

Abstract
Sixteen polyketides belonging to diverse structural classes, including monomeric/dimeric tetrahydroxanthones and resorcylic acid lactones, were isolated from an organic extract of a fungal culture Setophoma terrestris (MSX45109) using bioactivity-directed fractionation as part of a search for anticancer leads from filamentous fungi. Of these, six were new: penicillixanthone B (5), blennolide H (6), 11-deoxy blennolide D (7), blennolide I (9), blennolide J (10), and pyrenomycin (16). The known compounds were: secalonic acid A (1), secalonic acid E (2), secalonic acid G (3), penicillixanthone A (4), paecilin B (8), aigialomycin A (11), hypothemycin (12), dihydrohypothemycin (13), pyrenochaetic acid C (14), and nidulalin B (15). The structures were elucidated using a set of spectroscopic and spectrometric techniques; the absolute configurations of compounds 1–10 were determined using ECD spectroscopy combined with time-dependent density functional theory (TDDFT) calculations, while a modified Mosher’s ester method was used for compound 16. The cytotoxic activities of compounds (1–15) were evaluated using the MDA-MB-435 (melanoma) and SW-620 (colon) cancer cell lines. Compounds 1, 4, and 12 were the most potent with IC50 values ranging from 0.16 to 2.14 μM. When tested against a panel of bacteria and fungi, compounds 3 and 5 showed promising activity against the Gram-positive bacterium Micrococcus luteus with MIC values of 5 and 15 μg/mL, respectively.
Keywords: Cytotoxicity, fungus, secalonic acids, ergochromes, resorcylic acid lactones
Introduction
Structurally diverse cytotoxic secondary metabolites have been isolated and identified from filamentous fungi of the Mycosynthetix library, representing over 55,000 accessions, as part of ongoing bioactivity-directed studies for discovery of anticancer drug leads.[1] The organic extract of a solid phase culture of Setophoma terrestris (MSX45109), which was isolated from plant material collected in a mangrove habitat in 1989, showed potent cytotoxic activities against the SW-620 (colon) and MDA-MB-435 (melanoma) cancer cell lines (~91% and 100% inhibition of cell growth when tested at 20 μg/mL, respectively). Of the hundreds of cultures that have been investigated as part of this project, S. terrestris was intriguing due to the robust biosynthesis of 16 polyketides (1–16) of distinct structural classes. Over one third of the isolated compounds were new, while structural revisions and augmentations to the literature were developed for several others.
Compounds 1–10 were identified as a series of monomeric/dimeric tetrahydroxanthones, an important class of mycotoxins that are produced by a variety of microorganisms with remarkable biological activities, including antitumor, antibacterial, and anti-HIV.[2] In the current study, a series of new monomeric and homo/hetero dimeric tetrahydroxanthones were identified, thereby expanding the diversity of this class of natural products and updating the literature on structural elucidation and absolute configuration considerations of structurally related compounds. These points are also critical for accurate and comprehensive dereplication studies of fungal metabolites, an area of growing prominence in the field.[1a, 3] Interestingly, a recent and comprehensive study reported a similar series of compounds albeit with opposite absolute configuration.[4]
Compounds 11–13 were characterized as a series of structurally related resorcylic acid lactones (RALs), a family of benzannulated macrolides that are produced by a variety of fungi with a wide range of biological activities, including antitumor, antifungal, antibiotic, and antiviral.[5] Compounds 14 and 16, biogenetically related to RALs, were also isolated and identified.
The absolute configurations of compounds 1–10 were determined using time-dependent density functional theory (TDDFT) calculations of ECD spectra, while for 16 a modified Mosher’s ester method was used. The dimeric tetrahydroxanthone derivatives 1 and 4, and the RAL 12, all showed potent inhibition of MDA-MB-435 and SW-620 cancer cell lines. Alternatively, compounds 3 and 5 showed promising activity against the Gram-positive bacterium Micrococcus luteus.
