. Because a higher M30 level indicates the deterioration of hepatosteatosis 21–24, the results of the M30 assay suggested that larger concentrations of OA would increase the severity of NAFLD.
Fig 1.

Different concentrations of oleic acid (OA) have different effects on the induction of lipid accumulation, apoptosis and autophagy. HepG2 cells were treated with 400, 800 or 1200 μM OA for 24 h with and without BafA 1 pretreatment. (A) Oil Red O staining, apoptosis detection using Calcein AM/PI and anti-M30, and autophagy detection using plasmids encoding GFP-LC3. Original magnification, 400×. (B–D) Percentages of late apoptotic cells (B), early apoptotic cells (C) and cells with autophagosomes (D). Data (mean ± SEM) represent three independent experiments. (E) Western blotting analysis of LC3 I/II and p85 protein levels. (F) Percentages of cells with autophagosomes. The data (mean ± SEM) represent three independent experiments. (G) Western blotting analysis of the effect of BafA 1 on LC3 I/II formation.
OA at a concentration of 400 μM induces the highest level of autophagy, followed by the 800 and 1200 μM treatments
Studies have demonstrated that autophagy is critical for NAFLD deterioration 25. By transfecting HepG2 cells with plasmids encoding for GFP-LC3, we determined that 400 μM OA induced the highest rate of cells with GFP-LC3 puncta, followed by the 800 and 1200 μM treatments (Fig.1A and D). Immunoblot analysis was used to determine LC3 II formation, and the results showed that 400 μM OA induced the highest level of LC3 II, followed by the 800 and 1200 μM treatments (Fig.1E). Bafilomycin A1 (BafA 1), a vacuolar H+-ATPase inhibitor, is commonly used to prevent the fusion of autophagosomes with lysosomes. We observed that BafA 1 blocked OA-induced autophagic flux because BafA 1 induced a higher number of GFP-LC3 puncta (Fig.1F) and induced additional LC3 II formation (Fig.1G) compared with cells that were not treated with BafA 1. BafA 1 did not alter the effect of OA on the appearance of GFP-LC3 puncta: 400 μM OA still induced the highest level of GFP-LC3 puncta, followed by the 800 and 1200 μM treatments (Fig.1F and G). Thus, it appears that lower concentrations of OA induce higher levels of autophagy. Because autophagy can be either an anti-apoptotic factor or a pro-apoptotic factor (of type II, or autophagic, cell death) 11, the relationship between autophagy and apoptosis in response to an OA stimulus is unknown. Next, we attempted to determine the role of autophagy in HepG2 cells in response to various concentrations of OA.
Autophagy contributes to the greatest number of apoptotic deaths in HepG2 cells treated with 400 μM OA, followed by those treated with 800 and 1200 μM OA
Autophagy is associated with cellular apoptosis and is regarded as type II programmed cell death 12–14. To elucidate the role of OA in autophagy, we employed an autophagy inhibitor, LY294002 (50 μM). Autophagy inhibition failed to affect the observed OA-induced increase in lipid accumulation (Fig. S2A); however, this inhibition reduced LDH release in cells treated with 400 μM OA (Fig. S2B). Moreover, autophagy inhibition reduced the amount of apoptosis observed in cells treated with 400 μM OA, but had no effect on apoptosis in the 800 and 1200 μM treatments (Fig.2A–C). Using an immunoblot assay, we determined that when autophagy was inhibited by LY294002, the p85 level was significantly decreased in the 400 μM treatment compared with the 800 or 1200 μM treatments (Fig.2D). These results suggest that autophagy is a critical, but not sole, pro-apoptotic factor in OA-induced apoptosis in less severe cases of hepatosteatosis; however, in more severe cases, other pro-apoptotic factors are more important.
Fig 2.

Autophagy contributes to the most apoptotic deaths in HepG2 cells in response to a 400 μM OA stimulus. HepG2 cells were pretreated with LY294002 (LY) for 5 h and were subsequently cultured with 400, 800 or 1200 μM OA for 24 h. (A) Apoptosis detection by Calcein AM/PI and anti-M30. Original magnification, 400×. (B, C) Percentages of late apoptotic cells (B) and early apoptotic cells (C). The data (mean ± SEM) represent three independent experiments. (D) Western blotting analysis of LC3 I/II and p85 protein levels.
