

Abstract
Of the deaths attributed to cancer, 90% are due to metastasis, and treatments that prevent or cure metastasis remain elusive. Emerging data indicate that hypoxia and the extracellular matrix (ECM) might have crucial roles in metastasis. During tumour evolution, changes in the composition and the overall content of the ECM reflect both its biophysical and biological properties and these strongly influence tumour and stromal cell properties, such as proliferation and motility. Originally thought of as independent contributors to metastatic spread, recent studies have established a direct link between hypoxia and the composition and the organization of the ECM, which suggests a new model in which multiple microenvironmental signals might converge to synergistically influence metastatic outcome.
Rapid cancer cell proliferation, combined with structural and functional abnormalities in tumour blood vessels, results in regions within solid tumours that have reduced oxygen availability1. Oxygen availability decreases as the distance from the nearest blood vessel increases. Intratumoural hypoxia is associated with disorganized vascular networks with intercapillary distances that are greater than the diffusion distance of oxygen (which is ~100–200 μm, depending on the local oxygen concentration in the blood and the rates of oxygen consumption)2. The direct measurement of the tumour partial pressure of oxygen (PO2) using Eppendorf microelectrodes (which remains the gold standard for determining oxygen levels) has revealed that patients whose primary tumours are poorly oxygenated (those with PO2 <10 mmHg) have an increased risk of metastasis and mortality1,3,4. The best understood mechanism of how cancer cells adapt to a hypoxic environment is through the transcriptional activity of hypoxia-inducible factors (hypoxia-inducible factor 1 (HIF1) and HIF2; see BOX 1)5. The role of hypoxia and HIFs in reprogramming cancer cells by regulating the expression of multiple genes involved in angiogenesis, by regulating the metabolism of glucose and by regulating cancer cell invasion and metastasis has been extensively reviewed elsewhere6–10. Recent reviews also highlight the importance of hypoxia in recruiting the stromal cell components of the tumour microenvironment11,12. In this Opinion article, we focus on how hypoxia affects extracellular matrix (ECM) deposition, remodelling and degradation, which might potentiate cancer metastasis. Central to this emerging paradigm are three crucial findings: the ECM is a dynamic structure that influences tumour progression13–18; multiple cell types, including cancer cells, contribute to ECM production19–23; and the remodelled ECM within regions of intratumoural hypoxia could be a pathway rather than an obstacle for cancer metastasis24–27.
Box 1. Regulation of HIFs.
Hypoxia-inducible factors (HIFs) are transcription factors that function as heterodimers, which consist of an oxygen-regulated HIF1α (or HIF2α) subunit and a constitutively expressed HIF1β subunit178,179. HIFs bind to the consensus sequence 5′-RCGTG-3′ that is present within or near HIF-regulated genes180. HIF1α protein levels are regulated by oxygen-dependent prolyl hydroxylation, which is required for binding of the von Hippel–Lindau (VHL) tumour suppressor protein, leading to ubiquitylation and proteasomal degradation of HIF1α181. Hydroxylation of HIF1α residues Pro402 and Pro564 is catalysed by HIF prolyl hydroxylase domain-containing protein 1 (PHD1), PHD2 and PHD3 in a reaction that is dependent on the presence of cofactors, oxygen and α-ketoglutarate (also known as 2-oxoglutarate). Under low oxygen conditions, HIF1α hydroxylation, ubiquitylation and degradation are inhibited153. HIF2α, which shares 48% amino acid sequence identity with HIF1α, is also oxygen-regulated and binds to HIF1β to form HIF2, which activates the transcription of some, but not all, HIF target genes182,183. Many oncogenic alterations in cancers cells, including loss of function of VHL, PTEN and p53184–186, as well as activation of the PI3K–AKT187 pathway, also cause an increase in HIF activity. Data obtained from many recent studies that use a range of approaches have revealed unique roles for HIF1α and HIF2α in both normal and cancer cells152.
The ECM and cancer
The ECM is composed of approximately 300 proteins that regulate tissue homeostasis, organ development, inflammation and disease19. The major constituents of the ECM are fibrous proteins (such as collagens, elastins, fibronectins and laminins) and proteoglycans (such as chondroitin sulphate, heparan sulphate, keratan sulphate and hyaluronic acid) that are locally secreted and assembled into an organized mesh, which forms the structural framework for most tissues28. Molecular approaches aiming to correlate clinical outcomes with specific gene expression patterns within the primary tumour have highlighted genes that encode tumour-associated ECM components19,29–34. An increased expression of genes encoding proteins that mediate ECM remodelling has been associated with increased mortality in patients with breast, lung and gastric cancers35,36. These studies corroborate histological findings that show an excessive ECM deposition (also termed fibrosis) within solid tumours37–43.
The most well-recognized ECM alteration that occurs in the tumour tissue is increased collagen deposition44–53. Collagens are the most abundant ECM components, constituting up to 90% of the ECM and 30% of the total protein in humans, and they provide the structural integrity and the tensile strength of human tissues and organs54. In the context of cancer biology, collagens regulate the physical and the biochemical properties of the tumour microenvironment, which modulate cancer cell polarity, migration and signalling17,55–58. Collagen I deposition and cancer metastasis have been causally linked using mice engineered to express a collagenase-resistant α1 chain of type I collagen (Col1a1tm1jae mice)59. Col1a1tm1jae mice were crossed with mouse mammary tumour virus promoter-driven polyoma middle T antigen (MMTV-PyMT) transgenic mice to model increased type I collagen deposition during the progression of human breast cancer59–61. Col1a1tm1jae; MMTV-PyMT bitransgenic mice had a threefold increase in the incidence of tumour formation and metastasis compared with their wild-type littermates60. Furthermore, histological studies of human breast carcinomas have shown that fibrosis is localized to hypoxic regions within tumours and correlates with immunostaining of the HIF1 target gene product carbonic anhydrase IX (CAIX)51,52. Highly fibrotic tumours also have the highest CAIX immune reactivity, which can independently predict patient relapse rate and shorter disease-free survival51,53. In this Opinion article, we discuss emerging data that has provided experimental evidence linking the mechanisms of hypoxia-induced collagen deposition and remodelling to those of invasion and metastasis.
Tumour ECM synthesis and degradation
The current view of tumour fibrosis suggests that recruited and resident fibroblasts and myofibroblasts within the primary tumour are mediators of tumour fibrosis. These cells are activated by proteins that are secreted by cancer cells, most notably by transforming growth factor-β (TGFβ), which stimulates the synthesis of ECM proteins and the remodelling of the ECM by proteases produced by cancer-associated fibroblasts62. Fibroblasts that are isolated from the site of a healing wound or from fibrotic tissues secrete higher levels of normal ECM constituents and proliferate more than their normal counterparts that are isolated from healthy organs, which is an explanation for the increase in matrix deposition that occurs within a tumour63. Although it is well accepted that invasive carcinoma is often associated with increased ECM deposition in tumours64, there is also evidence for an increased deposition of ECM in hypoxic tumour regions. Recent studies have uncovered mechanisms of tumour fibrosis that specifically occur under hypoxic conditions and that involve not only fibroblasts but also other cell types, including cancer cells20,21,65–68.
Hypoxia induces increased collagen gene expression
Hypoxia and HIF1 have been implicated in renal, liver and adipose tissue fibrosis69–71. Dermal, cardiac and renal fibroblasts cultured under hypoxic conditions show increased type I procollagen α1 mRNA levels72–74. Furthermore, increased levels of type I, II and IV procollagen mRNA are present in the peripheral lung parenchyma and pulmonary artery of rats that have been exposed to hypoxia75. However, studies that describe the regulation of collagen gene expression in hypoxic cancer cells in vitro and during cancer progression in vivo are lacking. By contrast, the dramatic effect of hypoxia on the post-translational modification of collagen is a matter of considerable investigation, as described below.
HIF1 regulates the expression of intracellular collagen-modifying enzymes
Collagen biogenesis originates with gene transcription and is followed by the translation of mRNA into procollagen (pro-α-chains) (FIG. 1). At least 28 collagen subtypes, which are encoded by 42 genes that generate 42 distinct pro-α-chains, have been identified in vertebrates76. Within the endoplasmic reticulum, the pro-α-chains undergo multiple post-translational modifications, which include the hydroxylation of proline and lysine residues, followed by the glycosylation of hydroxylysine residues76. The modification of proline to 4-hydroxyproline is essential for the thermal stability of the collagen triple helix77. Procollagen α-chains that are not hydroxylated are improperly folded, which leads to proteolytic degradation and to reduced collagen deposition76,78. Three isoforms of the prolyl 4-hydroxylase α-subunit (P4HA) have been identified (P4HA isoform 1 (P4HA1), P4HA2 and P4HA3) that form A2B2 tetramers with P4HB, which results in the generation of P4H1 (from P4HA1), P4H2 (from P4HA2) and P4H3 (from P4HA3) holoenzymes79,80. Three procollagen-lysine 2-oxyglutarate 5-dioxygenase genes (PLOD1, PLOD2 and PLOD3) encode enzymes that mediate collagen lysine hydroxylation. Collagen crosslinks that are derived from hydroxylated lysine residues compared with non-hydroxylated lysine residues have increased stability, which leads to increased tissue stiffness81. Thus, stiff tissues, such as bones, cartilage and tendons, contain a higher percentage of hydroxylated lysine residues in collagen compared with soft tissues, such as the skin81.
Figure 1. Biosynthesis of fibrillar collagens.