Results and Discussion
The organic extract of a large-scale solid-substrate fungal culture of S. terrestris (MSX45109) showed potent cytotoxic activity against the SW-620 and MDA-MB-435 cancer cell lines, and thus, it was fractionated by silica gel flash chromatography to yield five fractions. The third fraction, which eluted with 10% MeOH/CHCl3, showed potent cytotoxic activity on the two cancer cell lines, and as such, it was purified using reversed-phase preparative and semipreparative HPLC to yield polyketides 1–16. The purity of the isolated compounds was verified using UPLC (Figure S1, Supporting Information).
Monomeric/dimeric tetrahydroxanthones
Compounds 1–10 were identified as a series of monomeric (6–8), homodimeric (1–5), and heterodimeric (9 and 10) tetrahydroxanthones by comparison of HRMS, NMR, and ECD data with each other and with structurally related compounds (Figure 1). Stereoisomeric compounds 1 (35.1 mg), 2 (51.3 mg), and 3 (9.5 mg) were all obtained as yellow powders and had molecular formulae of C32H30O14 as determined by HRESIMS. The 1H and 13C NMR spectra of compounds 1 and 2 showed the presence of only 15 protons and 16 carbons, indicating that these compounds were homodimers, whereas 3 was clearly a heterodimer (Figures S2–S4). A search of the “Dictionary of Natural Products”[6] for the molecular formula and a UV range of 325–345 nm resulted in twelve hits, five of which were excluded based on NMR data. The remaining seven hits were the secalonic acids A–G, all of which contained a 2,2′-linkage. The fact that compounds 1 and 2 were homodimeric cut down the number of possibilities for compounds 1 and 2 into secalonic acids A, B, D, or E, while the heterodimeric nature of 3 suggested either secalonic acid C, F, or G. Key differences between compounds 1 and 2 were the chemical shift values and splitting patterns of H-5, H-6, and H2-7. A proton doublet of doublets corresponding to H-5/H-5′ (δH 3.92, dd, J = 11.2, 0.5 Hz) in 1 was downfield shifted in 2 (δH 4.11, d, J = 1.2 Hz). These J values implied a pseudodiaxial trans orientation of H-5/H-6 in 1 and a pseudoaxial/pseudoequatorial cis orientation in 2 (Figures S2 and S3, Table S1). On the other hand, the 1H NMR spectrum of 3 showed similarity with the 1H NMR spectra of both 1 and 2, revealing two sets of protons corresponding to asymmetric monomers (Figure S4, Table S1). The 2,2′-linkage in 1–3 was confirmed by diagnostic HMBC correlations of H-3 and H-3′ with C-2′ and C-2, respectively. The absolute configurations of the 2,2′-secalonic acids have been determined by ECD spectroscopy.[7] The Cotton effect around 330 nm corresponds to the n–π* electronic transition, and it has been correlated with the configurations of C-10a and C-10a′.[7a, 8] Negative Cotton effects at 332 nm in the ECD spectra of compounds 1, 2, and 3 (Δε = −38.8, −24.6, and −31.2, respectively), indicated an S-configuration at both C-10a and C-10a′ (Figure 2). This allowed the assignment of the R-configuration at C-6 and C-6′ based on the trans orientation of C-6/6′ and C-10a/10a′ substituents deduced from the biosynthetic route for the formation of stereoisomeric tetrahydroxanthone precursors of secalonic acids, with no exception so far in literature.[9] Combining all the data together suggested the absolute configuration as (5S,6R,10aS,5′S,6′R,10a′S), (5R,6R,10aS,5′R,6′R,10a′S), and (5R,6R,10aS,5′S,6′R,10a′S) for compounds 1–3, respectively. Consequently, compounds 1–3 were identified as secalonic acid A,[10] secalonic acid E,[11] and secalonic acid G,[12] respectively, in accordance with published data (Table S2). Most literature pertaining to these compounds dates back to the 1960s and 1970s, and therefore, the 1H and 13C NMR data for 1–3 were provided in the Supporting Information (Figures S2–S4, Table S1).
Figure 1.

Compounds 1–16 from S. terrestris (MSX45109).
Figure 2.

Comparison of the experimental ECD spectra of secalonic acids A (1), E (2), and G (3) [0.03 mM CHCl3, cell length 2 cm].
