

Abstract
Fibroblast growth factor (FGF2) regulates endothelial and melanoma cell migration. The binding of FGF2 to its receptor requires N-sulfated heparan sulfate (HS) glycosamine. We have previously reported that Epac1, an exchange protein activated by cAMP, increases N-sulfation of HS in melanoma. Therefore, we examined whether Epac1 regulates FGF2-mediated cell–cell communication. Conditioned medium (CM) of melanoma cells with abundant expression of Epac1 increased migration of human umbilical endothelial cells (HUVEC) and melanoma cells with poor expression of Epac1. CM-induced increase in migration was inhibited by antagonizing FGF2, by the removal of HS and by the knockdown of Epac1. In addition, knockdown of Epac1 suppressed the binding of FGF2 to FGF receptor in HUVEC, and in vivo angiogenesis in melanoma. Furthermore, knockdown of Epac1 reduced N-sulfation of HS chains attached to perlecan, a major secreted type of HS proteoglycan that mediates the binding of FGF2 to FGF receptor. These data suggested that Epac1 in melanoma cells regulates melanoma progression via the HS–FGF2-mediated cell–cell communication.
Keywords: Epac, heparan sulfate, human umbilical vein endothelial cells, cell–cell communication, FGF2, migration, angiogenesis, paracrine signaling
Significance.
There is an emerging need for elucidating the mechanism of cell–cell interaction in melanoma progression. Our study provides information regarding FGF2-related cell–cell interaction between melanoma/endothelial and melanoma/melanoma cells which is regulated by melanoma cells with the higher expression of Epac1.
Introduction
Despite recent advances in melanoma therapies utilizing inhibitors of the ERK-signaling pathway, prognosis of advanced melanoma is still poor. In addition, acquired resistance becomes a critical problem with those inhibitors (Little et al., 2012; Maurer et al., 2011). Therefore, the development of a novel therapeutic strategy is an urgent demand for this life-threatening disease. cAMP signaling controls a variety of cellular functions in cancer cells. Exchange protein activated by cAMP (Epac), a guanine nucleotide exchange factor, was found as an additional target of cAMP apart from the conventional one, that is, protein kinase A (De Rooij et al., 1998). Two isoforms of Epac, Epac1 and Epac2, mediate cAMP signaling by the activation of a small-molecular-weight G protein, Rap1 (Bos, 2006). In cancer cells, reports have demonstrated following functions of Epacs such as cell adhesion in human ovarian carcinoma Ovcar3 cells (Quilliam et al., 2002), apoptosis (Tiwari et al., 2004) and growth arrest (Grandoch et al., 2009a) in B lymphoma cells, formation of embryonic vasculogenic networks in melanoma cells (Lissitzky et al., 2009), and proliferation of prostate carcinoma cells (Grandoch et al., 2009b). We have previously reported that Epac1 is expressed in various melanoma cell lines (Baljinnyam et al., 2011) and plays a role in cell migration via modification of heparan sulfate (HS) glycosaminoglycan (HSPG) chains. The increased migration by Epac1-enhanced metastasis to the lungs in mice (Baljinnyam et al., 2009). Recently, we have also found that, in addition to this HS-related mechanism, a Ca2+-dependent mechanism is also involved in Epac1-induced melanoma cell migration. Epac1 releases cytosolic Ca2+ from the endoplasmic reticulum (ER) via the phospholipase C (PLC)/inositol triphosphate (IP3)/IP3 receptor pathway (Baljinnyam et al., 2010). These data suggested that Epac1 plays a critical role in melanoma cell migration via at least two independent mechanisms, that is, the HS-related and the Ca2+-dependent mechanisms.
Fibroblast growth factor-2 (FGF2) is known to increase tumor growth and metastasis by the activation of migration of cancer and vascular endothelial cells (Hibino et al., 2005; Meier et al., 2003; Montesano et al., 1986; Moscatelli et al., 1986; Nugent et al., 2000; Ponta et al., 1998; Sola et al., 1997; Taylor et al., 1993). Binding of FGF2 to FGF receptor requires coordination with N-sulfated glucosamine (Faham et al., 1996; Kreuger et al., 1999; Maccarana et al., 1993; Schlessinger et al., 2000), a component of HS chain (Iozzo and San Antonio, 2001). In addition, perlecan, one of the HSPGs, attaches to FGF2 for its binding to FGF receptors (Knox et al., 2002; Sharma et al., 1998). We have previously reported that, in a human melanoma cell line, Epac1 increases NDST-1, which converts N-acetylated glucosamine into N-sulfated form (Baljinnyam et al., 2009). In addition, it was suggested that Epac1 overexpression increases N-sulfation of HS chain (Baljinnyam et al., 2009).These data led us to examine the hypothesis that Epac1 can control FGF2 signaling by modification of N-sulfation of HS, most probably on perlecan. Further, as secreted FGF2 can act in a paracrine fashion, it is possible that melanoma cells expressing Epac1 regulate migration of surrounding endothelial or other melanoma cells. In this study, we found that Epac1 in melanoma cells increases N-sulfation of secreted perlecan and activates migration of endothelial/melanoma cells by FGF2/HS-mediated cell-cell interaction. In addition, the Epac1 in melanoma cells activates angiogenesis in vivo, which may support the survival of other melanoma cells expressing lower amounts of Epac1. Therefore, in addition to our previous reports showing the role of Epac1 in melanoma cells, this study demonstrated that expression of Epac1 in melanoma cells plays a role in melanoma progression by controlling cell/cell communication with endothelial cells and other melanoma cells.
Results
Epac1 in melanoma cells increases migration of neighboring endothelial cells via cell/cell communication
It was suggested that Epac1-expressing melanoma cells can increase migration of neighboring endothelial cells via N-sulfation of HSPG, and subsequently, the activation of paracrine-acting FGF2 signaling. Therefore, we investigated whether melanoma cells with abundant Epac1 expression can increase migration of those with scarce Epac1 expression. According to our previous report (Baljinnyam et al., 2010, 2011), in this study, we have divided the cell lines into two groups: Epac1-rich cell lines, in which Epac1 expression is of the same or higher level than that in SK-Mel-2 (SK-Mel-2, SK-Mel-24, SK-Mel187, and C8161 cells). Epac1-poor cell lines, in which Epac1 expression is lower than a half of Epac1 expression in SK-Mel-2 (HEMA-LP, WM3248, WM1552C, and WM115 cells). Conditioned medium (CM) of C8161 cells, which expresses abundant Epac1 (Baljinnyam et al., 2011), increased migration of human umbilical vein endothelial cells (HUVEC) (Figure1A). Both a neutralizing antibody against FGF2 and heparitinase, a HS-cleaving enzyme, inhibited the CM-induced HUVEC migration. Knockdown of Epac1 in C8161 cells (Figure1B) suppressed the CM-induced HUVEC migration (Figure1A). Hence, these data suggested that Epac1 in melanoma cells can increase migration of endothelial cells via FGF2- and/or HS-dependent mechanisms.