OA-induced apoptosis is dependent on the p53 signalling pathway
Apoptosis mediated by p53 affects NAFLD deterioration by promoting BAX expression 14, and p53 can also promote autophagy-mediated apoptosis by increasing DRAM expression 16,17. Thus, we attempted to examine the role of p53 in our in vitro model by detecting the expression levels of p53 and its target genes. First, we determined that all three tested concentrations of OA induced greater expression of p53 and its target genes, PUMA, BAX and DRAM, when compared with time zero (Fig.3A,B). Next, we found that 1200 μM OA induced the highest expression levels of PUMA and BAX, followed by the 800 and 400 μM treatments (Fig.3A,B); however, the expression of DRAM was greatest in the presence of 400 μM OA, followed by 800 and 1200 μM OA (Fig.3A,B). Moreover, transfecting HepG2 cells with p53 siRNA to block p53 function did not affect OA-induced lipid accumulation (data not shown), but notably inhibited the OA-induced overexpression of BAX and DRAM (Fig.3C). Inhibiting the function of p53 also largely blocked LDH release (Fig.3G), apoptosis and autophagy development (Fig.3D,F). Treatment with control siRNA did not alter the OA-induced expression of p53, DRAM or BAX (Fig.3C), and OA-induced apoptosis and autophagy development were also unchanged (data not shown). These results suggest that the activation of the p53 signalling pathway is critical for OA-induced apoptosis, cell impairment and autophagy.
Fig 3.
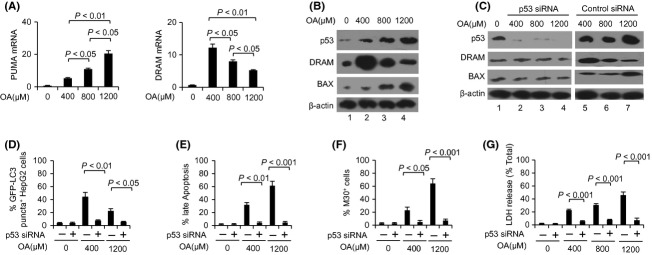
Oelic acid (OA)-induced apoptosis is dependent on p53. HepG2 cells were cultured with 400, 800 or 1200 μM OA for 24 h with or without p53 siRNA pretreatment. (A) Detection of PUMA and DRAM mRNA levels by real-time PCR. (B, C) Western blotting analysis of p53, damage-regulated autophagy modulator (DRAM) and BAX protein levels. (D–F) Percentages of cells with autophagosomes (D), late apoptotic cells (E) and early apoptotic cells (F). (G) The levels of LDH release were analyzed. The data (mean ± SEM) represent three independent experiments.
DRAM-mediated autophagy is a primary effector of apoptosis induced by 400 μM OA, whereas BAX expression is the primary effector in apoptosis induced by 1200 μM OA
DRAM, a p53-induced modulator of autophagy, can cause apoptosis 16. We found that specific DRAM siRNA largely inhibited autophagy induced by 400 and 1200 μM OA, suggested that the mechanism for this process was DRAM-dependent (Fig.4A,C). In the 400 μM treatment, the application of DRAM siRNA decreased BAX expression; however, this was not observed in the 1200 μM treatment (Fig.4A). Immunoblot analysis of p85 formation as well as PI and M30 staining also showed that DRAM siRNA largely inhibited apoptosis (Fig.4A,D and E). The application of control siRNA did not affect apoptosis, autophagy development (data not shown) or the expression of DRAM, LC3 II, p85 or BAX in cells treated with OA (Fig.4B). These results suggest that DRAM-mediated autophagy is a primary effector of apoptosis in cases of mild NAFLD; in severe cases, other pro-apoptotic factors, such as BAX, may be critical for the induction of apoptosis.
Fig 4.