Compound 4 (5.4 mg) was obtained as a yellow powder. The chemical formula was determined as C32H30O14 by HRESIMS and analysis of 1H, 13C and edited-HSQC NMR data (Figure S5 and Table S3). These data suggested an asymmetric secalonic acid analogue with structural similarity to 1. However, a key difference between 4 and 1 was the linkage point of the monomeric units, being a 2,2′ in 1 vs. 2,4′ in 4, which rendered 4 asymmetric, as evidenced by diagnostic HMBC correlations of H-3 and H-3′ with C-4′ and C-4, respectively (Figure S6). The two monomeric moieties in 4 were assigned the same configuration as 1, as evidenced by a negative Cotton effect at 331 nm in the ECD spectrum of 4 (Δε = −24.8), and negative values of optical rotation for both 1 and 4 (Figures 2 and S7, Table S2). The absolute configuration of 4 was established as (5S,6R,10aS,5′S,6′R,10a′S) by comparing experimental and calculated ECD spectra predicted by the time-dependent density functional theory/electronic circular dichroism (TDDFT-ECD) approach (Figure 3).[13] Briefly, the conformation analysis of the 3D models of compound (5S,6R,10aS,5′S,6′R,10a′S)-4 and its hypothetical enantiomer (5R,6S,10aR,5′R,6′S,10a′R)-4a gave twelve and eleven conformers, respectively, within a 2 kcal/mol energy window from the global minimum. All conformers were geometrically optimized at the B3LYP/DGDZVP level.[14] Relative free energies (ΔG∘) as well as the Boltzmann distribution for the most relevant optimized conformers were given in the supplementary data (Figure S8). The Boltzmann-averaged ECD spectra of re-optimized conformers of 4 using TDDFT showed excellent fit with the experimental data, with a negative Cotton effect around 320 nm (n–π* transition), whereas the 4a enantiomer showed a positive Cotton effect at the same wavelength (Figure 3). The NMR (Figure S5, Table S3) and optical rotation data (Table S2) were in agreement with those reported for penicillixanthone A, though the absolute configuration was not determined.[15] Interestingly, compound 4 was reported by Kurobane et al.[16] as a chemically rearranged analogue of 1 by dissolving the latter into polar organic solvents, such as pyridine, acetone, and acetonitrile, either at room or higher temperatures for hours to days, depending on the solvent used. This suggests that 4 may have been an artifact of the purification scheme.
Figure 3.

Experimental and calculated ECD spectra for penicillixanthone A (4) [0.05 mM, CHCl3, cell length 2 cm].
Compound 5 (2.1 mg), which was obtained as a yellow powder, had a chemical formula of C32H30O14 as determined by HRESIMS and analysis of 1H, 13C and edited-HSQC NMR data (Figure S9), establishing an index of hydrogen deficiency of 18. The NMR data suggested an asymmetric secalonic acid analogue with structural similarity to 2 (Figures 4 and S9, Table 1). However, as was observed in 1 vs. 4, the 2,2′-linkage in 2 was supplanted by a 2,4′-linkage in 5, as evidenced by diagnostic HMBC correlations of H-3 and H-3′ with C-4′ and C-4, respectively (Figure 4). Two singlets, corresponding to H-5/H-5′ (δH 4.14/3.99) in 5 implied a pseudoaxial/pseudoequatorial cis orientation of H-5/H-6 and H-5′/H-6′, similar to that in 2, which were further supported by NOESY correlations between H-5/H-6 and H-5′/H-6′ (Figure 4). The two monomeric moieties in 5 were assigned the same configuration as 2, as evidenced by a negative Cotton effect at 332 nm in the ECD spectrum of 5, Δε = −18.9 (Figure S7), and negative values of optical rotation for both compounds (Table S2). The absolute configuration of 5 was established as (5R,6R,10aS,5′R,6′R,10a′S) by comparing experimental and calculated ECD spectra predicted by TDDFT using the same protocol described for compound 4 (Figures 5 and S10).[13a–d] These data suggested the structure of 5, which was ascribed the trivial name penicillixanthone B. As noted for 4,[10] compound 5 was potentially an artifact produced by rearrangement of 2 in polar organic solvents.