Figure 1.
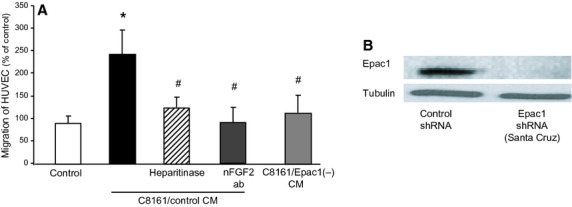
Epac1 in melanoma cells increases migration of endothelial cells via cell/cell communication. (A) CM of C8161 (C8161/control CM) increased migration of human umbilical vein endothelial cells (HUVEC). Epac1 knockdown in C8161 cells (C8161/Epac1(−) CM) inhibited the CM-induced migration. The CM-induced increase in migration was inhibited by the neutralizing antibody against FGF2 [nFGF2 ab (25 μg/ml)], and heparitinase (0.08 U/ml). *P < 0.05 versus control, #P < 0.05 versus C8161/control CM, n = 4. (B) Western blot of C8161 cells with stable knockdown of Epac1 performed with lentivirus-based shRNA induction.
Epac1 in melanoma cells induces tube formation of endothelial cells via cell/cell communication
As endothelial cell migration is fundamental for angiogenesis (Lamalice et al., 2007), we examined whether Epac1-expressing melanoma cells can stimulate endothelial tube formation, which mimics in vivo angiogenesis. As shown in Figure2A, B, CM of C8161 cells increased tube formation of HUVEC. Similar to migration (Figure1A), the CM-induced tube formation was inhibited by the neutralizing antibody against FGF2 and by heparitinase. In addition, CM of C8161 cells in which Epac1 was knocked down showed reduced tube formation (Figure2A, B). In vivo angiogenesis assay showed the same effect of Epac1 knockdown (Figure2C, D). These data suggested that Epac1 in melanoma cells have the ability to induce angiogenesis via FGF2- and/or HS-mediated cell/cell communication.
Figure 2.
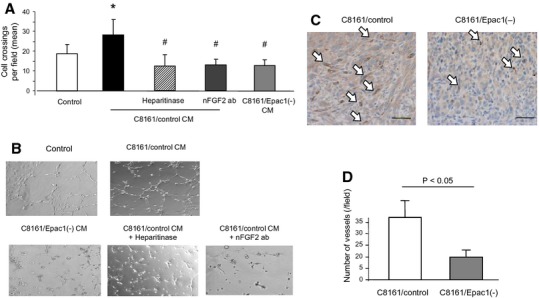
Epac1 in melanoma cells activates angiogenesis. (A) C8161/control CM increased tube formation of human umbilical vein endothelial cells (HUVEC). C8161/Epac1(−) CM showed reduced tube formation compared to C8161/control CM. The C8161/control CM-induced tube formation was inhibited by nFGF2 ab (25 μg/ml), and by heparitinase (0.08 U/ml). C8161/Epac1(−) CM showed reduced tube formation compared with C8161/control CM.*P < 0.01 versus control medium. #P < 0.01 versus C8161/control CM, n = 4. (B) Representative images of HUVEC tube formation described in A. (C and D) Epac1 knockdown reduces angiogenesis in vivo. C8161 cells with or without Epac1 knockdown (1 × 106 cells) were inoculated in the interscapular region of BALB/c mice. One week after the inoculation, tumor was removed. (C) Immunohistochemical images with anti-CD31 staining for the detection of endothelial cells are shown. White arrows indicate CD31-positive cells stained brown. Scale bar: 100 μm. (D) The number of microvessels in each mouse was counted with the positively stained cells in 10 different fields, n = 4.
Epac1 in melanoma cells increases migration of neighboring melanoma cells via cell/cell communication
Based on the increased HUVEC cell migration shown previously, we hypothesized that a similar cell/cell interaction may also exist among melanoma cells. To test this hypothesis, we examined whether CM derived from a melanoma cell line affects migration of other melanocyte/melanoma cells. CM from WM3248 or WM115 cells, both primary melanoma cell lines, did not change cell migration of HEMA-LP melanocyte cells (Figure3A). In contrast, CM sourced from SK-Mel-2 or C8161 cells, both metastatic melanoma cell lines, increased migration of HEMA-LP. Migration of WM1552C cells, a primary melanoma cell line of the radial growth phase (RGP), was examined next (Figure3B). CM of WM3248, a melanoma cell line of the vertical growth phase (VGP), SK-Mel-187, SK-Mel-2, or C8161 cells, all metastatic melanoma cell lines, increased WM1552C cell migration (Figure S3). In contrast, migration of the metastatic melanoma cell line, C8161 cells, was not affected by CM of SK-Mel-2. Epac1 overexpression (OE) in Epac1-poor melanoma cells indeed increased cell migration in both WM115 and WM3248 cells (Figure S1), suggesting that Epac1's effect on migration is saturated in Epac1-rich melanoma cells such as C8161 and SK-Mel-2 cells. Epac1 knockdown by two different Epac1 shRNAs (from Santa Cruz Biotechnology and Sigma Aldrich) in C8161 cells inhibited the CM-induced migration of HEMA-LP and WM1552C cells (Figure3A, B and S2). Similar result was obtained in Epac1 knockdown in SK-Mel-2 cells (Figure3B). These data suggested the specific role of Epac1 in the CM-induced migration.
Figure 3.