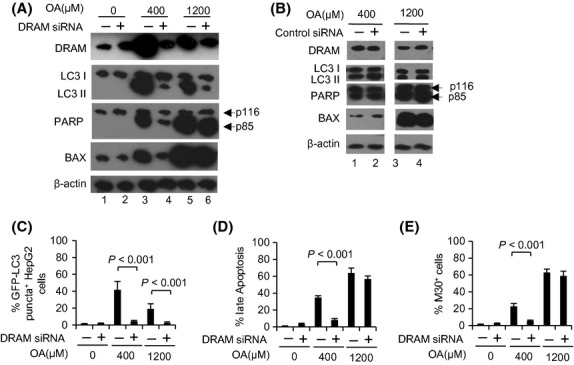
Damage-regulated autophagy modulator (DRAM)-mediated autophagy is the primary effector of apoptosis induced by 400 μM oleic acid (OA), whereas BAX is the primary effector in apoptosis induced by 1200 μM OA. HepG2 cells were cultured with either 400 or 1200 μM OA for 24 h with or without DRAM siRNA pretreatment. (A, B) Western blotting analysis of DRAM, LC3I/II, p85 and BAX protein levels. (C–E) Percentages of cells with autophagosomes (C), late apoptotic cells (D) and early apoptotic cells (E). The data (mean ± SEM) represent three independent experiments.
DRAM-mediated autophagy primarily occurs at the mitochondria
By isolating mitochondria and the endoplasmic reticulum and performing immunoblot assays, we investigated the subcellular localization of LC3 II, DRAM and BAX. Our results showed that OA-induced autophagy primarily took place at the mitochondria (autophagy at this location is termed mitophagy), and DRAM localized to mitochondria (Fig.5). Moreover, 400 μM OA induced the highest levels of mitophagy, followed by the 800 and 1200 μM treatments (Fig.5). The 800 and 1200 μM treatments induced additional BAX expression, and BAX was also primarily localized to mitochondria (Fig.5). Taken together with our previous results, this subcellular localization analysis suggests that the activation of p53 promotes greater DRAM expression in mild hepatosteatosis. DRAM then localizes to mitochondria, inducing mitophagy and promoting apoptosis. In contrast, in severe hepatosteatosis, p53 promotes greater expression of BAX, which also localizes to mitochondria and induces apoptosis.
Fig 5.
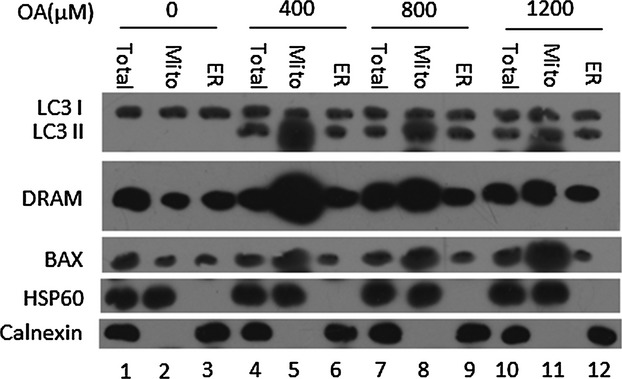
Damage-regulated autophagy modulator (DRAM)-mediated autophagy primarily occurs at the mitochondria. HepG2 cells cultured with 400, 800 or 1200 μM oleic acid (OA) for 24 h were separated into fractions mainly containing mitochondria (‘mito’) and endoplasmic reticulum (‘ER’). Total: total lysate of the HepG2 cells. Western blotting analysis was used to detect the level of autophagy and DRAM expression in the total lysate as well as in the mito- and ER-enriched fractions. HSP60 and calnexin were used as controls for the fractions mainly containing mitochondria and endoplasmic reticulum respectively.