Figure 4.

Key COSY, HMBC, and NOESY correlations of 5–10 and 16. Based on the NOESY correlations, Gaussian 09 was used for ground state mechanics optimization to generate the structures shown on the right.
Table 1.
NMR data for 5 (500 MHz for 1H, 125 MHz for 13C; chemical shifts in δ, coupling constants in Hz, CDCl3).
| Position | δC | δH mult (J in Hz) | Position | δC | δH mult (J in Hz) |
|---|---|---|---|---|---|
| 1 | 159.4 | 1′ | 161.9 | ||
| 2 | 119.4 | 2′ | 110.8 | 6.60, d (8.6) | |
| 3 | 139.6 | 7.47, d (8.6) | 3′ | 140.1 | 7.33, d (8.6) |
| 4 | 107.9 | 6.58, d (8.6) | 4′ | 115.9 | |
| 4a | 157.2 | 4a′ | 154.5 | ||
| 5 | 71.5 | 4.14, s | 5′ | 70.9 | 3.99, s |
| 6 | 28.6 | 2.12, m | 6′ | 28.5 | 2.12, m |
| 7 | 32.7 | 2.38, m | 7′ | 32.6 | 2.38, m |
| 2.49, m | 2.49, m | ||||
| 8 | 180.2 | 8′ | 179.4 | ||
| 8a | 99.9 | 8a′ | 99.7 | ||
| 9 | 187.6 | 9′ | 187.9 | ||
| 9a | 107.0 | 9a′ | 106.9 | ||
| 10a | 84.9 | 10a′ | 84.4 | ||
| 11 | 17.6 | 1.17, dd (6.9, 0.6) | 11′ | 17.6 | 1.13, dd (6.9, 0.6) |
| 12 | 171.4 | 12′ | 170.9 | ||
| 13 | 53.7 | 3.75, s | 13′ | 53.4 | 3.64, s |
| 1-OH | 11.91, s | 1′-OH | 11.56, s | ||
| 5-OH | 2.55, brs | 5′-OH | 2.77, brs |
Figure 5.

Experimental and calculated ECD spectra for penicillixanthone B (5) [0.06 mM, CHCl3, cell length 2 cm].
Compound 6 (0.8 mg) was obtained as a yellow gum. The molecular formula was deduced as C16H16O6 by HRESIMS and analysis of 1H, 13C and edited-HSQC NMR data (Figure S11), indicating an index of hydrogen deficiency of 9. Inspection of the HRMS and NMR data suggested 6 as a tetrahydroxanthone derivative with structural similarity to the monomeric unit of 1. However, key differences were the replacement of the C-2 quaternary carbon in 1 (δC 118.3) by an aromatic methine in 6 (δH/δC 6.54/110.5), and the replacement of the hydrogen-bonded phenolic proton in 1 (δH 13.78) by an olefinic proton in 6 (δH 7.23) (Table 2, Figure S11). COSY data identified two spin systems as H-2/H-3/H-4 and H-5/H-6/H2-7/H-8 (Figure 4). Further examination of the NMR spectra, including HMBC data (Figure 4), yielded the planar structure of 6, which was ascribed the trivial name blennolide H. The relative configuration of 6 was found to be (5S,6R,10aS), the same as that of the monomeric units of 1, deduced from NOESY correlations and coupling constants for both compounds (Figure 4). However, the ECD calculations suggested the opposite configuration (5R,6S,10aR) (Figure S12). Since this finding was inconsistent with biosynthetic considerations, the calculations were repeated yielding identical results. As such, we consider the absolute configuration of 6 as tentative.
Table 2.