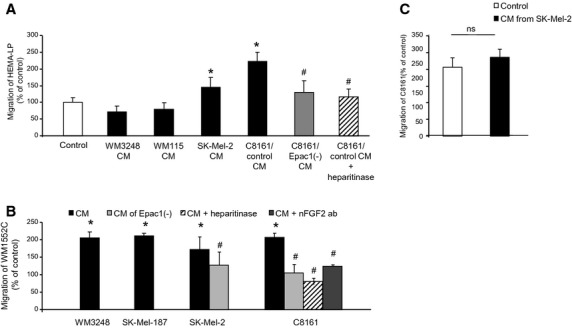
Epac1 in melanoma cells increases migration of melanocytes/other melanoma cells. (A) Conditioned media of indicated melanoma cell lines were used for the Boyden chamber migration assay of HEMA-LP cells. Conditioned media from SK-Mel-2 and C8161 cells, but not those from WM3248 and WM115 cells, increased migration of HEMA-LP cells. Knockdown of Epac1 (C8161/Epac1(−) CM) as well as heparitinase inhibited the CM-induced migration. *P < 0.05 versus control medium, #P < 0.05 versus C8161/control CM, n = 4. (B) Conditioned media of indicated melanoma cell lines were used for the Boyden chamber assay of WM1552C cells. Conditioned media of all cell lines examined increased migration of WM1552C cells. Knockdown of Epac1 inhibited migration induced by CM derived from SK-Mel-2 and C8161 cells. Heparitinase and the nFGF-2 antibody suppressed migration induced by CM of C8161 cells.*P < 0.05 versus control medium, #P < 0.05 versus CM, n = 4. (C) The Boyden chamber assay showed that CM of SK-Mel-2 cells did not increase migration of C8161 cells, n = 4.
The CM-induced migration of HEMA-LP and WM1552C cells were inhibited by heparitinase (Figure3A and B), and the CM-induced migration of WM1552C cells was suppressed by the neutralizing FGF2 antibody (Figure3B). The neutralizing FGF2 antibody inhibited CM-induced migration in other combinations of CM and cell lines used for migration (Figure S3). In addition, Epac1 OE in WM3248 cells increased their migration, and it was reduced by neutralizing FGF2 antibody (Figure S4).These data suggested that CM-induced migration was regulated by Epac1, HS and/or FGF2 signaling.
Epac1 augments the binding of FGF2 to FGF receptor
We next investigated the effects of Epac1 on HS including N-sulfation and FGF2 signaling. It has been demonstrated that perlecan interacts with FGF2 via its HS chains (Knox et al., 2002; Sharma et al., 1998). We thus examined perlecan expression of CM by isolation with chromatography. N-sulfated HS chains of perlecan were detected by the anti-HS antibody (clone 10E4) (Figure4A). The N-sulfation of HS bound to the perlecan was significantly reduced by Epac1 knockdown. In addition, both the amount of N-sulfation and the number of FGF receptors bound to FGF2 were decreased by knockdown of Epac1 (Figure4B). In contrast, neither the expression of total HS bound to FGF2 nor FGF2 itself in CM were changed by Epac1 knockdown (Figure4B), suggesting that Epac1 enhances FGF2-binding to FGF receptor via N-sulfation of HS. The binding assay showed that CM from C8161 cells increases FGF2 binding to FGF receptor expressed in HUVEC cells. The CM-induced FGF2 binding was inhibited by the FGF2 antibody and by Epac1 knockdown in C8161 cells (Figure4C). Taken together, these data demonstrated that Epac1-expressing melanoma cells regulate paracrine-acting FGF2 signaling in neighboring cells such as endothelial and melanoma cells by modification of HS.
Figure 4.
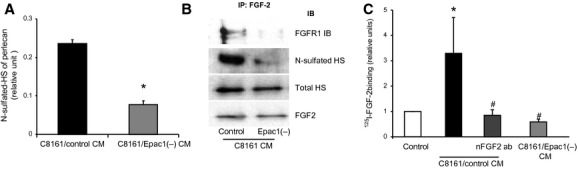
Epac1 enhances the binding of fibroblast growth factor (FGF2) to FGF receptor via N-sulfation of HS. (A) Perlecan was isolated from the DEAE chromatography fractions using a polyclonal antiperlecan antibody. The presence of HS chains on perlecan was detected using an anti-HS-specific antibody (10E4). Epac1 knockdown reduced the amount of N-sulfated HS attached to perlecan. *P < 0.05 versus C8161/control CM, n = 8. (B) CM of C8161 was subjected to immunoprecipitation with the antibody against FGF2 followed by Western blot for indicated antibodies. Both N-sulfated HS and FGF receptor 1 (FGFR1) attached to FGF2 were reduced by Epac1 knockdown whereas the amount of FGF2 in the CM was not different. (C) The binding assay for FGF2 in human umbilical vein endothelial cells (HUVEC) was performed with indicated CM. C8161/control CM increased the binding of FGF2 to HUVEC. The neutralizing antibody for FGF2 (nFGF2 ab) and knockdown of Epac1 inhibited the CM-induced FGF2 binding. *P < 0.05 versus control medium, #P < 0.05 versus C8161/control CM, n = 4.
Epac1-rich melanoma cells support proliferation of Epac1-poor melanoma cells in vivo
Increased angiogenesis by Epac1 (Figure2) suggested that Epac1-rich melanoma cells can support proliferation not only of Epac1-rich melanoma cells themselves but also of Epac1-poor melanoma cells via newly supplied blood flow. If this is the case, melanoma cells expressing low Epac1 that cannot survive in vivo are rescued by coexistence of Epac1-rich melanoma cells. Therefore, we examined whether coinoculation of melanoma cells with high Epac1 expression and those with low Epac1 expression enables the second type of cells to survive in mice. To show this, we used SK-Mel-2 cells, which abundantly express Epac1, and WM1552C cells, which poorly express Epac1 (Baljinnyam et al., 2011). In addition, we used green fluorescent protein (GFP) – or red fluorescent protein (RFP) to distinguish WM1552C cells from SK-Mel-2 cells. Our study showed that SK-Mel-2 cells inoculated in athymic nude mice, but not WM1552C cells, formed a tumor (Figure5A), suggesting that WM1552C cells alone cannot survive in mice. A tumor was formed by WM1552C cells coinoculated with SK-Mel-2 cells, but not with WM1552C cells inoculated alone (Figure5A–C). The tumor formed by the coinoculation showed both GFP- and RFP-fluorescent signal (Figure5D). In addition, fluorescence-activated cell sorting (FACS) analysis demonstrated that individual cells isolated from the tumor have either RFP signal or GFP signal (Table1). These data showed the existence of both WM1552C and SK-Mel-2 cells in the tumor and thus suggested that Epac1-rich melanoma cells can support the survival of Epac1-poor melanoma cells. As the percentages of GFP- and RFP-positive cells are not equal even in the same SK-Mel-2 cells (Table1) under in vivo conditions, it seems that one of the two inoculated cell lines becomes dominant. As CM of SK-Mel-2 cells did not increase proliferation of WM1552C cells (data not shown), these data suggest that SK-Mel-2 cells enable WM-1552C to survive in vivo most probably by modification of the extracellular matrix and enhanced angiogenesis.
Figure 5.