Autophagy-induced apoptosis triggered by DRAM primarily occurs in mild hepatosteatosis, whereas BAX-induced apoptosis primarily occurs in severe hepatosteatosis
To determine the effects of DRAM-mediated autophagy and BAX expression on the induction of apoptosis in vivo, mice were fed a HFD for 20 or 40 weeks. Immunohistochemical analysis demonstrated that the mice fed a HFD for 40 weeks exhibited additional lipid accumulation (presenting with severe hepatosteatosis) than mice fed a HFD for only 20 weeks (presenting with mild hepatosteatosis) (Fig.6A). An immunoblot assay was performed to determine the amount of autophagy and the expression levels of p53, p85, DRAM and BAX in liver tissues. We found that mice with both mild (fed a HFD for 20 weeks) and severe (fed a HFD for 40 weeks) hepatosteatosis exhibited greater p53 expression than control mice, which were fed a normal diet (Fig.6B). A HFD could induce autophagy development, increasing the expression levels of DRAM and BAX (Fig.6B). Moreover, mice with mild hepatosteatosis had higher levels of both autophagy and DRAM expression when compared with mice with severe hepatosteatosis (Fig.6B). However, mice with severe hepatosteatosis had higher BAX expression levels (Fig.6B). These in vivo results combined with our in vitro results suggest that DRAM-mediated autophagy may be critical for the induction of apoptosis in mild hepatosteatosis; however, other pro-apoptotic factors, such as BAX, were more important causes of apoptosis in severe hepatosteatosis (Fig.6C).
Fig 6.
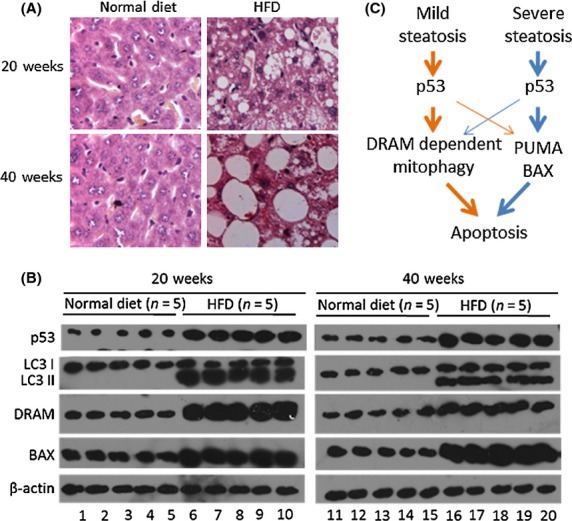
Detection of p53, damage-regulated autophagy modulator (DRAM), LC3I/II and BAX expression in vivo. Test mice were fed a high-fat diet (HFD) for 20 or 40 weeks, and control mice were given a normal diet. (A) The severity of hepatosteatosis was analyzed by immunohistochemistry. (B) Western blotting analysis of p53, LC3I/II, DRAM and BAX protein levels in liver tissues. (C) Model. At different stages of hepatosteatosis, p53 uses different strategies to induce apoptosis: In mild cases, p53-induced apoptosis is primarily dependent on DRAM-mediated autophagy; in severe cases, other p53-dependent apoptotic signals, such as PUMA and BAX, are the primary inducers of apoptosis.
Discussion
Apoptosis is correlated with progressive inflammation and fibrosis in NAFLD; thus, its induction by excessive FFAs is a critical histological feature of the disease 5,6. However, the mechanisms of apoptosis induction remain elusive. In this study, 1200 μM OA induced the most severe lipotoxicity, followed by 800 and 400 μM OA treatments. M30 stains for keratin 18 that has been cleaved by caspase 3, hence identifying apoptotic cells 19. Recent studies have demonstrated that M30 can predict steatosis and hepatocytic injury 21–24. Our results suggest that a higher concentration of OA will increase the severity of NAFLD. Taken together with the findings of other groups, our results demonstrate that the induction of a more severe NAFLD pathology leads to higher levels of apoptosis.
Autophagy is a biological process that maintains cellular homeostasis, recycling impaired organelles or longevity proteins in response to stress; examples of this include mitochondrial impairment and amino acid starvation 26,27. Some in vivo studies have demonstrated that impairing the autophagy function exacerbates NAFLD by causing ER stress and increasing insulin resistance 11. However, other studies have demonstrated that FFAs can induce autophagy in vitro. For example, OA induces autophagy in HepG2 cells, including in the progress of NAFLD 8. However, the underlying mechanisms associated with autophagy and the pathological progression of NAFLD are not clear. In this study, our results show that OA-induced autophagy is a pro-apoptotic factor. This result is consistent with other data indicating that apoptosis can be induced by autophagy, which is regarded as a type of programmed cell death (type II) 12–14.