The biosynthesis of type I collagen and other fibrillar collagens can be divided into intracellular (parts a–c) and extracellular (parts d–f) steps. The first intracellular step involves the synthesis of procollagen polypeptides from any of 42 distinct collagen gene transcripts (part a). Procollagens are post-translationally modified within the cisternae of the endoplasmic reticulum (ER) by prolyl 4-hydroxylase α-subunit isoform 1 (P4HA1), P4HA2 and P4HA3 and by procollagen-lysine 2-oxyglutarate 5-dioxygenase 1 (PLOD1), PLOD2 and PLOD3 lysyl hydroxylase enzymes (part b). Hydrolysine residues can be further modified to galactosyl hydroxylysine and to glucosylgalactosyl hydroxylysine by collagen galactosyltransferase and glucosyltransferase, respectively. The carboxyl termini of three properly hydroxylated procollagen molecules will associate and spontaneously propagate a procollagen triple helix from the carboxyl terminus to the amino terminus. The triple helical procollagen will be transported from the ER to the extracellular space via the Golgi (part c). Two metalloproteinases, a procollagen N-terminal proteinase and a procollagen C-terminal proteinase, cleave the non-helical termini (part d) and the mature collagen proteins spontaneously aggregate to form a collagen fibril (part e). The final step, collagen fibre formation, is initiated by collagen crosslinking, which is catalysed by lysyl oxidase (LOX) family members and occurs via the lysine aldehyde- or hydroxylysine aldehyde-initiated pathway (part f). The number and the proportion of the various crosslinks are tissue specific and are regulated by the steric relationship between localized collagen molecules, the type of collagens co-polymerized and the glycosylation and the hydroxylation of the participating amino acid residues. For example, lysine aldehyde-initiated crosslinks are found in soft connective tissue, in contrast to hydroxylysine aldehyde-initiated crosslinks, which are found in stiff connective tissues. Many non-fibrillar collagens retain a non-collagenous N- or C-terminal, which prevents the spontaneous formation of collagen fibrils, and in these collagens cysteine crosslinks might be the only source of covalent intermolecular bonds. Enzymes highlighted in red are induced under hypoxic conditions. LOXL, LOX-like protein.
HIF1 regulates the expression of P4HA1, P4HA2, PLOD1 and PLOD2 in cancer cells, fibroblasts, chondrocytes and endothelial cells20,21,67,82–86. Abrogating the expression of HIF1α, P4HA1 or P4HA2 through the stable transfection of cells with short hairpin RNA (shRNA) vectors inhibits collagen deposition from both breast cancer cells and fibroblasts in vitro21,82. Reducing the levels of HIF1α, P4HA1 or P4HA2 in vivo results in decreased fibrosis and decreased tissue stiffness in orthotopic tumours that are formed by the injection of human breast cancer cells into the mammary fat pads of immunodeficient mice21,68. Decreasing the levels of HIF2α expression in breast cancer cells had no effect21. Importantly, P4HA1 or P4HA2 knockdown inhibited the spontaneous metastasis of breast cancer cells to the lungs and to the lymph nodes of mice by reducing the formation of collagen fibres, which are required for cancer cell adhesion, spreading and invasion21,68. In contrast to P4HA1 and P4HA2, the depletion of PLOD2 in breast cancer cells did not suppress collagen deposition in vitro or in vivo, but reduced tumour stiffness by reducing fibrillar collagen content20. PLOD2 knockdown also significantly impaired the invasion of cancer cells into the adjacent normal tissue of the mouse mammary fat pad, reduced the number of circulating tumour cells and prevented the spontaneous metastasis of breast cancer cells to the lungs and to the lymph nodes of mice20. In murine models of sarcoma, abrogating HIF1-dependent PLOD2 expression disrupted collagen modification, cell migration, and pulmonary metastasis67. Taken together, the studies described above indicate that hypoxia might regulate ECM deposition by multiple cell types within the tumour microenvironment20,21,67,68,82,83,86. In addition to the marked effects of collagen prolyl and lysyl hydroxylase expression in experimental mouse models of metastasis, P4HA1, P4HA2 and PLOD2 expression have also been suggested as biomarkers for human cancer progression in several independent studies (BOX 2).
Box 2. Collagen hydroxylases and cancer.
Procollagen-lysine 2-oxyglutarate 5-dioxygenase 2 (PLOD2) was included among the genes shown to be upregulated in gene expression screens of cervical cancer188, glioblastoma189 and gastric cancer190, and was 1 of 17 genes that predicted breast cancer metastasis to the brain191. Gene expression studies also revealed increased PLOD2 mRNA expression in primary sarcoma samples from patients with metastatic compared to non-metastatic sarcomas67. Moreover, human osteosarcoma samples have two to three times more hydroxylysine content than normal bone collagen, which indicates that PLOD activity is increased in these patients44. Increased prolyl 4-hydroxylase α-subunit isoform 1 (P4HA1) expression was revealed by a meta-analysis that was used to identify genes that are upregulated across many different cancer types192. P4HA2 was determined to be a metastasis-associated protein in oral cavity squamous cell carcinoma using comparative tissue proteomics193. Increased P4HA2 expression levels also discriminated papillary thyroid cancer from normal thyroid tissue194. Increased P4HA1, P4HA2 or PLOD2 mRNA expression is predictive of breast cancer patient survival; the predictive power is improved when the expression of all three genes is evaluated and determined to be greater than the median expression level20,21.
HIFs regulate extracellular collagen-modifying enzymes
Following enzymatic modification of type I collagen by hydroxylation, two α1(I)-chains and one α2(I)-chain associate to form a triple helix that is secreted into the extracellular space (FIG. 1). Collagen peptidases cleave the carboxy- and amino-terminal peptides, and type I collagen fibrils form spontaneously, are covalently crosslinked on hydroxylysine and lysine residues and form structurally stable collagen I fibres76,87. Fibrillar collagens, such as type I collagen, establish the interstitial matrix and contribute to tissue stiffness with extensive post-translational modifications that increase tensile strength54. Non-fibrillar collagens, such as type IV collagen, constitute a key component of the basement membrane, which is a compact sheet-like structure that functions as a barrier to separate tumour cells from the adjacent stroma88.
Collagen crosslinking is extracellularly initiated by the lysyl oxidase (LOX) family of secreted enzymes that oxidatively deaminate lysine or hydroxylysine collagen residues87. Three LOX enzymes — LOX, LOX-like protein 2 (LOXL2) and LOXL4 — are important hypoxia-induced and HIF-regulated target gene products that are involved in collagen crosslinking and tumour fibrosis65,89–93. In addition to collagen crosslinking within the primary tumour, secreted LOX has been shown to localize within the lungs and to remodel existing collagen to establish a premetastatic niche containing bone marrow-derived cells (BMDCs), which facilitates colonization of the niche by cancer cells in murine models of breast cancer90–91. LOX family members are upregulated to varying levels and in different combinations in human breast cancers93. Similarly, breast cancer cell lines show different patterns of LOX family member expression in response to hypoxia, but in each case the expression is HIF dependent89. Consideration of the specific LOX family members that are induced by hypoxia is therefore essential to prevent collagen remodelling, BMDC recruitment and metastasis in the lungs of tumour-bearing mice89,90. The pharmacological inhibition of LOX by β-aminopropionitrile (βAPN) has been reported to inhibit metastasis in experimental mouse models; however, βAPN might not inhibit the activity of all LOX family members93, which suggests that HIFs or pan-LOX inhibitors could represent broader targets than currently available drugs or antibodies that target only a subset of LOX and LOXL proteins.
HIF1 and HIF2 can regulate ECM degradation
In addition to collagen deposition, collagen degradation also contributes to ECM remodelling and is mediated by several families of proteinases that have been suggested to promote cancer cell invasion; for example, the matrix metalloproteinases (MMPs) are a family of zinc-dependent enzymes that are divided into several subgroups (collagenases, gelatinases, stromelysins and cell membrane-bound MMPs) with different substrate specificities. Hypoxia is associated with an increase in the expression and the activity of type IV collagen-degrading enzymes (MMP2 and MMP9) in vitro94–96. MMP2 and MMP9 are upregulated by hypoxia in breast and colon cancer cells via a HIF1-dependent mechanism94–96, whereas membrane-bound membrane-type 1 MMP (MT1-MMP; also known as MMP14) is upregulated in a HIF2-dependent manner94,97. In addition to collagen degradation by MMPs, hypoxic cancer cells also show increased proteolytic activity as a result of HIF-dependent increases in their expression of urokinase plasminogen activator surface receptor98,99 (PLAUR). PLAUR promotes cell invasion by altering the interactions between integrins and the ECM. When PLAUR expression levels are depleted by the expression of shRNAs, cells with reduced levels of PLAUR are incapable of intravasation100. Thus, HIFs activate a transcriptional programme that results in the degradation of the basement membrane while simultaneously increasing the de novo synthesis of fibrillar collagens to function as a physical pathway for tumour invasion (FIG. 2).
Figure 2. Hypoxia promotes ECM remodelling to facilitate metastasis.

Extracellular matrix (ECM) remodelling is tightly controlled to maintain tissue integrity. Cancer cells and associated stromal cells that have been exposed to hypoxia are transcriptionally reprogrammed to produce: matrix metalloproteinases (MMPs) and other proteases, which degrade the basement membrane surrounding a tumour (part a); aligned collagen fibres within the interstitial matrix, which function as a highway for local invasion, intravasation and metastasis (part b); and growth factors, which might be retained in the fibrotic microenvironment and function as chemotactic signals that recruit and activate stromal cells to further promote cancer progression (part c).