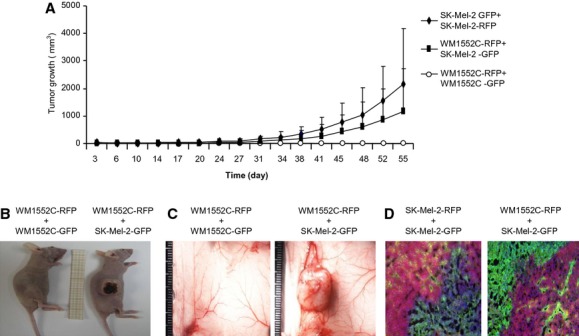
Epac1-rich melanoma cells support survival of Epac1-poor melanoma cells. (A) Tumor growth of WM1552C and SK-Mel-2 cells expressing Red Fluorescent Protein (RFP) and Green Fluorescent Protein (GFP) is shown. A mixture of indicated cells was injected in the right dorsolateral flank region in athymic BALB/c nude mice. Tumor size was measured twice a week to calculate tumor volume. Tumor failed to grow in the mixture of RFP- and GFP-labeled WM1552C. (B and C) Representative images of the tumors in the 12 weeks after the inoculation are shown. The mixture of RFP-labeled WM1552C cells and GFP-labeled SK-Mel-2 cells formed a tumor. (D) Representative images of coimmunostaining for RFP and GFP of the tumors formed by the indicated cell mixtures. Blue indicates 6-diamidino-2-phenylindole (DAPI) staining.
Table 1.
Fluorescence activated cell sorting (FACS) analyses for the population of red fluorescent protein (RFP)- and green fluorescent protein (GFP)-positive cells in melanoma tumor
| Cell lines coinoculated | Fluorescent signal used | % of total sorted cells in tumor | SD |
|---|---|---|---|
| WM1552C-RFP + SK-Mel-2-GFP | RFP | 0.26 | 0.21 |
| GFP | 85.9 | 4.72 | |
| SK-Mel-2-RFP + SK-Mel-2-GFP | RFP | 3.22 | 1.8 |
| GFP | 42 | 1.6 |
Formed tumors with coinoculation of indicated cell lines were isolated, dissected, and subjected to FACS analyses, n = 4.
Discussion
Our previous reports showed that Epac1 increases migration of melanoma cells themselves (Baljinnyam et al., 2009, 2010, 2011). Epac1 in melanoma cells may regulate the cell–cell communication, which could lead to an augmented migration of neighboring endothelial and melanoma cells. Our findings suggest that Epac1-rich melanoma cells play a major role in melanoma progression through migration of the Epac1-rich melanoma cells themselves, but also through increasing migration of neighboring Epac1-poor melanoma cells and more importantly, by the increased migration of neighboring endothelial cells that can accelerate tumor growth via angiogenesis. Therefore, it is plausible that Epac1-rich population in the melanoma tumor critically regulates tumor growth rate.
Although a number of reports demonstrated the role of FGF2 in melanoma progression (Gartside et al., 2009; Hibino et al., 2005; Meier et al., 2000; Ozen et al., 2004), little attention was focused on the role of paracrine-acting FGF2. Using B16F10, an invasive mouse melanoma cell line, CM-activated capillary formation of bovine aortic endothelial cells (Garrido et al., 1995). CM from A375, a human melanoma cell line, but not from normal melanocytes, increased migration and invasion of human mesenchymal stem cells. The CM-induced migration was inhibited by neutralization of FGF2 (Watts and Cui, 2012). Our results are consistent with these studies showing that CM of human melanoma cells increased migration of human endothelial cells via FGF2 signaling (Fig.1). Furthermore, we have demonstrated the role of Epac1 in migration of endothelial cells via paracrine-acting FGF2 signaling, which subsequently results in increased angiogenesis (Figure2). In addition, our results indicated the existence of FGF2-dependent cell/cell communication not only between melanoma and endothelial cells but also between melanoma and melanoma cells. This melanoma/melanoma cell communication in migration was obvious between Epac1-rich and Epac1-poor melanoma cells, but unclear between Epac1-rich and Epac1-rich melanoma cells (Figure3C). This lacking of cell/cell communication is probably explained by saturated migration via abundant expression of Epac1 in the same cells as we have previously shown (Baljinnyam et al., 2011) and by the minimal effect of autocrine FGF2 signaling. Regarding WM1552C migration (Figure3B), although Epac1's expression varies between the cell lines used for the study, the degree of migration did not directly reflect the degree of Epac1 expression. This was attributable, at least in part, to saturation of paracrine-acting FGF2 signaling and is supported by the data showing that FGF2 receptor expression is much higher in WM1552C cells compared with HEMA-LP (data not shown) in which the effects of CM are variable. Altogether, in terms of melanoma progression, Epac1's role in migration affects three types of cells: 1) Epac1-rich melanoma cells themselves, 2) Neighboring endothelial cells, 3) Neighboring Epac1-poor melanoma cells. Accordingly, targeting Epac1 would be an inhibitory mechanism for melanoma progression.
Perlecan is necessary for the binding of FGF2 to FGF receptor in human melanoma cells (Aviezer et al., 1997). N-sulfation of HS chains is critical for this interaction (Faham et al., 1996; Kreuger et al., 1999; Maccarana et al., 1993; Schlessinger et al., 2000). Although N-sulfation is largely regulated by NDSTs, little is known about how the expression/activity of NDSTs is regulated. We have shown that Epac1 can increase NDST-1 expression in melanoma cells (Baljinnyam et al., 2009). In addition, N-sulfation of HS was increased in the mixture of medium and cell lysate (Baljinnyam et al., 2011). In the present study, N-sulfation of secreted perlecan in the CM was reduced by Epac1 knockdown (Figure4A). Furthermore, FGF2 binding to FGF receptor was inhibited by Epac1 knockdown (Figure4B, C). Therefore, it is proposed that Epac1-rich melanoma cells can affect FGF2 signaling in neighboring cells via modification of N-sulfation of HS on perlecan. Meanwhile, knockdown of Epac1 reduced the amount of perlecan as demonstrated by Western blot analysis with a perlecan-specific antibody (CCN-1) (data not shown). Interestingly, expression of perlecan is regulated by the cAMP response element (CRE) as its promoter (Furuta et al., 2000). Thus, Epac1 potentially may regulate perlecan expression itself in addition to N-sulfation of HS, suggesting multiple roles of Epac1 on biosynthesis HSPG. However, further studies would be required to confirm this because another study found that Epac1 does not regulate transcription through CREB transcription factors and that the best characterized route for Epac1 to regulate transcription is through C/EBP transcription factors (Yarwood et al., 2008, JBC).