A previous study demonstrated that Bax/Bak double knockout mice are resistant to apoptosis although their cells still undergo autophagy-mediated cell death 12. When apoptosis dysfunction is caused by a Bax/Bak knockout, JNK activation is crucial for autophagic death 28. Thus, it appears that cells have the ability to choose between apoptotic and autophagic processes to execute cell death. The pathways of apoptosis and autophagy are closely associated. Beclin-1, an important inducer of autophagy, can control the level of p53 29. The Bcl-2 family of proteins can also control the non-apoptotic programmed cell death that depends on autogenes 12. Thus, it is reasonable that a cell would respond to stress using two strategies that cause cell death (i.e., the apoptotic and autophagic processes). In this study, our data demonstrate that the level of OA-induced lipotoxicity determines the primary strategy that HepG2 cells use to induce apoptosis.
A previous study demonstrated that an autophagic process could be involved in the mitochondrial dysfunction-induced apoptotic death of striatal neurons 30. In our study, both DRAM-mediated mitophagy and the subcellular localization of BAX to the mitochondria may induce mitochondrial apoptosis. Mitophagy, a cellular ‘quality control system,’ can reduce the overproduction of ROS and RNS by degrading impaired mitochondria, maintaining cellular homeostasis 31. However, a recent study has indicated that tumour progression is abrogated by mitophagy but not by apoptosis 32. In hepatosteatosis, the relationship between mitophagy and apoptosis is still unknown. Our results demonstrate that mitophagy, a pro-apoptotic factor for inducing hepatocytic apoptosis, plays a role in exacerbating NAFLD.
Our results also show that the activation of the p53 signalling pathway is critical for OA-induced autophagy, apoptosis and cell impairment but not for OA-induced lipotoxicity. p53 acts as a master regulator, with pleiotropic effects on metabolism, antioxidant defence, genomic stability, proliferation, senescence and cell death 33. Previous data have shown that p53 pathway activation is involved in cell apoptosis in NAFLD 18,34, regulating the balance of Bcl-2 and BAX function 35. In the present study, the p53 signalling pathway is activated, as shown by the upregulation of PUMA, BAX and DRAM expression. In response to DNA impairment, p53 promotes the expression of PUMA, which interacts with Bcl-2 family members and frees BAX to signal cells to undergo apoptosis 36,37. This p53-PUMA-Bax system is a classic signal for the induction of cell death via apoptosis 38. DRAM is a p53-induced modulator of autophagy 16, which can co-localize with Beclin-1, a critical mediator of autophagy development 39. DRAM-mediated autophagy is considered critical for p53-induced apoptosis 40. Recent studies have shown that in rat striatum, p53 mediates autophagy activation and neurodegeneration via DRAM expression 40. However, our results demonstrate that p53 can induce hepatocytic apoptosis via different mechanisms in different stages of hepatosteatosis: DRAM-mediated autophagy is primarily mitophagy, and mitophagy is a critical pro-apoptotic factor in mild cases. However, the PUMA/BAX signalling pathway appears to play an important role in more severe cases.
There are no established therapeutic strategies for hepatosteatosis, and effective treatments are urgently needed. Our findings that DRAM-mediated autophagy and BAX-induced apoptotic processes can induce hepatocytic death via p53 provide a promising new avenue for studying hepatosteatosis.
Acknowledgments
Financial support: This work was supported in part by National Natural Science Foundation of China (81071843, 81272266, 30910103915), and Basic-Clinical Collaborative Research Fund, CCMU (12JL-L05), Fund of Beijing Institute of Hepatology (BJIH-01201).
Conflicts of interest: The authors do not have any disclosures to report.
Supporting Information
Additional Supporting Information may be found in the online version of this article
Different concentrations of OA induce lipid accumulation and LDH release differently. HepG2 cells were cultured in 24-well plates for 24 h with 400, 800 or 1200 μM OA. The levels of intracellular triglycerides and released LDH in the HepG2 cells in response to the OA stimulus were analyzed. Data (mean ± SEM) represent three independent experiments. Significance levels were P < 0.05 and P < 0.01 at 24 h.