Growth factors and ECM deposition
Tumours have long been described as ‘wounds that won’t heal’ (REF. 101). Similarly, hypoxia is known to have a role in both normal and pathological wound healing. In normal cutaneous wounds, HIF1 is important for appropriate angiogenic responses, for mobilization of circulating angiogenic cells, such as endothelial precursor cells and mesenchymal stem cells (MSCs), and for normal wound contraction102. Partial reduction of HIF1α expression is consequently sufficient to impair wound healing103. During wound healing, angiogenesis and ECM deposition occur in parallel104; therefore, it is not surprising that some of the same factors that stimulate angiogenesis also promote fibrosis. Hypoxia-induced angiogenic growth factor production has been well established105. HIF1 has been shown to bind to a cis-acting hypoxia-response element in the genes that encode vascular endothelial growth factor (VEGF), stromal cell-derived factor 1 (SDF1; also known as CXCL12), angiopoietin 2 (ANG2), platelet-derived growth factor B (PDGFB), placental growth factor (PGF), connective tissue growth factor (CTGF) and stem cell factor (SCF)106–114 and can also indirectly promote fibroblast growth factor 2 (FGF2)115 production in a variety of cell types (TABLE 1).
Table 1.
Factors induced by HIFs and their role in fibrosis
| Factor induced by HIFs | Role in fibrosis |
|---|---|
| latelet-derived growth factor | Stimulates the replication, the survival and the migration of myofibroblasts118 |
| Connective tissue growth factor | Promotes collagen deposition by myofibroblasts195 |
| Fibroblast growth factor 2 | Promotes the proliferation and the differentiation of endothelial cells, smooth muscle cells and fibroblasts, and stimulates collagen deposition196 |
| Endothelin | Promotes fibroblast activation, proliferation and differentiation into myofibroblasts197 |
| Angiotensin | Stimulates TGFβ production198 and promotes collagen I and collagen III deposition199 |
| Insulin growth factor 2 | Increases connective tissue growth factor-stimulated collagen deposition200 |
| CXC-chemokine ligand 2 | Promotes fibrocyte recruitment119 |
HIFs, hypoxia-inducible factors; TGFβ, transforming growth factor-β.
Although well-known for their influence on tumour angiogenesis, many of these growth factors also contribute to fibrosis116– 118, potentially by attracting fibroblasts to the primary tumour and/or by activating resident fibroblasts. Experimental evidence indicates that the recruitment of fibroblasts or myofibroblasts to sites of pathological fibrosis is driven by hypoxia119–121. Similarly, hypoxia increases the recruitment of bone marrow-derived MSCs in murine models of breast cancer, which results in increased lymphatic and vascular metastasis112,122; for example, VEGF — which is released by hypoxic cancer cells but more often by endothelial cells, fibroblasts and inflammatory cells — has been implicated in fibrosis because of its role in stromal cell activation and because it leads to the production of an ECM that is rich in fibronectin and type I collagen63. VEGF also induces microvascular permeability, which in turn mediates an influx of fibroblasts, inflammatory cells and endothelial cells to the primary tumour123.
Hypoxia and macrophage recruitment
Hypoxia-induced growth factor secretion in the primary tumour also promotes the accumulation of macrophages, which rapidly respond to the hypoxic microenvironment by altering their gene expression patterns124–126. The importance of hypoxia in stimulating macrophage infiltration during wound healing has been shown in heterozygous HIF1α-deficient mice, which show considerable delays in myeloid cell infiltration127. Recent studies highlight a potential mechanism of macrophage recruitment into hypoxic regions involving the release of semaphorin 3A by hypoxic cancer cells, which functions as an attractant for macrophages that express neuropilin 1 (NRP1)128. Once in the region of hypoxia, macrophages stimulate fibrosis by producing growth factors such as TGFα, TGFβ1, VEGF, FGF, PDGF, tumour necrosis factor-α (TNFα), interleukin-1 (IL-1) and IL-8, which can attract additional macrophages and mesenchymal cells, such as fibroblasts and endothelial cells, and can further activate stromal cells129. Macrophages also directly promote the process of cancer cell intravasation into nearby blood vessels130. In addition, they contribute to ECM turnover by secreting MMPs, which suggests that the identification of a specific macrophage subpopulation and/or soluble mediator that preferentially promotes or degrades the ECM might be an important determinant of the extent of fibrosis within a tumour129. Taken together, the studies described above suggest that hypoxic signalling engages multiple cell types that contribute to ECM remodelling within the tumour microenvironment (FIG. 3).
Figure 3. Hypoxia recruits and reprogrammes cells to produce fibrillar collagen.
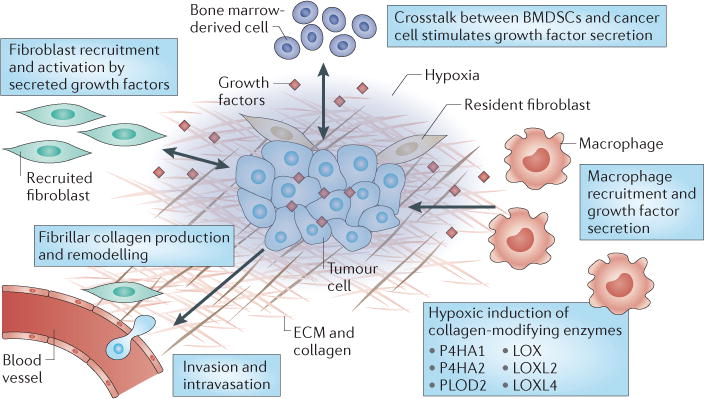
Hypoxia-induced and hypoxia-inducible factor (HIF)-regulated growth factor secretion by tumour cells promotes the recruitment of macrophages and fibroblasts to hypoxic regions of the primary tumour. Macrophages produce growth factors such as transforming growth factor β1 (TGFβ1) and platelet-derived growth factor (PDGF) that activate recruited and resident fibroblasts to stimulate collagen deposition. Hypoxic cancer cells also signal to mesenchymal stem cells, which might participate in collagen deposition. HIFs regulate the production of collagen-modifying enzymes, including prolyl 4-hydroxylase α-subunit isoform 1 (P4HA1), P4HA2, procollagen-lysine 2-oxyglutarate 5-dioxygenase 2 (PLOD2), lysyl oxidase (LOX), LOX-like protein 2 (LOXL2) and LOXL4 to facilitate the proper maturation of collagen fibres. Together, these signalling pathways promote the production of a fibrillar collagen network (that is produced by multiple cell types), which increases the ability of cancer cells to invade blood vessels. BMDSCs, bone marrow-derived stem cells; ECM, extracellular matrix.
Physical properties of tumour ECM
The physical properties of the tumour ECM refer to its stiffness, topography, porosity and solubility131. The physical properties of the tumour-associated ECM are not only fundamentally different from the ECM of normal tissues but are also continuously remodelled17,132, which reflects the dynamic changes that occur in the tumour microenvironment, including changes in oxygen availability.
Tumour Stiffness
Tumour stroma is typically stiffer than normal stroma; for example, breast tumours can be ten times stiffer than normal breast tissue57,133 and expression of collagen-modifying enzymes, such as P4HA1, P4HA2, PLOD2 and LOX, that can be induced by hypoxia promote tumour stiffness20,21,58,65,66,82. Stiffening of the ECM causes a reciprocal increase in the traction forces that are exerted by a cell134,135. Intracellular contraction in response to ECM stiffening results in an increase in the stiffness of the actin cytoskeleton and an increase in cell migration57,58,136–138. Increased tumour stiffness might regulate tumour progression in several ways; for example, increasing matrix stiffness increases RHO-generated cytoskeletal tension to promote focal adhesion assembly and to increase growth factor-dependent ERK activation123,135. Moreover, matrix stiffness facilitates integrin clustering, leading to the activation of focal adhesion kinase 1 (FAK1), which in turn activates the MAP/ERK kinase (MEK; also known as MAP2K)–ERK pathway and leads to increased cell survival, migration, invasion and proliferation57,138,139. Depletion of FAK1 in mouse tumour models inhibits local invasion and metastasis, which indicates that FAK1 activation might be an important mediator of stiffness-induced tumour metastasis140–142. Interestingly, matrix stiffening can lead to a feed-forward signalling mechanism that further increases matrix stiffening; for example, YAP1 is required for matrix stiffening by cancer-associated fibroblasts during tumour progression143. Conversely, stiff matrices and the contractile actin cytoskeleton further increase YAP1 activation143.
Tumour topography
In addition to the changes in matrix stiffness that occur during tumour progression, the topography of the ECM is also highly dynamic. For example, invasive breast cancers often contain type I collagen fibres that are oriented perpendicular to the tumour margin at the invasive front, in contrast to the non-oriented fibrils that are often seen in less aggressive breast cancers25,26,144,145. Straightened and aligned collagen fibres are found at sites of breast cancer invasion — a histological pattern that is termed tumour-associated collagen signature 3 (REF. 146), which is associated with decreased patient survival25. Similarly to breast cancer, in early melanomas, collagen is localized to the periphery of the tumour144,145,147. By contrast, metastatic melanomas have a less compact ECM structure with no barrier between the cancer cells and the adjacent normal tissue. Highly aligned collagen fibres within a tumour might not just be predictive of the metastatic potency of the tumour but may also be causative given the finding that cancer cells preferentially invade along straightened and aligned collagen bundles24,26,148.