Our data showed that melanomas formed by coinoculation of Epac1-rich and Epac1-poor melanoma cells involved both melanoma populations (Figure5D and Table1). These data suggest that cell/cell communication within melanomas may support the survival of melanoma cells with lower malignancy potential. To confirm that Epac1 in Epac1-rich melanoma cells affect proliferation of another Epac1-poor melanoma cells, it is necessary to examine whether Epac1 knockdown decreases the number of Epac1-poor melanoma cells in vivo. However, inhibition of Epac1 itself affects angiogenesis as shown in our data (Figure2), which may result in decreased proliferation of Epac1-rich (SK-Mel-2) cells themselves. Indeed, knockdown of Epac1 reduced tumor growth in vivo (data not shown). Therefore, knockdown of Epac1 itself may affect the local blood supply and thus survival and proliferation of Epac1-poor melanoma cells. Therefore, when Epac1 is knocked down in Epac1-rich melanoma cells, multiple factors may affect proliferation of Epac1-poor melanoma cell, suggesting difficulty of interpretation of the acquired data. Recently, specific Epac1 inhibitors have become commercially available. These inhibitors, HJC-0350 and ESI-09, indeed suppressed CM-induced migration in WM3248 cells (Figure S5), suggesting potential usage of these inhibitors for melanoma therapy, which will be addressed in our future study. Finally, HS binds to and regulates the activity of extracellular superoxide dismutase (EC-SOD), which results in increased protection against oxidative stress (Yamamoto et al., 2000). In addition, a device containing HS to deliver FGF2 enhanced FGF2's antioxidative property (Galderisi et al., 2013). Accordingly, one could argue that Epac1 has antioxidative stress effects via the modification of HS-FGF2 signaling. Indeed, CM of SK-Mel-2 cells inhibited H2O2-induced apoptosis of WM1552C cells (data not shown). This antiapoptotic effect of the CM may modify the survival of WM1552C cells coinoculated with SK-Mel-2 cells in vivo (Figure5), whereas rigorous examination for the protection against antioxidative stress should be performed to obtain conclusive evidence.
In summary, this study for the first time demonstrated Epac1-mediated cell/cell communication by modification of FGF2–HS interaction. Our findings may lead to a new strategy for the melanoma therapy targeting a certain population of melanoma cells, that is, Epac1-rich melanoma cells. Future research should attempt to examine the effect of Epac1-specific inhibitors on melanoma progression.
Methods
Reagents and cell lines
HEMA-LP was purchased from Invitrogen (Carlsbad, CA, USA), HUVEC was from VEC Technologies. WM1552C was from Dr. Meenhard Herlyn, Wistar Institute. C8161 cell line was provided by Dr. Mary JC Hendrix. SK-Mel-2 cells (ATCC) were maintained in MEM containing 10% FBS, 1% penicillin/streptomycin. WM1552C and C8161 cells were maintained in RPMI with 10% FBS, 1% penicillin/streptomycin. HEMA-LP and HUVEC cells were maintained in EndoGRO medium (EMD Millipore, Billerica, MA, USA) containing 5% FBS. Antibodies against Epac1, FGF2, and FGFR-1 were from Cell Signaling, anti-NDST-1 antibody was from Abnova and anti-α-tubulin antibody was purchased from Abcam (Cambridge, MA, USA).
Short hairpin RNA transduction
Short hairpin RNA (shRNA) transductions with lentivirus (Santa Cruz Biotechnology) were performed as we previously described (Baljinnyam et al., 2010). C8161 cells were incubated with 8 μg/ml of Polybrene and lentiviral particles harboring shRNA were selected with puromycin dihydrochloride for 1 week. Fresh puromycin-containing medium was replaced every 3–4 days. Established cell lines are as follows: C8161 cells with control shRNA (C8161/control), C8161 cells with Epac1 shRNA [C8161/Epac1(−)].
Migration assay
Migration assay was performed using the 24-well Boyden chambers (8 μm pores, BD Biosciences, San Jose, CA, USA) as we previously described (Baljinnyam et al., 2009). The cells were plated at a density of 1 × 106 cells/100 μl of medium in the inserts and incubated for 16 h at 37°C in the conditioned media. The insert membranes were stained using the Diff-Quick kit (Dade Behring). Pictures were taken and migrated cells were counted with Image J software using 10 randomly chosen fields.
Purification of human perlecan
About 2 L of conditioned medium for 72 h by confluent cultures of human melanoma cells was purified by DEAE–Sepharose chromatography (Whitelock et al., 1999) (100 ml bed volume, flow rate 1 ml/min) which had been equilibrated with 250 mM NaCl (20 mM Tris, 10 mM Methylenediaminetetraacetic acid, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, pH 7.5). The column was washed extensively with the buffer, and bound proteins were eluted using 1 M NaCl, 20 mM Tris, 10 mM Methylenediaminetetraacetic acid. The presence of HS-bound perlecan was monitored in column fractions using antibodies to HS (10E4) in an enzyme-linked immunosorbent assay (ELISA). The protein concentration was measured using the Coomassie Plus assay (Pierce), and aliquots were stored at −70°C until used for further Western blot analyses.
Western blot analysis
Western blot analysis was performed as previously described (Iwatsubo et al., 2003, 2004). Briefly, cells were lysed and sonicated in RIPA lysis buffer. Equal amounts of protein were subjected to SDS-PAGE, were transferred to Millipore Immobilon-P membrane, and immunoblotting with respective antibodies was performed.
Tube formation assay
Human umbilical vein endothelial cells under seven passages were used in all experiments. In vitro angiogenesis tube formation assay was performed as we previously described with some modifications (De Lorenzo et al., 2004; Movafagh et al., 2006). HUVEC (5000/well) were seeded in 24-well plates coated with Matrigel (Biosciences Discovery), incubated in CM for 4 h at 37°C. The tube formation was quantified by counting the number of connecting branches between two discrete endothelial cells.
Immunoprecipitation
Dynabeads-Protein G for immunoprecipitation (Life Technologies, Carlsbad, CA, USA) were incubated with the primary antibodies and added to the soluble cell lysate fraction. These antibody-coated Dynabeads™, Life Technologies, Carlsbad, CA, USA bound to the target proteins were separated by the magnet and after repeated washing three times, the isolated protein complexes were subjected to SDS-PAGE and immunoblotting with respective antibodies.