NMR data for 6 (500 MHz for 1H, 175 MHz for 13C; chemical shifts in δ, coupling constants in Hz, CDCl3).
| Position | δC | δH mult (J in Hz) |
|---|---|---|
| 1 | 163.0 | |
| 2 | 110.5 | 6.54, dd (8.6, 0.6) |
| 3 | 138.5 | 7.39, dd (8.6, 8.0) |
| 4 | 107.7 | 6.56, dd (8.0, 0.6) |
| 4a | 159.5 | |
| 5 | 77.5 | 3.92, dd (11.5, 2.9) |
| 6 | 30.0 | 2.36, m |
| 7 | 33.8 | 2.16, ddd (20.6, 10.3, 2.9) |
| 2.72, dt (20.6, 5.2) | ||
| 8 | 141.9 | 7.23, dd (5.2, 2.9) |
| 8a | 129.9 | |
| 9 | 184.7 | |
| 9a | 107.7 | |
| 10a | 85.8 | |
| 11 | 17.7 | 1.15, d (6.3) |
| 12 | 169.1 | |
| 13 | 53.3 | 3.69, s |
| 1-OH | 12.00, s | |
| 5-OH | 2.74, s |
Diastereoisomers 7 (5.4 mg) and 8 (1.0 mg) were obtained as colorless oils and both had molecular formulae of C16H16O7, as determined by HRESIMS and analysis of 1H, 13C and edited-HSQC NMR data (Figures S13 and S14), establishing an index of hydrogen deficiency of 9. Compounds 7 and 8 showed similar 1H and 13C NMR spectra (Table 3) with small differences in four chemical shift values. Interestingly, compounds 7 and 8 displayed opposite signs for optical rotation data; while compound 7 was levorotatory (−26.14°), compound 8 was dextrorotary (+42.05°) (Table S2). Moreover, the ECD spectra of 7 and 8 were mirror images (Figure 6). Analysis of the HRMS and NMR data indicated 7 and 8 as monomers and structurally related to the monomeric units of compounds 1–5. In comparison to the chromanone moiety in compounds 1–5, rings A and B, were preserved in 7 and 8, as evidenced from comparable 13C NMR data and the hydrogen-bonded phenolic proton (δH 11.43), except for a quaternary aromatic carbon that was replaced by an aromatic methine in 7 and 8. In contrast, however, ring C experienced significant changes, as evidenced from 1D- and 2D-NMR data. For example, the 13C NMR spectra displayed signals characteristic of ester functionalities (δC 175.3 and 175.7 for 7 and 8, respectively; Table 3). Thus, ring C was established as a γ-lactone moiety, as evidenced from the COSY spin system of H-9/H-10/(H3-13)/H-11 and HMBC correlations from H3-13 to C-11 and C-9, H-9 to C-12, and H-11 to C-9 (Figure 4). On the other hand, HMBC correlations from H2-3 to C-9 and C-14 and from H-9 to C-14 indicated a 2,9-linkage between the chromanone and the γ-lactone moieties, establishing the planar structures of 7 and 8 (Figure 4). The relative configuration of ring C in compounds 7 and 8 was similar based on NOESY correlations between H3-13 and H-9 and coupling constant values of 4.0 and 3.4 ppm for H-9 in compounds 7 and 8, respectively, implying a pseudodiaxial trans orientation of H-9/H-10 (Figure 4, Table 3). The spatial arrangement of 7 and 8 were different, as observed via NOESY data, particularly between H-10 to H-11α and H-3α, as well as H-9 to H2-3 in 7 and from H-9 to H-3α in 8 (Figure 4). Putting these data together, and taking into consideration the fact that compounds 7 and 8 could not be enantiomers, since they were separated using non-enantioselective methods, compounds 7 and 8 could either be epimers in C-2 (2R9S10S or 2S9S10S) or have the same configuration at C-2 but the opposite configuration of ring C. The absolute configuration of 7 and 8 was established by comparing the experimental ECD data with those obtained through molecular modeling calculations (Figures S15 and S16). The excellent fit between the observed and calculated ECD plots (Figure 7) established the absolute configuration of 7 as (2S,9S,10S) and for 8 as (2R,9S,10S), supporting epimerization at C-2. The NMR (Table S4) and optical rotation data of 8 (Table S2) indicated similarity with paecilin B, which was isolated from the mangrove endophytic fungus, Paecilomyces sp., and its planar structure was published in 2007.[17] However, significant differences in the 1H and 13C NMR data of compound 8 and paecilin B were observed (Table S4). Recently, paecilin B was reported as a monomer and as a subunit of a dimer from the seagrass-derived fungus Bipolaris sp.[18] Our data were in agreement with the latter study, including the absolute configuration. With respect to compound 7, two structurally related compounds, blennolide D and blennolide E, where C-11 is hydroxylated, were reported previously from Blennoria sp.[4] Hence, the trivial name 11-deoxyblennolide D was ascribed to 7. The planar structure of compounds 7 and 8 was reported previously by synthesis but without the reporting of NMR data.[19]
Table 3.