The ECM isolated from wild-type fibroblasts that have been exposed to hypoxia is more highly aligned than the ECM deposited by fibroblasts that have been cultured under ambient conditions, and collagen fibre alignment under hypoxic conditions is abrogated in fibroblasts transfected with shRNA against HIF1α82. Breast cancer cells that have been plated on ECM produced by hypoxic cells are highly aligned and migrate with directional persistence along ECM fibres, in contrast to cells that have been plated on ECM produced by non-hypoxic cells, which migrate in a random manner21. Similarly, in an orthotopic mouse model of breast cancer, aligned ECM fibres are present in the perinecrotic (hypoxic) region of control tumours; by contrast, tumours that are derived from breast cancer cells expressing shRNA against HIF1α have a disorganized ECM comprised of almost no fibrillar collagen21.
One potential mechanism of collagen alignment in hypoxia might involve the activity of the small GTPase RHOA, which interacts with RHO-associated protein kinase 1 (ROCK1) to mediate myosin II phosphorylation, resulting in cell contraction. RHOA-mediated ROCK1 activity is required for caveolin 1-induced cell contraction, which enables cancer cells to align with and potentially to migrate along the pre-existing collagen matrix in vitro149. Experiments in vivo also indicate that caveolin 1-dependent regulation of RHOA is required for fibroblasts to produce an aligned matrix149. In renal clear cell carcinoma, caveolin 1 is a direct transcriptional target of HIF1 and HIF2 (REF. 150). Moreover, hypoxia coordinately regulates the expression of RHOA and ROCK1 through HIF1- and HIF2-dependent transcription in breast cancer cells, which results in increased cell-induced matrix contraction151. An alternative or an additional mechanism of collagen alignment could involve LOX expression. Second harmonic generation (SHG) imaging of mammary glands that have been preconditioned with LOX-expressing fibroblasts shows that they contain more linearized collagen than the mammary glands of control mice58. Taken together, these data suggest a model in which hypoxic cells can generate and organize an aligned ECM through multiple mechanisms (FIG. 2).
Biochemical properties of the ECM
The structure of the ECM influences the stability and the bioavailability of growth factors and cytokines14, many of which are generated under hypoxic conditions (TABLE 1). The balance between ECM-mediated confinement or distribution of growth factors and their concentration will determine their availability to cell surface receptors and consequently will regulate intracellular signalling14. A highly aligned ECM might more readily establish a chemoattractive gradient that potentiates hypoxic signalling. Conversely, a dense collagen network could function as a sink for growth factors and thereby could reduce their rate of diffusion. Future studies to determine the ECM arrangement and composition that supports the optimal distribution of growth factors to mediate metastasis might lead to a better understanding of how the ECM influences cancer cell motility and dissemination.
Potential therapeutic interventions
HIF inhibitors
Increased expression of HIF1α and HIF2α has been observed in a broad range of human cancers and has been associated (in most but not all cases) with a poor prognosis152–154, which suggests that use of HIF inhibitors has the potential to improve patient survival not only by blocking ECM deposition but also by blocking dozens of other HIF target genes that encode proteins involved in cell survival, angiogenesis, metabolic reprogramming, immortalization, epithelial-to-mesenchymal transition (EMT), stem cell maintenance, resistance to radiation and chemotherapy, invasion and metastasis155. Although considerable work has been done to characterize the role of HIFs in experimental cancers with regards to tumour incidence and growth152, the direct requirement for HIFs in metastasis has only recently been shown in both orthotopic models and autochthonous breast tumour models156,157. Conditional knockout models of HIFs have also aided our understanding of how the hypoxic tumour environment affects different cell types to drive cancer progression; for example, loss of either HIF1α or HIF2α in mouse vascular endothelial cells has been shown to reduce tumour growth because of impaired angiogenesis158,159. Conversely, haplodeficiency of prolyl hydroxylase domain-containing protein 2 (PHD2; also known as EGLN1) increases the HIF-driven upregulation of expression of VEGF receptor 1 (VEGFR1) in endothelial cells and decreases intratumoural hypoxia, resulting in decreased HIF1α expression in cancer cells, which reduces pulmonary metastasis160,161. The studies described above suggest that clinical trials are warranted for HIF inhibitors that show efficacy in preclinical models. It will also be crucial to determine how the activity of each HIFα subunit is affected by the potential inhibitor, given the reported functional differences between HIF1α and HIF2α162,163.
Two inhibitors of HIF1α accumulation that have shown anticancer effects in preclinical models are the topoisomerase I inhibitor topotecan151 and the cardiac glycoside digoxin90,106,157. In addition to reducing the expression of many HIF target genes, treatment of tumour-bearing mice with digoxin reduces tumour fibrosis, as well as lymph node and lung metastasis90,106,157. A Phase II clinical trial for digoxin is currently being carried out for men with recurrent prostate cancer (ClinicalTrials.gov, number: NCT01162135). In addition, a pilot clinical trial of topotecan (ClinicalTrials.gov, number: NCT00182676) in patients with advanced cancer and HIF1α overexpression shown on tumour biopsy was recently reported in which HIF1α protein levels were undetectable in the post-treatment biopsy samples from four of seven patients who were studied, and decreased tumour blood flow was observed in 70% of patients by contrast-enhanced dynamic magnetic resonance imaging164. Neither trial assessed treatment-induced changes to the tumour ECM. However, preclinical use of the HIF1α inhibitor PX-478 or overexpression of a dominant-negative HIF1α mutant showed that the increased fibrotic response identified in fat pads from mice that were fed a high-fat diet could be effectively prevented by treatment with PX-478. The preclinical effectiveness of PX-478 has previously been established in tumour models where treatment reduces tumour growth165 and it will be interesting to determine the effect of PX-478 on tumour fibrosis.
Targeting fibrosis
Blocking collagen hydroxylases or lysyl hydroxylases might also provide a strategy to reduce tumour fibrosis. P4Hs have been regarded as attractive targets for the pharmacological inhibition of collagen accumulation in fibrotic diseases and severe scarring. P4Hs belong to a superfamily of dioxygenases that use oxygen and α-ketoglutarate (also known as 2-oxoglutarate) as substrates. P4Hs are competitively inhibited by α-ketoglutarate analogues, including N-oxalylglycine, pyridine 2,4-dicarboxylate and pyridine 2,5-dicarboxylate, coumalic acid and 3,4-ethyl dihydroxybenzoate (EDHB)166. As these agents are not selective for collagen hydroxylases, it is probable that they will also inhibit the HIF PHDs and will potentially promote HIF expression. Preclinical testing will have to be carried out to determine their potential usefulness in preventing metastasis. Minoxidil has been shown to decrease the expression of PLOD mRNAs and the activity of PLOD proteins and thereby to inhibit fibrosis167. In a mouse model of sarcoma, minoxidil treatment reduced tumour fibrosis and suppressed lung metastasis67.
Studies that target LOX family members have focused on blocking the enzymatic activity of these proteins using competitive inhibitors such as βAPN168 or using neutralizing antibodies, which abrogate lung and liver metastases in xenograft and transgenic mouse models93. D-penicillamine (DPEN), which is a LOXL2 inhibitor, was developed and used to treat rheumatoid arthritis and biliary cirrhosis but it does have some unintended side effects169. A more selective inhibitory monoclonal antibody (AB0023) against LOXL2 has been developed and was effective in reducing fibrosis in primary and metastatic xenografts as well as in liver and lung fibrosis models in mice170. The ECM of tumours from mice that had been treated with AB0023 showed a marked reduction in crosslinked collagen compared with results in mice that had been treated with the lysyl oxidase inhibitor βAPN170. Treatment with AB0023 also resulted in a marked reduction in the number of activated fibroblasts and endothelial cells and led to a decreased production of growth factors and cytokines170. The safety of the humanized version of AB0023, AB0024 (also known as simtuzumab) has been tested in Phase I dose escalation trials in patients with advanced solid tumours171 (ClinicalTrials.gov, number: NCT01323933) and with idiopathic pulmonary fibrosis (ClinicalTrials.gov, number: NCT01362231). Enrolment for a Phase II clinical trial in patients with idiopathic pulmonary fibrosis (ClinicalTrials.gov, number: NCT01759511) has begun. Additional non-selective inhibitors of lysyl oxidases also include p-halobenzylamines, ethylenediamine and homocysteine thiolactone93,172.
Conclusions
Although tumours from two different patients might have similar genetic alterations, these tumours will develop in different microenvironmental contexts13, which suggests that hypoxia and the ECM are important in contributing to tumour heterogeneity, which might influence metastatic outcome. Hypoxic regions within the tumour microenvironment can simultaneously relay signals to cancer cells and cells that have been recruited to the local environment directly (for example, by transcriptional reprogramming), through paracrine signalling events and, as highlighted in this Opinion article, by establishing a hypoxia-induced ECM that is fibrotic, stiff and aligned, which are all properties that promote metastatic dissemination11,173. Further studies are needed to investigate the mechanisms by which the hypoxia-induced ECM might have a role in dynamically maintaining and distributing growth factors that provide chemotactic signals to recruit cells to the primary tumour and that promote the intravasation of cancer cells for dissemination to distant organs. Advances in imaging techniques, such as intravital microscopy, have the potential to shed light on this issue and might direct our research to appropriate targeting strategies that will be most beneficial to prevent metastasis174.