FGF2-binding assay
FGF2-binding assay was performed as previously described (Reiland and Rapraeger, 1993). Briefly, HUVEC cells were plated in 24-well plate with 1.5 × 105 cells density and incubated with and without indicated. The cells were pulsed with 50 pM 125I-bFGF for 2 h at 4°C in binding buffer, washed three times with 20 mM HEPES (pH 7.4) containing 150 mM NaCl and 0.2% BSA at 4°C. Low-affinity HSPG-binding sites were detected by two collected 1-ml washes of 20 mM HEPES (pH 7.4) containing 2 M NaCl and 0.2% BSA at 4°C. High-affinity FGFR complex binding sites were detected by two collected 1-ml washes of 20 mM sodium acetate (pH 4.0) containing 2 M NaCl and 0.2% BSA at 4°C. Collected washes were counted in a Cobra 5003 counter (Packard/Perkin Elmer, Waltham, MA, USA). Control experiments were performed with unlabeled FGF2 to determine non-specific binding. Results were reported as the relative binding of experimental condition compared with untreated controls.
Generation of GFP- and RFP-labeled melanoma cells
Cells were incubated with lentiviral particles for GFP and RFP expression (Biogenova, Potomac, MD, USA) and were selected with FACS before the inoculation to obtain the cells homogenously expressing RFP or GFP. FACS cell sorting was performed by a FACS Caliburs (BD Biosciences). In vivo imaging of RFP- and GFP-labeled tumor cells were carried out by in vivo imaging system (IVIS).
Tumor growth assay
BALB/c athymic (nu/nu) mice were inoculated in the right flank with C8161 cells with or without Epac1 shRNA deletions (106 cells/0.1 ml culture medium) (n = 6/group). In another series of experiments, prelabeled SK-Mel-2 cells (MM, high Epac1 expression) and WM1552C cells (RGP, low Epac1 expression) were used: (a) SK-Mel-2-GFP + SK-Mel-2-RFP injected mice n = 8/group; (b) SK-Mel-2-GFP+WM-1552C-RFP cells injected mice n = 8/group; c. WM-1552C-GFP+ WM-1552C-RFP cells injected mice, n = 4/group. Tumor growth was assessed twice a week by caliper measurement of tumor diameter in the longest dimension (L) and at right angles to that axis (W) (De Lorenzo et al., 2011). Tumor volumes were estimated using the formula, L × W × W × π/6. At the end of the experiment, half of each tumor was fixed by immersion in 10% phosphate-buffered formalin, dehydrated, and embedded in paraffin. Major organs were subjected to gross pathology and histology analysis to determine metastases. Studies were approved by the Animal Care and Use Committee of New Jersey Medical School.
Immunofluorescent staining
The paraffin-embedded slides of melanomas from BALB/c mice were subjected to deparaffinization in xylene, followed by treatment with a graded series of alcohols (100%, 95%, and 80% ethanol [v/v] in double-distilled H2O) and rehydration in PBS (pH 7.5). For antigen retrieval, the sections were submerged in a boiling temperature citrate buffer (pH 6.0) for 15 m. The samples were blocked with the Image-iT FX signal enhancer (Invitrogen) to prevent non-specific staining and incubated with primary antibodies and respective secondary antibodies. Alexa Fluor 488– and 594-conjugated goat anti-rabbit and anti-mouse antibodies (Molecular Probes, Life Technologies) were used as secondary antibodies. The slides were mounted using Prolong Gold mounting media with 4', 6-diamidino-2-phenylindole (DAPI).
For the study of RFP- and GFP-labeled cells in tumors, tissue sections from tumors were immunostained with rabbit antibody against GFP (dilution 1:100, Abcam), mouse antibody against RFP (dilution 1:200; Abcam). Negative controls without the primary antibody were performed to show specificity of the antibody.
Immunohistochemical staining
Tumor angiogenesis was evaluated by immunostaining for CD31 (dilution1:250, Santa Cruz Biotechnologies, Santa Cruz, CA, USA). Tissue sections were cut and immunostained with the primary antibody for CD31 using the standard VectaStain ABC kit (Vector Laboratories, Burlingame, CA, USA). Microvessel density was assessed by counting the number of microvessels positive for CD31 at ×400 magnification. Negative control without the primary antibody was performed at the same time.
Overexpression of Epac1
Adenoviral OE of Epac1 in melanoma cells was performed as we previously described (Baljinnyam et al., 2009).
Data analysis and statistics
Statistical comparisons among groups were performed using one-factor anova with Bonferroni post hoc test. Statistical significance was set at the 0.05 level.
Acknowledgments
This study was supported by the American Heart Association (SDG 0835596D), the Foundation of UMDNJ and Melanoma Research Foundation (K. Iwatsubo).
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Epac1 overexpression (OE) increases migration of primary melanoma cell lines. (A) Western blot of Epac1 OE in WM115 and WM3248 cells 24 h after adenoviral infection. (B) Epac1 OE increased migration of WM115 and WM3248 cells. *P < 0.05 versus control, n = 4.
Figure S2. Epac1 regulates CM-induced migration of primary melanoma cells. (A) Western blot of C8161 cells with or without Epac1shRNA (Sigma Aldrich) transduction. (B) Epac1 knockdown with Epac1 shRNA (Sigma) in C8161 cells inhibited the CM-induced migration of WM1552C cells.
Figure S3. FGF2 is involved CM-induced migration of primary melanoma. Indicated combinations of cells for the evaluation of migration and CM preparation were examined. The neutralizing FGF2 antibody reduced cell migration in all examined combinations. #, P < 0.05 versus CM, n = 4.
Figure S4. FGF2 is involved in Epac1 OE-mediated CM migration. CM of WM3248 cells with adenoviral Epac1 OE increased migration of SK-Mel-2 cells. The nFGF2 antibody inhibited the Epac1 OE-induced migration, n = 4.
Figure S5. Epac1 inhibitors reduce CM-induced migration. Migration of WM3248 cells was inhibited by CM of SK-Mel-24 cells were treated with indicated Epac inhibitors, n = 4.