NMR data for 7 and 8 (500 MHz for 1H, 125 MHz for 13C; chemical shifts in δ, coupling constants in Hz, CDCl3).
| Position | 7 | 8 | ||
|---|---|---|---|---|
| δC | δH mult (J in Hz) | δC | δH mult (J in Hz) | |
| 2 | 84.3 | 84.3 | ||
| 3 | 39.8 | 3.04, dd (17.2, 0.6) | 40.7 | 3.07, d (17.2) |
| 3.17, d (17.2) | 3.48, d (17.2) | |||
| 4 | 194.1 | 194.9 | ||
| 4a | 107.7 | 107.5 | ||
| 5 | 162.0 | 161.9 | ||
| 6 | 110.7 | 6.55, dd (8.6, 0.6) | 110.6 | 6.54, dd (8.6, 0.6) |
| 7 | 139.2 | 7.42, t (8.6) | 139.0 | 7.40, t (8.6) |
| 8 | 107.7 | 6.54, dd (8.6, 0.6) | 107.5 | 6.49, dd (8.6, 0.6) |
| 8a | 159.2 | 159.3 | ||
| 9 | 87.6 | 4.43, dd (4.0, 0.6) | 86.5 | 4.36, dd (3.4, 0.6) |
| 10 | 30.1 | 2.83, m | 29.7 | 2.85, m |
| 11 | 36.2 | 2.21, dd (17.8, 4.6) | 36.4 | 2.21, dd (17.8, 4.6) |
| 2.89, dd (17.8, 9.2) | 3.01, dd (17.8, 9.2) | |||
| 12 | 175.3 | 175.7 | ||
| 13 | 20.9 | 1.28, d (6.9) | 20.7 | 1.18, d (6.9) |
| 14 | 168.9 | 169.2 | ||
| 15 | 53.8 | 3.72, s | 53.7 | 3.74, s |
| 5-OH | 11.43, s | 11.45, s | ||
Figure 6.

Experimental ECD spectra of 11-deoxyblennolide D (7) [0.16 mM] and paecilin B (8) [0.56 mM, CHCl3, cell length 2 cm].
Figure 7.

Experimental and calculated ECD spectra for 11-deoxyblennolide D (7) [0.16 mM] and paecilin B (8) [0.56 mM, CHCl3, cell length 2 cm].
Compound 9 (1.1 mg) was isolated as a yellow gum with a molecular formula of C32H30O14 as deduced by HRESIMS, 1H, 13C and edited-HSQC NMR data (Figure S17), indicating an index of hydrogen deficiency of 18. Analysis of the NMR data, including HMBC and NOESY spectra, suggested 9 as a heterodimer, with one monomeric moiety similar to a monomeric unit of 2 and the other similar to 7 (Table 4, 9Figure 4). A key difference between a monomeric moiety in that was related to 7 was replacement of the C-6 aromatic methine (δH/δC 6.55/110.7) in 7 by a quaternary carbon in 9 (δC-6′ 118.1). A 2,6′-linkage in 9 was evident by diagnostic HMBC correlations of H-3 to C-6′ and H-7′ to C-2, respectively, establishing the planar structure of 9 (Figure 4). The absolute configuration was deduced based on the monomeric constituents. The secalonic acid moiety was assigned the same configuration as 2, evidenced from similar NOESY correlations and coupling constant values of H-5 and preservation of the configuration at C-10a through the series. As well, the moiety similar to 7 was assigned the same configuration as 7 based on consistencies in the 1H, 13C, and NOESY NMR data (Tables 3 and 4, 9Figure 4). Hence, the absolute structure of was assigned as (5R,6R,10aS,2′S,9′S,10′S) and was confirmed by ECD calculations (Figures 8, S18 and S19). The NMR data of 9 were found to be in agreement with those for blennolide G, which was reported previously from Blennoria sp.[4] However, compound 9 and blennolide G showed opposite ECD and optical rotation data, and opposite absolute configuration at four chiral centers, indicating 9 as a diastereoisomer of blennolide G (Table S2, Figure 9). The trivial name blennolide I was ascribed to 9.