It is also important to consider that the collagen-modifying enzymes discussed in this Opinion article might have alternative roles in cancer progression that are not limited to fibrosis93. For example, LOX has a role in PDGF and insulin growth factor 1 (IGF1) signalling, but its precise mechanisms of action remain to be elucidated175. Additional regulators of the collagen hydroxylases and lysyl oxidases remain to be determined; for example, TGFβ1 has been shown to influence LOX expression93. Furthermore, the role of collagen in the regulation of ECM composition and assembly (and vice versa) is also unknown; for example, collagen I-containing fibrils do not form in the absence of fibronectin in vivo and fibronectin fibril assembly has a reciprocal requirement for collagen176. Whether fibronectin–procollagen interactions are established before the molecules are secreted is unknown and suggests that the complex regulation and dynamics of the ECM need to be carefully investigated in order to design strategies that target the ECM. Another important consideration will be the receptors that interact with the ECM molecules. A recent study regarding the fibrillar collagen receptor discoidin domain receptor 2 (DDR2) has shown that DDR2 is required for breast cancer cell invasion and migration in vitro and for metastasis in vivo by promoting the stabilization of SNAIL1 (REF. 177).
Future preclinical studies are warranted to identify new inhibitors and/or to identify optimal combinations of existing inhibitors that can block hypoxic changes to the ECM while maintaining the integrity of the ECM in healthy tissues. One major obstacle in the field of cancer therapeutics for metastasis is the definition of success. Many agents effectively target tumour growth but fail to prevent metastasis, which is the major cause of cancer mortality. For metastasis inhibitors to be tested in early phase clinical trials patients that do not already have metastatic disease will have to be included in order to have meaningful end points and to establish efficacy in metastasis prevention.
Acknowledgments
D.M.G. is supported by funding from the National Cancer Institute (NCI) (K99CA181352) and is a Susan G. Komen postdoctoral fellow. Cancer research in the Wirtz laboratory is supported by the US National Insititutes of Health (grants U54-CA143868 and R01-CA174388). Cancer Research in the Semenza laboratory is supported by the American Cancer Society, NCI grant U54-CA143868, the Department of Defense Breast Cancer Research Program and the Johns Hopkins Institute for Cell Engineering. G.L.S. is the C. Michael Armstrong Professor at the Johns Hopkins University School of Medicine, USA, and an American Cancer Society Research Professor. D.W. is the T.H. Smoot Professor and Vice Provost for Research at Johns Hopkins University.
Footnotes
Competing interests statement
The authors declare no competing interests.
Contributor Information
Daniele M. Gilkes, Vascular Program, Institute for Cell Engineering, and McKusick-Nathans, Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA Johns Hopkins Physical Sciences-Oncology Center, The Johns Hopkins University, Baltimore, Maryland 21218, USA.
Gregg L. Semenza, Vascular Program, Institute for Cell Engineering, and McKusick-Nathans, Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA Johns Hopkins Physical Sciences-Oncology Center, The Johns Hopkins University, Baltimore, Maryland 21218, USA; Departments of Pediatrics, Oncology, Medicine, Radiation Oncology and Biological Chemistry, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA.
Denis Wirtz, Johns Hopkins Physical Sciences-Oncology Center, The Johns Hopkins University, Baltimore, Maryland 21218, USA; Department of Chemical and Biomolecular Engineering, The Johns Hopkins University, Baltimore, Maryland 21218, USA; Departments of Oncology and Pathology and Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA.
References
- 1.Vaupel P, Mayer A, Hockel M. Tumour hypoxia and malignant progression. Methods Enzymol. 2004;381:335–354. doi: 10.1016/S0076-6879(04)81023-1. [DOI] [PubMed] [Google Scholar]
- 2.Sorg BS, Hardee ME, Agarwal N, Moeller BJ, Dewhirst MW. Spectral imaging facilitates visualization and measurements of unstable and abnormal microvascular oxygen transport in tumours. J Biomed Opt. 2008;13:014026. doi: 10.1117/1.2837439. [DOI] [PubMed] [Google Scholar]
- 3.Vaupel P. Prognostic potential of the pre-therapeutic tumour oxygenation status. Adv Exp Med Biol. 2009;645:241–246. doi: 10.1007/978-0-387-85998-9_36. [DOI] [PubMed] [Google Scholar]
- 4.Vaupel P, Hockel M, Mayer A. Detection and characterization of tumour hypoxia using pO2 histography. Antioxid Redox Signal. 2007;9:1221–1235. doi: 10.1089/ars.2007.1628. [DOI] [PubMed] [Google Scholar]
- 5.Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014;9:47–71. doi: 10.1146/annurev-pathol-012513-104720. [DOI] [PubMed] [Google Scholar]
- 6.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nature Rev Cancer. 2008;8:967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumour microenvironment in metastatic disease. Cancer Metastasis Rev. 2010;29:285–293. doi: 10.1007/s10555-010-9224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semenza GL. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol Med. 2012;18:534–543. doi: 10.1016/j.molmed.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207–214. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semenza GL. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene. 2013;32:4057–4063. doi: 10.1038/onc.2012.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casazza A, et al. Tumour stroma: a complexity dictated by the hypoxic tumour microenvironment. Oncogene. 2014;33:1743–1754. doi: 10.1038/onc.2013.121. [DOI] [PubMed] [Google Scholar]
- 13.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 2002;70:537–546. doi: 10.1046/j.1432-0436.2002.700907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cretu A, Brooks PC. Impact of the non-cellular tumour microenvironment on metastasis: potential therapeutic and imaging opportunities. J Cell Physiol. 2007;213:391–402. doi: 10.1002/jcp.21222. [DOI] [PubMed] [Google Scholar]
- 17.van Kempen LC, Ruiter DJ, van Muijen GN, Coussens LM. The tumour microenvironment: a critical determinant of neoplastic evolution. Eur J Cell Biol. 2003;82:539–548. doi: 10.1078/0171-9335-00346. [DOI] [PubMed] [Google Scholar]
- 18.Lochter A, Bissell MJ. Involvement of extracellular matrix constituents in breast cancer. Semin Cancer Biol. 1995;6:165–173. doi: 10.1006/scbi.1995.0017. [DOI] [PubMed] [Google Scholar]
- 19.Naba A, et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumour extracellular matrices. Mol Cell Proteomics. 2012;11:M111.014647. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilkes DM, et al. Procollagen lysyl hydroxylase 2 is essential for hypoxia-induced breast cancer metastasis. Mol Cancer Res. 2013;11:456–466. doi: 10.1158/1541-7786.MCR-12-0629. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Gilkes DM, et al. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res. 2013;73:3285–3296. doi: 10.1158/0008-5472.CAN-12-3963. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Hielscher AC, Qiu C, Gerecht S. Breast cancer cell-derived matrix supports vascular morphogenesis. Am J Physiol Cell Physiol. 2012;302:C1243–C1256. doi: 10.1152/ajpcell.00011.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langness U, Udenfriend S. Collagen biosynthesis in nonfibroblastic cell lines. Proc Natl Acad Sci USA. 1974;71:50–51. doi: 10.1073/pnas.71.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wyckoff JB, et al. Direct visualization of macrophage-assisted tumour cell intravasation in mammary tumours. Cancer Res. 2007;67:2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 25.Conklin MW, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Provenzano PP, et al. Collagen reorganization at the tumour-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys J. 2008;95:5374–5384. doi: 10.1529/biophysj.108.133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumours. Nature Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 30.Eckhardt BL, et al. Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol Cancer Res. 2005;3:1–13. [PubMed] [Google Scholar]
- 31.Allinen M, et al. Molecular characterization of the tumour microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 32.Finak G, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nature Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 33.Qiu TH, et al. Global expression profiling identifies signatures of tumour virulence in MMTV-PyMT-transgenic mice: Correlation to human disease. Cancer Res. 2004;64:5973–5981. doi: 10.1158/0008-5472.CAN-04-0242. [DOI] [PubMed] [Google Scholar]
- 34.Ma XJ, Dahiya S, Richardson E, Erlander M, Sgroi DC. Gene expression profiling of the tumour microenvironment during breast cancer progression. Breast Cancer Res. 2009;11:R7. doi: 10.1186/bcr2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang HY, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci USA. 2005;102:3738–3743. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang HY, et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumours and wounds. PLoS Biol. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Artinian V, Kvale PA. Cancer and interstitial lung disease. Curr Opin Pulm Med. 2004;10:425–434. doi: 10.1097/00063198-200409000-00017. [DOI] [PubMed] [Google Scholar]
- 38.Bartow SA, Pathak DR, Mettler FA. Radiographic microcalcification and parenchymal patterns as indicators of histologic ‘high-risk’ benign breast disease. Cancer. 1990;66:1721–1725. doi: 10.1002/1097-0142(19901015)66:8<1721::aid-cncr2820660812>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 39.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bissell DM. Chronic liver injury, TGFβ, and cancer. Exp Mol Med. 2001;33:179–190. doi: 10.1038/emm.2001.31. [DOI] [PubMed] [Google Scholar]