The effects of autophagy inhibition by LY294002 pretreatment on OA-induced lipid accumulation and LDH release. HepG2 cells were pretreated with LY294002 for 5 h to inhibit autophagy and then treated with 400, 800 or 1200 μM OA for 24 h. The levels of intracellular triglycerides and released LDH were analyzed. Data (mean cells ± SEM) represent three independent experiments. The significance level was P < 0.05 at 24 h.
Supplementary
References
- 1.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–23. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 3.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–5. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 4.Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–64. doi: 10.1038/nrgastro.2010.41. [DOI] [PubMed] [Google Scholar]
- 5.Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–43. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 6.Lea RG, Riley SC, Antipatis C, et al. Cytokines and the regulation of apoptosis in reproductive tissues: a review. Am J Reprod Immunol. 1999;42:100–9. [PubMed] [Google Scholar]
- 7.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Mei S, Ni HM, Manley S, et al. Differential Roles of Unsaturated and Saturated Fatty Acids on Autophagy and Apoptosis in Hepatocytes. J Pharmacol Exp Ther. 2011;339:487–98. doi: 10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pagliassotti MJ. Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annu Rev Nutr. 2012;32:17–33. doi: 10.1146/annurev-nutr-071811-150644. [DOI] [PubMed] [Google Scholar]
- 10.Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–5. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rautou PE, Mansouri A, Lebrec D, et al. Autophagy in liver diseases. J Hepatol. 2010;53:1123–34. doi: 10.1016/j.jhep.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu S, Kanaseki T, Mizushima N, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Bio. 2004;6:1221–8. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 13.Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 14.Farrell GC, Larter CZ, Hou JY, et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J Gastroeneterol Hepatol. 2009;24:443–52. doi: 10.1111/j.1440-1746.2009.05785.x. [DOI] [PubMed] [Google Scholar]
- 15.Leibowitz BJ, Qiu W, Liu H, et al. Uncoupling p53 functions in radiation-induced intestinal damage via PUMA and p21. Mol Cancer Res. 2011;9:616–25. doi: 10.1158/1541-7786.MCR-11-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crighton D, Wilkinson S, O'Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 17.Maiuri MC, Malik SA, Morselli E, et al. Stimulation of autophagy by the p53 target gene sestrin2. Cell Cycle. 2009;8:1571–6. doi: 10.4161/cc.8.10.8498. [DOI] [PubMed] [Google Scholar]
- 18.Tomita K, Teratani T, Suzuki T, et al. p53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J Hepatol. 2012;57:837–43. doi: 10.1016/j.jhep.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 19.Kramer G, Erdal H, Mertens HJ, et al. Differentiation between cell death modes using measurements of different soluble forms of extracellular cytokeratin 18. Cancer Res. 2004;64:1751–6. doi: 10.1158/0008-5472.can-03-2455. [DOI] [PubMed] [Google Scholar]
- 20.Pandit B, Gartel AL. Proteasome inhibitors induce p53-independent apoptosis in human cancer cells. Am J Pathol. 2011;178:355–60. doi: 10.1016/j.ajpath.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabuchi M, Tomioka K, Kawakami T, et al. Serum cytokeratin 18 M30 antigen level and its correlation with nutritional parameters in middle-aged Japanese males with nonalcoholic fatty liver disease (NAFLD) J Nutr Sci Vitaminol (Tokyo) 2010;56:271–8. doi: 10.3177/jnsv.56.271. [DOI] [PubMed] [Google Scholar]
- 22.Lebensztejn DM, Wierzbicka A, Socha P, et al. Cytokeratin-18 and hyaluronic acid levels predict liver fibrosis in children with nonalcoholic fatty liver disease. Acta Biochem Pol. 2011;58:563–6. [PubMed] [Google Scholar]