References
- Aviezer D, Iozzo RV, Noonan DM. Yayon A. Suppression of autocrine and paracrine functions of basic fibroblast growth factor by stable expression of perlecan antisense cDNA. Mol. Cell. Biol. 1997;17:1938–1946. doi: 10.1128/mcb.17.4.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baljinnyam E, Iwatsubo K, Kurotani R, Wang X, Ulucan C, Iwatsubo M, Lagunoff D. Ishikawa Y. Epac increases melanoma cell migration by a heparan sulfate-related mechanism. Am. J. Physiol. Cell Physiol. 2009;297:C802–C813. doi: 10.1152/ajpcell.00129.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baljinnyam E, De Lorenzo MS, Xie LH, Iwatsubo M, Chen S, Goydos JS, Nowycky MC. Iwatsubo K. Exchange protein directly activated by cyclic AMP increases melanoma cell migration by a Ca2 + -dependent mechanism. Cancer Res. 2010;70:5607–5617. doi: 10.1158/0008-5472.CAN-10-0056. [DOI] [PubMed] [Google Scholar]
- Baljinnyam E, Umemura M, De Lorenzo MS, Iwatsubo M, Chen S, Goydos JS. Iwatsubo K. Epac1 promotes melanoma metastasis via modification of heparan sulfate. Pigment Cell Melanoma Res. 2011;24:680–687. doi: 10.1111/j.1755-148X.2011.00863.x. [DOI] [PubMed] [Google Scholar]
- Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem. Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- De Lorenzo MS, Farina HG, Alonso DF. Gomez DE. Role of protein kinase C-dependent signaling pathways in the antiangiogenic properties of nafoxidine. Anticancer Res. 2004;24:1737–1744. [PubMed] [Google Scholar]
- De Lorenzo MS, Baljinnyam E, Vatner DE, Abarzúa P, Vatner SF. Rabson AB. Caloric restriction reduces growth of mammary tumors and metastases. Carcinogenesis. 2011;32:1381–1387. doi: 10.1093/carcin/bgr107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A. Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Faham S, Hileman RE, Fromm JR, Linhardt RJ. Rees DC. Heparin structure and interactions with basic fibroblast growth factor. Science. 1996;271:1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- Furuta GT, Dzus AL, Taylor CT. Colgan SP. Parallel induction of epithelial surface-associated chemokine and proteoglycan by cellular hypoxia: implications for neutrophil activation. J. Leukoc. Biol. 2000;68:251–259. [PubMed] [Google Scholar]
- Galderisi U, Peluso G, Di Bernardo G, Calarco A, D'apolito M, Petillo O, Cipollaro M, Fusco FR. Melone MA. Efficient cultivation of neural stem cells with controlled delivery of FGF-2. Stem Cell Res. 2013;10:85–94. doi: 10.1016/j.scr.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Garrido T, Riese HH, Aracil M. Perez-Aranda A. Endothelial cell differentiation into capillary-like structures in response to tumour cell conditioned medium: a modified chemotaxis chamber assay. Br. J. Cancer. 1995;71:770–775. doi: 10.1038/bjc.1995.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartside MG, Chen H, Ibrahimi OA, et al. Loss-of-function fibroblast growth factor receptor-2 mutations in melanoma. Mol. Cancer Res. 2009;7:41–54. doi: 10.1158/1541-7786.MCR-08-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandoch M, Lopez De Jesus M, Oude Weernink PA, Weber AA, Jakobs KH. Schmidt M. B cell receptor-induced growth arrest and apoptosis in WEHI-231 immature B lymphoma cells involve cyclic AMP and Epac proteins. Cell. Signal. 2009a;21:609–621. doi: 10.1016/j.cellsig.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Grandoch M, Rose A, Ter Braak M, Jendrossek V, Rubben H, Fischer JW, Schmidt M. Weber AA. Epac inhibits migration and proliferation of human prostate carcinoma cells. Br. J. Cancer. 2009b;101:2038–2042. doi: 10.1038/sj.bjc.6605439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino S, Shibuya M, Hoffman MP, Engbring JA, Hossain R, Mochizuki M, Kudoh S, Nomizu M. Kleinman HK. Laminin alpha5 chain metastasis- and angiogenesis-inhibiting peptide blocks fibroblast growth factor 2 activity by binding to the heparan sulfate chains of CD44. Cancer Res. 2005;65:10494–10501. doi: 10.1158/0008-5472.CAN-05-0314. [DOI] [PubMed] [Google Scholar]
- Iozzo RV. San Antonio JD. Heparan sulfate proteoglycans: heavy hitters in the angiogenesis arena. J. Clin. Invest. 2001;108:349–355. doi: 10.1172/JCI13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsubo K, Toya Y, Fujita T, Ebina T, Schwencke C, Minamisawa S, Umemura S. Ishikawa Y. Ischemic preconditioning prevents ischemia-induced beta-adrenergic receptor sequestration. J. Mol. Cell. Cardiol. 2003;35:923–929. doi: 10.1016/s0022-2828(03)00173-1. [DOI] [PubMed] [Google Scholar]
- Iwatsubo K, Minamisawa S, Tsunematsu T, Nakagome M, Toya Y, Tomlinson JE, Umemura S, Scarborough RM, Levy DE. Ishikawa Y. Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J. Biol. Chem. 2004;279:40938–40945. doi: 10.1074/jbc.M314238200. [DOI] [PubMed] [Google Scholar]
- Knox S, Merry C, Stringer S, Melrose J. Whitelock J. Not all perlecans are created equal: interactions with fibroblast growth factor (FGF) 2 and FGF receptors. J. Biol. Chem. 2002;277:14657–14665. doi: 10.1074/jbc.M111826200. [DOI] [PubMed] [Google Scholar]
- Kreuger J, Prydz K, Pettersson RF, Lindahl U. Salmivirta M. Characterization of fibroblast growth factor 1 binding heparan sulfate domain. Glycobiology. 1999;9:723–729. doi: 10.1093/glycob/9.7.723. [DOI] [PubMed] [Google Scholar]
- Lamalice L, le Boeuf F. Huot J. Endothelial cell migration during angiogenesis. Circ. Res. 2007;100:782–794. doi: 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- Lissitzky JC, Parriaux D, Ristorcelli E, Verine A, Lombardo D. Verrando P. Cyclic AMP signaling as a mediator of vasculogenic mimicry in aggressive human melanoma cells in vitro. Cancer Res. 2009;69:802–809. doi: 10.1158/0008-5472.CAN-08-2391. [DOI] [PubMed] [Google Scholar]
- Little AS, Smith PD. Cook SJ. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene. 2012;32:1207–1215. doi: 10.1038/onc.2012.160. [DOI] [PubMed] [Google Scholar]
- Maccarana M, Casu B. Lindahl U. Minimal sequence in heparin/heparan sulfate required for binding of basic fibroblast growth factor. J. Biol. Chem. 1993;268:23898–23905. [PubMed] [Google Scholar]