- 41.Boyd NF, Jensen HM, Cooke G, Han HL. Relationship between mammographic and histological risk factors for breast cancer. J Natl Cancer Inst. 1992;84:1170–1179. doi: 10.1093/jnci/84.15.1170. [DOI] [PubMed] [Google Scholar]
- 42.Boyd NF, et al. Mammographic densities and the prevalence and incidence of histological types of benign breast disease. Reference Pathologists of the Canadian National Breast Screening Study. Eur J Cancer Prev. 2000;9:15–24. doi: 10.1097/00008469-200002000-00003. [DOI] [PubMed] [Google Scholar]
- 43.Boyd NF, et al. Heritability of mammographic density, a risk factor for breast cancer. N Engl J Med. 2002;347:886–894. doi: 10.1056/NEJMoa013390. [DOI] [PubMed] [Google Scholar]
- 44.Shapiro FD, Eyre DR. Collagen polymorphism in extracellular matrix of human osteosarcoma. J Natl Cancer Inst. 1982;69:1009–1016. [PubMed] [Google Scholar]
- 45.Coussens LM, et al. Inflammatory mast cells upregulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jussila T, Kauppila S, Risteli L, Risteli J, Stenback F. Collagen formation in extracellular matrix of transplants of human transformed keratinocyte cell lines. Anticancer Res. 2002;22:1705–1711. [PubMed] [Google Scholar]
- 47.Gould VE, Koukoulis GK, Virtanen I. Extracellular matrix proteins and their receptors in the normal, hyperplastic and neoplastic breast. Cell Differ Dev. 1990;32:409–416. doi: 10.1016/0922-3371(90)90057-4. [DOI] [PubMed] [Google Scholar]
- 48.Zhu GG, et al. Immunohistochemical study of type I collagen and type I pN-collagen in benign and malignant ovarian neoplasms. Cancer. 1995;75:1010–1017. doi: 10.1002/1097-0142(19950215)75:4<1010::aid-cncr2820750417>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 49.Kauppila S, Stenback F, Risteli J, Jukkola A, Risteli L. Aberrant type I and type III collagen gene expression in human breast cancer in vivo. J Pathol. 1998;186:262–268. doi: 10.1002/(SICI)1096-9896(1998110)186:3<262::AID-PATH191>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 50.Huijbers IJ, et al. A role for fibrillar collagen deposition and the collagen internalization receptor endo180 in glioma invasion. PLoS ONE. 2010;5:e9808. doi: 10.1371/journal.pone.0009808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colpaert CG, et al. The presence of a fibrotic focus in invasive breast carcinoma correlates with the expression of carbonic anhydrase IX and is a marker of hypoxia and poor prognosis. Breast Cancer Res Treat. 2003;81:137–147. doi: 10.1023/A:1025702330207. [DOI] [PubMed] [Google Scholar]
- 52.Trastour C, et al. HIF1α and CA IX staining in invasive breast carcinomas: prognosis and treatment outcome. Int J Cancer. 2007;120:1451–1458. doi: 10.1002/ijc.22436. [DOI] [PubMed] [Google Scholar]
- 53.Hasebe T, Tsuda H, Tsubono Y, Imoto S, Mukai K. Fibrotic focus in invasive ductal carcinoma of the breast: a histopathological prognostic parameter for tumour recurrence and tumour death within three years after the initial operation. Jpn J Cancer Res. 1997;88:590–599. doi: 10.1111/j.1349-7006.1997.tb00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van der Rest M, Garrone R. Collagen family of proteins. FASEB J. 1991;5:2814–2823. [PubMed] [Google Scholar]
- 55.Fraley SI, et al. A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nature Cell Biol. 2010;12:598–604. doi: 10.1038/ncb2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fraley SI, Feng Y, Giri A, Longmore GD, Wirtz D. Dimensional and temporal controls of three-dimensional cell migration by zyxin and binding partners. Nature Commun. 2012;3:719. doi: 10.1038/ncomms1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paszek MJ, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 58.Levental KR, et al. Matrix crosslinking forces tumour progression by enhancing integrin signalling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X, et al. A targeted mutation at the known collagenase cleavage site in mouse type I collagen impairs tissue remodelling. J Cell Biol. 1995;130:227–237. doi: 10.1083/jcb.130.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Provenzano PP, et al. Collagen density promotes mammary tumour initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin EY, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumour microenvironment and breast cancer. Cancer Metastasis Rev. 2013;32:303–315. doi: 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 64.Shekhar MP, Pauley R, Heppner G. Host microenvironment in breast cancer development: extracellular matrix-stromal cell contribution to neoplastic phenotype of epithelial cells in the breast. Breast Cancer Res. 2003;5:130–135. doi: 10.1186/bcr580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cox TR, et al. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013;73:1721–1732. doi: 10.1158/0008-5472.CAN-12-2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Erler JT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 67.Eisinger-Mathason TS, et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 2013;3:1190–1205. doi: 10.1158/2159-8290.CD-13-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xiong G, Deng L, Zhu J, Rychahou PG, Xu R. Prolyl-4-hydroxylase α–subunit 2 promotes breast cancer progression and metastasis by regulating collagen deposition. BMC Cancer. 2014;14:1. doi: 10.1186/1471-2407-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Halberg N, et al. Hypoxia-inducible factor 1α induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009;29:4467–4483. doi: 10.1128/MCB.00192-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Higgins DF, et al. Hypoxia promotes fibrogenesis in vivo via HIF1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moon JO, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1α-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G582–G592. doi: 10.1152/ajpgi.90368.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Falanga V, et al. Low oxygen tension increases mRNA levels of α1 (I) procollagen in human dermal fibroblasts. J Cell Physiol. 1993;157:408–412. doi: 10.1002/jcp.1041570225. [DOI] [PubMed] [Google Scholar]
- 73.Tamamori M, Ito H, Hiroe M, Marumo F, Hata RI. Stimulation of collagen synthesis in rat cardiac fibroblasts by exposure to hypoxic culture conditions and suppression of the effect by natriuretic peptides. Cell Biol Int. 1997;21:175–180. doi: 10.1006/cbir.1997.0130. [DOI] [PubMed] [Google Scholar]
- 74.Norman JT, Clark IM, Garcia PL. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000;58:2351–2366. doi: 10.1046/j.1523-1755.2000.00419.x. [DOI] [PubMed] [Google Scholar]
- 75.Berg JT, Breen EC, Fu Z, Mathieu-Costello O, West JB. Alveolar hypoxia increases gene expression of extracellular matrix proteins and platelet-derived growth factor-B in lung parenchyma. Am J Respir Crit Care Med. 1998;158:1920–1928. doi: 10.1164/ajrccm.158.6.9804076. [DOI] [PubMed] [Google Scholar]
- 76.Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 77.Kivirikko KI, Pihlajaniemi T. Collagen hydroxylases and the protein disulphide isomerase subunit of prolyl 4-hydroxylases. Adv Enzymol Relat Areas Mol Biol. 1998;72:325–398. doi: 10.1002/9780470123188.ch9. [DOI] [PubMed] [Google Scholar]
- 78.Myllyharju J. Prolyl 4-hydroxylases, the key enzymes of collagen biosynthesis. Matrix Biol. 2003;22:15–24. doi: 10.1016/s0945-053x(03)00006-4. [DOI] [PubMed] [Google Scholar]
- 79.Annunen P, et al. Cloning of the human prolyl 4-hydroxylase-α subunit isoform-α(II) and characterization of the type II enzyme tetramer. The-α(I) and-α(II) subunits do not form a mixed-α(I)α(II) β2 tetramer. J Biol Chem. 1997;272:17342–17348. doi: 10.1074/jbc.272.28.17342. [DOI] [PubMed] [Google Scholar]
- 80.Kukkola L, Hieta R, Kivirikko KI, Myllyharju J. Identification and characterization of a third human, rat, and mouse collagen prolyl 4-hydroxylase isoenzyme. J Biol Chem. 2003;278:47685–47693. doi: 10.1074/jbc.M306806200. [DOI] [PubMed] [Google Scholar]
- 81.van der Slot AJ, et al. Increased formation of pyridinoline crosslinks due to higher telopeptide lysyl hydroxylase levels is a general fibrotic phenomenon. Matrix Biol. 2004;23:251–257. doi: 10.1016/j.matbio.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 82.Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 (HIF1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem. 2013;288:10819–10829. doi: 10.1074/jbc.M112.442939. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 83.Hofbauer KH, et al. Oxygen tension regulates the expression of a group of procollagen hydroxylases. Eur J Biochem. 2003;270:4515–4522. doi: 10.1046/j.1432-1033.2003.03846.x. [DOI] [PubMed] [Google Scholar]
- 84.Elvidge GP, et al. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF1α, HIF2α, and other pathways. J Biol Chem. 2006;281:15215–15226. doi: 10.1074/jbc.M511408200. [DOI] [PubMed] [Google Scholar]
- 85.Aro E, et al. Hypoxia-inducible factor 1 (HIF1) but not HIF2 is essential for hypoxic induction of collagen prolyl 4-hydroxylases in primary newborn mouse epiphyseal growth plate chondrocytes. J Biol Chem. 2012;287:37134–37144. doi: 10.1074/jbc.M112.352872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bentovim L, Amarilio R, Zelzer E. HIF1α is a central regulator of collagen hydroxylation and secretion under hypoxia during bone development. Development. 2012;139:4473–4483. doi: 10.1242/dev.083881. [DOI] [PubMed] [Google Scholar]
- 87.Gordon MK, Hahn RA. Collagens. Cell Tissue Res. 2010;339:247–257. doi: 10.1007/s00441-009-0844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tanjore H, Kalluri R. The role of type IV collagen and basement membranes in cancer progression and metastasis. Am J Pathol. 2006;168:715–717. doi: 10.2353/ajpath.2006.051321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wong CC, et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci USA. 2011;108:16369–16374. doi: 10.1073/pnas.1113483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wong CC, et al. Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J Mol Med. 2012;90:803–815. doi: 10.1007/s00109-011-0855-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Erler JT, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schietke R, et al. The lysyl oxidases LOX and LOXL2 are necessary and sufficient to repress E-cadherin in hypoxia: insights into cellular transformation processes mediated by HIF1. J Biol Chem. 2010;285:6658–6669. doi: 10.1074/jbc.M109.042424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barker HE, Cox TR, Erler JT. The rationale for targeting the LOX family in cancer. Nature Rev Cancer. 2012;12:540–552. doi: 10.1038/nrc3319. [DOI] [PubMed] [Google Scholar]