- 23.Yilmaz Y, Dolar E, Ulukaya E, et al. Soluble forms of extracellular cytokeratin 18 may differentiate simple steatosis from nonalcoholic steatohepatitis. World J Gastroenterol. 2007;13:837–44. doi: 10.3748/wjg.v13.i6.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Younossi ZM, Page S, Rafiq N, et al. A biomarker panel for non-alcoholic steatohepatitis (NASH) and NASH-related fibrosis. Obes Surg. 2011;21:431–43. doi: 10.1007/s11695-010-0204-1. [DOI] [PubMed] [Google Scholar]
- 25.Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142:711–25. doi: 10.1053/j.gastro.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Xiao-Ming Yin, Wen-Xing Ding, Wentao Gao. Autophagy in the liver. Hepatology. 2008;47:1773–85. doi: 10.1002/hep.22146. [DOI] [PubMed] [Google Scholar]
- 27.Tian Y, Chen WL, Ou JH. Effects of interferon-α/β on HBV replication determined by viral load. PLoS Pathog. 2011;7:e1002159. doi: 10.1371/journal.ppat.1002159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimizu S, Konishi A, Nishida Y, et al. Involvement of JNK in the regulation of autophagic cell death. Oncogene. 2010;29:2070–82. doi: 10.1038/onc.2009.487. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–34. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang XD, Wang Y, Wu JC, et al. Down-regulation of Bcl-2 enhances autophagy activation and cell death induced by mitochondrial dysfunction in rat striatum. J Neurosci Res. 2009;87:3600–10. doi: 10.1002/jnr.22152. [DOI] [PubMed] [Google Scholar]
- 31.Taylor EB, Rutter J. Mitochondrial quality control by the ubiquitin-proteasome system. Biochem Soc Trans. 2011;39:1509–13. doi: 10.1042/BST0391509. [DOI] [PubMed] [Google Scholar]
- 32.Gargini R, García-Escudero V, Izquierdo M. Therapy mediated by mitophagy abrogates tumor progression. Autophagy. 2011;7:466–76. doi: 10.4161/auto.7.5.14731. [DOI] [PubMed] [Google Scholar]
- 33.Purvis JE, Karhohs KW, Mock C, et al. Science. 2012;336:1440–4. doi: 10.1126/science.1218351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–83. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 35.Panasiuk A, Dzieciol J, Panasiuk B, Prokopowicz D. Expression of p53, Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World J Gastroenterol. 2006;12:6198–202. doi: 10.3748/wjg.v12.i38.6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bedelbaeva K, Snyder A, Gourevitch D, et al. Lack of p21 expression links cell cycle control and appendage regeneration in mice. Proc Natl Acad Sci USA. 2010;107:5845–50. doi: 10.1073/pnas.1000830107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008;27:s71–83. doi: 10.1038/onc.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skírnisdóttir I, Seidal T. The apoptosis regulators p53, bax and PUMA: relationship and impact on outcome in early stage (FIGO I-II) ovarian carcinoma after post-surgical taxane-based treatment. Oncol Rep. 2012;27:741–7. doi: 10.3892/or.2011.1578. [DOI] [PubMed] [Google Scholar]
- 39.Hong MY, Gao JL, Cui JZ, et al. Effect of c-Jun NH - terminal kinase-mediated p53 expression on neuron autophagy following traumatic brain injury in rats. Chin Med J (Engl) 2012;125:2019–24. [PubMed] [Google Scholar]
- 40.Zhang XD, Wang Y, Wang Y, et al. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy. 2009;5:339–50. doi: 10.4161/auto.5.3.8174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Different concentrations of OA induce lipid accumulation and LDH release differently. HepG2 cells were cultured in 24-well plates for 24 h with 400, 800 or 1200 μM OA. The levels of intracellular triglycerides and released LDH in the HepG2 cells in response to the OA stimulus were analyzed. Data (mean ± SEM) represent three independent experiments. Significance levels were P < 0.05 and P < 0.01 at 24 h.
The effects of autophagy inhibition by LY294002 pretreatment on OA-induced lipid accumulation and LDH release. HepG2 cells were pretreated with LY294002 for 5 h to inhibit autophagy and then treated with 400, 800 or 1200 μM OA for 24 h. The levels of intracellular triglycerides and released LDH were analyzed. Data (mean cells ± SEM) represent three independent experiments. The significance level was P < 0.05 at 24 h.
Supplementary