- Maurer G, Tarkowski B. Baccarini M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene. 2011;30:3477–3488. doi: 10.1038/onc.2011.160. [DOI] [PubMed] [Google Scholar]
- Meier F, Nesbit M, Hsu MY, et al. Human melanoma progression in skin reconstructs: biological significance of bFGF. Am. J. Pathol. 2000;156:193–200. doi: 10.1016/S0002-9440(10)64719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier F, Caroli U, Satyamoorthy K, et al. Fibroblast growth factor-2 but not Mel-CAM and/or beta3 integrin promotes progression of melanocytes to melanoma. Exp. Dermatol. 2003;12:296–306. doi: 10.1034/j.1600-0625.2003.120310.x. [DOI] [PubMed] [Google Scholar]
- Montesano R, Vassalli JD, Baird A, Guillemin R. Orci L. Basic fibroblast growth factor induces angiogenesis in vitro. Proc. Natl Acad. Sci. USA. 1986;83:7297–7301. doi: 10.1073/pnas.83.19.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscatelli D, Presta M, Joseph-Silverstein J. Rifkin DB. Both normal and tumor cells produce basic fibroblast growth factor. J. Cell. Physiol. 1986;129:273–276. doi: 10.1002/jcp.1041290220. [DOI] [PubMed] [Google Scholar]
- Movafagh S, Hobson JP, Spiegel S, Kleinman HK. Zukowska Z. Neuropeptide Y induces migration, proliferation, and tube formation of endothelial cells bimodally via Y1, Y2, and Y5 receptors. FASEB J. 2006;20:1924–1926. doi: 10.1096/fj.05-4770fje. [DOI] [PubMed] [Google Scholar]
- Nugent MA. Iozzo RV. Fibroblast growth factor-2. Int. J. Biochem. Cell Biol. 2000;32:115–120. doi: 10.1016/s1357-2725(99)00123-5. [DOI] [PubMed] [Google Scholar]
- Ozen M, Medrano EE. Ittmann M. Inhibition of proliferation and survival of melanoma cells by adenoviral-mediated expression of dominant negative fibroblast growth factor receptor. Melanoma Res. 2004;14:13–21. doi: 10.1097/00008390-200402000-00003. [DOI] [PubMed] [Google Scholar]
- Ponta H, Wainwright D. Herrlich P. The CD44 protein family. Int. J. Biochem. Cell Biol. 1998;30:299–305. doi: 10.1016/s1357-2725(97)00152-0. [DOI] [PubMed] [Google Scholar]
- Quilliam LA, Rebhun JF. Castro AF. A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family GTPases. Prog. Nucleic Acid Res. Mol. Biol. 2002;71:391–444. doi: 10.1016/s0079-6603(02)71047-7. [DOI] [PubMed] [Google Scholar]
- Reiland J. Rapraeger AC. Heparan sulfate proteoglycan and FGF receptor target basic FGF to different intracellular destinations. J. Cell Sci. 1993;105(Pt 4):1085–1093. doi: 10.1242/jcs.105.4.1085. [DOI] [PubMed] [Google Scholar]
- Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, Linhardt RJ. Mohammadi M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell. 2000;6:743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- Sharma B, Handler M, Eichstetter I, Whitelock JM, Nugent MA. Iozzo RV. Antisense targeting of perlecan blocks tumor growth and angiogenesis in vivo. J. Clin. Invest. 1998;102:1599–1608. doi: 10.1172/JCI3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola F, Gualandris A, Belleri M, et al. Endothelial cells overexpressing basic fibroblast growth factor (FGF-2) induce vascular tumors in immunodeficient mice. Angiogenesis. 1997;1:102–116. doi: 10.1023/A:1018309200629. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Greenberg AH, Turley EA. Wright JA. Cell motility, invasion, and malignancy induced by overexpression of K-FGF or bFGF. Exp. Cell Res. 1993;204:295–301. doi: 10.1006/excr.1993.1036. [DOI] [PubMed] [Google Scholar]
- Tiwari S, Felekkis K, Moon EY, Flies A, Sherr DH. Lerner A. Among circulating hematopoietic cells, B-CLL uniquely expresses functional EPAC1, but EPAC1-mediated Rap1 activation does not account for PDE4 inhibitor-induced apoptosis. Blood. 2004;103:2661–2667. doi: 10.1182/blood-2003-06-2154. [DOI] [PubMed] [Google Scholar]
- Watts TL. Cui R. Malignant melanoma induces migration and invasion of adult mesenchymal stem cells. Laryngoscope. 2012;122:2769–2772. doi: 10.1002/lary.23652. [DOI] [PubMed] [Google Scholar]
- Whitelock JM, Graham LD, Melrose J, Murdoch AD, Iozzo RV. Underwood PA. Human perlecan immunopurified from different endothelial cell sources has different adhesive properties for vascular cells. Matrix Biol. 1999;18:163–178. doi: 10.1016/s0945-053x(99)00014-1. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Hara H. Adachi T. Effects of homocysteine on the binding of extracellular-superoxide dismutase to the endothelial cell surface. FEBS Lett. 2000;486:159–162. doi: 10.1016/s0014-5793(00)02260-2. [DOI] [PubMed] [Google Scholar]
- Yarwood SJ, Borland G, Sands WA. Palmer TM. Identification of CCAAT/enhancer-binding proteins as exchange protein activated by cAMP-activated transcription factors that mediate the induction of the SOCS-3 gene. J. Biol. Chem. 2008;283:6843–6853. doi: 10.1074/jbc.M710342200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Epac1 overexpression (OE) increases migration of primary melanoma cell lines. (A) Western blot of Epac1 OE in WM115 and WM3248 cells 24 h after adenoviral infection. (B) Epac1 OE increased migration of WM115 and WM3248 cells. *P < 0.05 versus control, n = 4.
Figure S2. Epac1 regulates CM-induced migration of primary melanoma cells. (A) Western blot of C8161 cells with or without Epac1shRNA (Sigma Aldrich) transduction. (B) Epac1 knockdown with Epac1 shRNA (Sigma) in C8161 cells inhibited the CM-induced migration of WM1552C cells.
Figure S3. FGF2 is involved CM-induced migration of primary melanoma. Indicated combinations of cells for the evaluation of migration and CM preparation were examined. The neutralizing FGF2 antibody reduced cell migration in all examined combinations. #, P < 0.05 versus CM, n = 4.
Figure S4. FGF2 is involved in Epac1 OE-mediated CM migration. CM of WM3248 cells with adenoviral Epac1 OE increased migration of SK-Mel-2 cells. The nFGF2 antibody inhibited the Epac1 OE-induced migration, n = 4.
Figure S5. Epac1 inhibitors reduce CM-induced migration. Migration of WM3248 cells was inhibited by CM of SK-Mel-24 cells were treated with indicated Epac inhibitors, n = 4.