- 94.Munoz-Najar UM, Neurath KM, Vumbaca F, Claffey KP. Hypoxia stimulates breast carcinoma cell invasion through MT1-MMP and MMP2 activation. Oncogene. 2006;25:2379–2392. doi: 10.1038/sj.onc.1209273. [DOI] [PubMed] [Google Scholar]
- 95.Krishnamachary B, et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003;63:1138–1143. [PubMed] [Google Scholar]
- 96.Choi JY, Jang YS, Min SY, Song JY. Overexpression of MMP9 and HIF1α in breast cancer cells under hypoxic conditions. J Breast Cancer. 2011;14:88–95. doi: 10.4048/jbc.2011.14.2.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Petrella BL, Lohi J, Brinckerhoff CE. Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor 2α in von Hippel–Lindau renal cell carcinoma. Oncogene. 2005;24:1043–1052. doi: 10.1038/sj.onc.1208305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Graham CH, Forsdike J, Fitzgerald CJ, Macdonald-Goodfellow S. Hypoxia-mediated stimulation of carcinoma cell invasiveness via upregulation of urokinase receptor expression. Int J Cancer. 1999;80:617–623. doi: 10.1002/(sici)1097-0215(19990209)80:4<617::aid-ijc22>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 99.Buchler P, et al. Transcriptional regulation of urokinase-type plasminogen activator receptor by hypoxia-inducible factor 1 is crucial for invasion of pancreatic and liver cancer. Neoplasia. 2009;11:196–206. doi: 10.1593/neo.08734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim J, Yu W, Kovalski K, Ossowski L. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell. 1998;94:353–362. doi: 10.1016/s0092-8674(00)81478-6. [DOI] [PubMed] [Google Scholar]
- 101.Dvorak HF. Tumours: wounds that do not heal. Similarities between tumour stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 102.Lokmic Z, Musyoka J, Hewitson TD, Darby IA. In: International Review of Cell and Molecular Biology. Kwang WJ, editor. Vol. 296. Academic Press; 2012. pp. 139–185. Ch.3. [DOI] [PubMed] [Google Scholar]
- 103.Zhang X, et al. Impaired angiogenesis and mobilization of circulating angiogenic cells in HIF-1α heterozygous-null mice after burn wounding. Wound Repair Regen. 2010;18:193–201. doi: 10.1111/j.1524-475X.2010.00570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Velnar T, Bailey T, Smrkolj V. The wound healing process: an overview of the cellular and molecular mechanisms. J Int Med Res. 2009;37:1528–1542. doi: 10.1177/147323000903700531. [DOI] [PubMed] [Google Scholar]
- 105.Rey S, Semenza GL. Hypoxia-inducible factor 1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc Res. 2010;86:236–242. doi: 10.1093/cvr/cvq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schito L, et al. Hypoxia-inducible factor 1-dependent expression of platelet-derived growth factor B promotes lymphatic metastasis of hypoxic breast cancer cells. Proc Natl Acad Sci USA. 2012;109:E2707–E2716. doi: 10.1073/pnas.1214019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Manalo DJ, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 108.Kelly BD, et al. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93:1074–1081. doi: 10.1161/01.RES.0000102937.50486.1B. [DOI] [PubMed] [Google Scholar]
- 109.Forsythe JA, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ceradini DJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF1 induction of SDF1. Nature Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 111.Simon MP, Tournaire R, Pouyssegur J. The angiopoietin-2 gene of endothelial cells is upregulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA2 and ETS1. J Cell Physiol. 2008;217:809–818. doi: 10.1002/jcp.21558. [DOI] [PubMed] [Google Scholar]
- 112.Chaturvedi P, et al. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signalling promotes metastasis. J Clin Invest. 2013;123:189–205. doi: 10.1172/JCI64993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kondo S, et al. Connective tissue growth factor increased by hypoxia may initiate angiogenesis in collaboration with matrix metalloproteinases. Carcinogenesis. 2002;23:769–776. doi: 10.1093/carcin/23.5.769. [DOI] [PubMed] [Google Scholar]
- 114.Higgins DF, et al. Hypoxic induction of CTGF is directly mediated by HIF1. Am J Physiol Renal Physiol. 2004;287:F1223–F1232. doi: 10.1152/ajprenal.00245.2004. [DOI] [PubMed] [Google Scholar]
- 115.Le YJ, Corry PM. Hypoxia-induced bFGF gene expression is mediated through the JNK signal transduction pathway. Mol Cell Biochem. 1999;202:1–8. doi: 10.1023/a:1007059806016. [DOI] [PubMed] [Google Scholar]
- 116.Lappi-Blanco E, Soini Y, Kinnula V, Paakko P. VEGF and bFGF are highly expressed in intraluminal fibromyxoid lesions in bronchiolitis obliterans organizing pneumonia. J Pathol. 2002;196:220–227. doi: 10.1002/path.1038. [DOI] [PubMed] [Google Scholar]
- 117.Lasky JA, et al. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am J Physiol. 1998;275:L365–371. doi: 10.1152/ajplung.1998.275.2.L365. [DOI] [PubMed] [Google Scholar]
- 118.Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 119.Phillips RJ, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hayashida K, et al. Bone marrow-derived cells contribute to pulmonary vascular remodeling in hypoxia-induced pulmonary hypertension. Chest. 2005;127:1793–1798. doi: 10.1378/chest.127.5.1793. [DOI] [PubMed] [Google Scholar]
- 121.Mehrad B, Burdick MD, Strieter RM. Fibrocyte CXCR4 regulation as a therapeutic target in pulmonary fibrosis. Int J Biochem Cell Biol. 2009;41:1708–1718. doi: 10.1016/j.biocel.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Karnoub AE, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 123.Brown LF, et al. Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin Cancer Res. 1999;5:1041–1056. [PubMed] [Google Scholar]
- 124.Wels J, Kaplan RN, Rafii S, Lyden D. Migratory neighbours and distant invaders: tumour-associated niche cells. Genes Dev. 2008;22:559–574. doi: 10.1101/gad.1636908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumours and other ischemic tissues. Blood. 2004;104:2224–2234. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- 126.Cramer T, et al. HIF1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rezvani HR, et al. HIF1α in epidermis: oxygen sensing, cutaneous angiogenesis, cancer, and non-cancer disorders. J Invest Dermatol. 2011;131:1793–1805. doi: 10.1038/jid.2011.141. [DOI] [PubMed] [Google Scholar]
- 128.Casazza A, et al. Impeding macrophage entry into hypoxic tumour areas by SEMA3A/NRP1 signalling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24:695–709. doi: 10.1016/j.ccr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 129.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nature Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wirtz D, Konstantopoulos K, Searson PC. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nature Rev Cancer. 2011;11:512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Clarijs R, Ruiter DJ, De Waal RM. Pathophysiological implications of stroma pattern formation in uveal melanoma. J Cell Physiol. 2003;194:267–271. doi: 10.1002/jcp.10214. [DOI] [PubMed] [Google Scholar]
- 133.Lopez JI, Kang I, You WK, McDonald DM, Weaver VM. In situ force mapping of mammary gland transformation. Integr Biol. 2011;3:910–921. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kim DH, et al. Actin cap associated focal adhesions and their distinct role in cellular mechanosensing. Sci Rep. 2012;2:555. doi: 10.1038/srep00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Samuel MS, et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumour growth. Cancer Cell. 2011;19:776–791. doi: 10.1016/j.ccr.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nature Cell Biol. 2003;5:711–719. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- 138.Kim DH, Wirtz D. Focal adhesion size uniquely predicts cell migration. FASEB J. 2013;27:1351–1361. doi: 10.1096/fj.12-220160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Provenzano PP, Keely PJ. The role of focal adhesion kinase in tumour initiation and progression. Cell Adh Migr. 2009;3:347–350. doi: 10.4161/cam.3.4.9458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Provenzano PP, Inman DR, Eliceiri KW, Beggs HE, Keely PJ. Mammary epithelial-specific disruption of focal adhesion kinase retards tumour formation and metastasis in a transgenic mouse model of human breast cancer. Am J Pathol. 2008;173:1551–1565. doi: 10.2353/ajpath.2008.080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lahlou H, et al. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumour progression. Proc Natl Acad Sci USA. 2007;104:20302–20307. doi: 10.1073/pnas.0710091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Baker AM, Bird D, Lang G, Cox TR, Erler JT. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene. 2013;32:1863–1868. doi: 10.1038/onc.2012.202. [DOI] [PubMed] [Google Scholar]
- 143.Calvo F, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nature Cell Biol. 2013;15:637–646. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tamimi SO, Ahmed A. Stromal changes in early invasive and non-invasive breast carcinoma: an ultrastructural study. J Pathol. 1986;150:43–49. doi: 10.1002/path.1711500108. [DOI] [PubMed] [Google Scholar]
- 145.Tamimi SO, Ahmed A. Stromal changes in invasive breast carcinoma: an ultrastructural study. J Pathol. 1987;153:163–170. doi: 10.1002/path.1711530209. [DOI] [PubMed] [Google Scholar]
- 146.Conklin MW, Keely PJ. Why the stroma matters in breast cancer: insights into breast cancer patient outcomes through the examination of stromal biomarkers. Cell Adh Migr. 2012;6:249–260. doi: 10.4161/cam.20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ruiter D, Bogenrieder T, Elder D, Herlyn M. Melanoma–stroma interactions: structural and functional aspects. Lancet Oncol. 2002;3:35–43. doi: 10.1016/s1470-2045(01)00620-9. [DOI] [PubMed] [Google Scholar]
- 148.Wang W, et al. Single cell behaviour in metastatic primary mammary tumours correlated with gene expression patterns revealed by molecular profiling. Cancer Res. 2002;62:6278–6288. [PubMed] [Google Scholar]
- 149.Goetz JG, et al. Biomechanical remodeling of the microenvironment by stromal caveolin 1 favours tumour invasion and metastasis. Cell. 2011;146:148–163. doi: 10.1016/j.cell.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang Y, et al. Hypoxia promotes ligand-independent EGF receptor signalling via hypoxia-inducible factor-mediated upregulation of caveolin 1. Proc Natl Acad Sci USA. 2012;109:4892–4897. doi: 10.1073/pnas.1112129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gilkes DM, et al. Hypoxia-inducible factors mediate coordinated RhoA-ROCK1 expression and signalling in breast cancer cells. Proc Natl Acad Sci USA. 2014;111:E384–E393. doi: 10.1073/pnas.1321510111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 152.Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nature Rev Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Semenza GL. Targeting HIF1 for cancer therapy. Nature Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 154.Shen C, et al. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discov. 2011;1:222–235. doi: 10.1158/2159-8290.CD-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Semenza GL. HIF1 mediates metabolic responses to intratumoural hypoxia and oncogenic mutations. J Clin Invest. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liao D, Corle C, Seagroves TN, Johnson RS. Hypoxia-inducible factor 1α is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 2007;67:563–572. doi: 10.1158/0008-5472.CAN-06-2701. [DOI] [PubMed] [Google Scholar]
- 157.Zhang H, et al. HIF1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene. 2012;31:1757–1770. doi: 10.1038/onc.2011.365. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 158.Tang N, et al. Loss of HIF1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell. 2004;6:485–495. doi: 10.1016/j.ccr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 159.Yamashita T, et al. Hypoxia-inducible transcription factor 2α in endothelial cells regulates tumour neovascularization through activation of ephrin A1. J Biol Chem. 2008;283:18926–18936. doi: 10.1074/jbc.M709133200. [DOI] [PubMed] [Google Scholar]
- 160.Mazzone M, et al. Heterozygous deficiency of PHD2 restores tumour oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–851. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Leite de Oliveira R, et al. Gene-targeting of PHD2 improves tumour response to chemotherapy and prevents side-toxicity. Cancer Cell. 2012;22:263–277. doi: 10.1016/j.ccr.2012.06.028. [DOI] [PubMed] [Google Scholar]
- 162.Acker T, et al. Genetic evidence for a tumour suppressor role of HIF2α. Cancer Cell. 2005;8:131–141. doi: 10.1016/j.ccr.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 163.Mazumdar J, et al. HIF2α deletion promotes KRAS-driven lung tumour development. Proc Natl Acad Sci USA. 2010;107:14182–14187. doi: 10.1073/pnas.1001296107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kummar S, et al. Multihistology, target-driven pilot trial of oral topotecan as an inhibitor of hypoxia-inducible factor 1α in advanced solid tumours. Clin Cancer Res. 2011;17:5123–5131. doi: 10.1158/1078-0432.CCR-11-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sun K, Tordjman J, Clément K, Scherer Philipp E. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013;18:470–477. doi: 10.1016/j.cmet.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rose NR, McDonough MA, King ON, Kawamura A, Schofield CJ. Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc Rev. 2011;40:4364–4397. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- 167.Murad S, Walker LC, Tajima S, Pinnell SR. Minimum structural requirements for minoxidil inhibition of lysyl hydroxylase in cultured fibroblasts. Arch Biochem Biophys. 1994;308:42–47. doi: 10.1006/abbi.1994.1006. [DOI] [PubMed] [Google Scholar]
- 168.Tang SS, Trackman PC, Kagan HM. Reaction of aortic lysyl oxidase with β-aminopropionitrile. J Biol Chem. 1983;258:4331–4338. [PubMed] [Google Scholar]
- 169.Siegel RC. Collagen crosslinking. Effect of D-penicillamine on crosslinking in vitro. J Biol Chem. 1977;252:254–259. [PubMed] [Google Scholar]
- 170.Barry-Hamilton V, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nature Med. 2010;16:1009–1017. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]
- 171.Zardavas D, Baselga J, Piccart M. Emerging targeted agents in metastatic breast cancer. Nature Rev Clin Oncol. 2013;10:191–210. doi: 10.1038/nrclinonc.2013.29. [DOI] [PubMed] [Google Scholar]
- 172.Liu G, Nellaiappan K, Kagan HM. Irreversible inhibition of lysyl oxidase by homocysteine thiolactone and its selenium and oxygen analogues: Implications for homocystinuria. J Biol Chem. 1997;272:32370–32377. doi: 10.1074/jbc.272.51.32370. [DOI] [PubMed] [Google Scholar]
- 173.Gilkes DM, Semenza GL. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol. 2013;9:1623–1636. doi: 10.2217/fon.13.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Entenberg D, et al. Current Protocols in Cell Biology. Wiley; 2013. pp. 19.7.1–19.7.19. Ch. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Giampuzzi M, et al. Downregulation of lysyl oxidase-induced tumorigenic transformation in NRK-49F cells characterized by constitutive activation of RAS proto-oncogene. J Biol Chem. 2001;276:29226–29232. doi: 10.1074/jbc.M101695200. [DOI] [PubMed] [Google Scholar]
- 176.Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20:495–501. doi: 10.1016/j.ceb.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang K, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nature Cell Biol. 2013;15:677–687. doi: 10.1038/ncb2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix–loop–helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 180.Semenza GL, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 181.Kaelin WG, Jr, Ratcliffe P. J Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 182.Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- 183.Wiesener MS, et al. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1α. Blood. 1998;92:2260–2268. [PubMed] [Google Scholar]
- 184.Maxwell PH, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 185.Zundel W, et al. Loss of PTEN facilitates HIF1-mediated gene expression. Genes Dev. 2000;14:391–396. [PMC free article] [PubMed] [Google Scholar]
- 186.Akakura N, et al. Constitutive expression of hypoxia-inducible factor-1α renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 2001;61:6548–6554. [PubMed] [Google Scholar]
- 187.Zhong H, et al. Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumour angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- 188.Rajkumar T, et al. Identification and validation of genes involved in cervical tumorigenesis. BMC Cancer. 2011;11:80. doi: 10.1186/1471-2407-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Dong S, et al. Histology-based expression profiling yields novel prognostic markers in human glioblastoma. J Neuropathol Exp Neurol. 2005;64:948–955. doi: 10.1097/01.jnen.0000186940.14779.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Arao T, et al. ZD6474 inhibits tumour growth and intraperitoneal dissemination in a highly metastatic orthotopic gastric cancer model. Int J Cancer. 2006;118:483–489. doi: 10.1002/ijc.21340. [DOI] [PubMed] [Google Scholar]
- 191.Bos PD, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kavak E, Unlu M, Nister M, Koman A. Meta-analysis of cancer gene expression signatures reveals new cancer genes, SAGE tags and tumour associated regions of co-regulation. Nucleic Acids Res. 2010;38:7008–7021. doi: 10.1093/nar/gkq574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chang KP, et al. Identification of PRDX4 and P4HA2 as metastasis-associated proteins in oral cavity squamous cell carcinoma by comparative tissue proteomics of microdissected specimens using iTRAQ technology. J Proteome Res. 2011;10:4935–4947. doi: 10.1021/pr200311p. [DOI] [PubMed] [Google Scholar]
- 194.Jarzab B, et al. Gene expression profile of papillary thyroid cancer: sources of variability and diagnostic implications. Cancer Res. 2005;65:1587–1597. doi: 10.1158/0008-5472.CAN-04-3078. [DOI] [PubMed] [Google Scholar]
- 195.Brigstock DR. Connective tissue growth factor (CCN2, CTGF) and organ fibrosis: lessons from transgenic animals. J Cell Commun Signal. 2010;4:1–4. doi: 10.1007/s12079-009-0071-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Vlodavsky I, et al. Extracellular matrix-resident growth factors and enzymes: possible involvement in tumour metastasis and angiogenesis. Cancer Metastasis Rev. 1990;9:203–226. doi: 10.1007/BF00046361. [DOI] [PubMed] [Google Scholar]
- 197.Swigris J, Brown K. The role of endothelin 1 in the pathogenesis of idiopathic pulmonary fibrosis. BioDrugs. 2010;24:49–54. doi: 10.2165/11319550-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kagami S, Border WA, Miller DE, Noble NA. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-β expression in rat glomerular mesangial cells. J Clin Invest. 1994;93:2431–2437. doi: 10.1172/JCI117251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wang Z, et al. Hypoxia-inducible factor 1α contributes to the profibrotic action of angiotensin II in renal medullary interstitial cells. Kidney Int. 2011;79:300–310. doi: 10.1038/ki.2010.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Grotendorst GR, Rahmanie H, Duncan MR. Combinatorial signalling pathways determine fibroblast proliferation and myofibroblast differentiation. FASEB J. 2004;18:469–479. doi: 10.1096/fj.03-0699com. [DOI] [PubMed] [Google Scholar]