Table 4.
NMR data for 9 and 10 (700 MHz for 1H, 175 MHz for 13C; chemical shifts in δ, coupling constants in Hz, CDCl3).
| Position | 9 | 10 | ||
|---|---|---|---|---|
| δC | δH mult (J in Hz) | δC | δH mult (J in Hz) | |
| 1 | 159.4 | 159.3 | ||
| 2 | 118.3 | 117.8 | ||
| 3 | 139.6 | 7.42, d (8.3) | 140.1 | 7.45, d (8.5) |
| 4 | 107.5 | 6.57, d (8.3) | 107.5 | 6.62, d (8.5) |
| 4a | 157.2 | 158.4 | ||
| 5 | 71.3 | 4.12, s | 78.0 | 3.93, dd (11.2, 2.7) |
| 6 | 28.5 | 2.11, m | 29.2 | 2.41, m |
| 7 | 32.6 | 2.40, dd (18.9, 6.1) | 36.2 | 2.32, dd (19.4, 10.6) |
| 2.52, dd (18.9, 11.2) | 2.73, dd (19.4, 6.5) | |||
| 8 | 179.9 | 177.6 | ||
| 8a | 99.9 | 101.5 | ||
| 9 | 187.5 | 187.1 | ||
| 9a | 107.0 | 106.9 | ||
| 10a | 84.8 | 84.8 | ||
| 11 | 17.5 | 1.17, d, (7.1) | 18.0 | 1.17, d, (6.5) |
| 12 | 171.2 | 170.2 | ||
| 13 | 53.5 | 3.71, s | 53.3 | 3.71, s |
| 1-OH | 11.85, s | 11.72, s | ||
| 5-OH | 2.52, brs | 2.77, d (2.7) | ||
| 8-OH | 13.95, s | 13.76, s | ||
| 2′ | 84.2 | 84.2 | ||
| 3′ | 39.7 | 3.05, d (17.0) | 39.7 | 3.05, d (17.0) |
| 3.21, d (17.0) | 3.21, d (17.0) | |||
| 4′ | 194.0 | 194.0 | ||
| 4a′ | 107.6 | 107.6 | ||
| 5′ | 159.2 | 159.2 | ||
| 6′ | 118.1 | 118.1 | ||
| 7′ | 141.3 | 7.52, d (8.3) | 141.3 | 7.51, d (8.5) |
| 8′ | 107.3 | 6.61, d (8.3) | 107.3 | 6.62, d (8.5) |
| 8a′ | 158.6 | 158.6 | ||
| 9′ | 87.5 | 4.45, d (3.8) | 87.5 | 4.46, d (4.1) |
| 10′ | 30.0 | 2.84, m | 30.0 | 2.84, m |
| 11′ | 36.0 | 2.91, dd (18.0, 9.3) | 36.0 | 2.92, dd (17.9, 9.4) |
| 2.23, dd (18.0, 4.8) | 2.22, dd (17.9, 4.6) | |||
| 12′ | 175.1 | 175.1 | ||
| 13′ | 20.9 | 1.28, d (7.1) | 20.9 | 1.28, d (6.8) |
| 14′ | 168.8 | 168.8 | ||
| 15′ | 53.7 | 3.76, s | 53.7 | 3.75, s |
| 5′-OH | 11.88, s | 11.89, s | ||
Figure 8.

Experimental and calculated ECD spectra for blennolide I (9) [0.06 mM, CHCl3, cell length 2 cm].
Figure 9.

Experimental and calculated ECD spectra for blennolide J (10) [0.17 mM, CHCl3, cell length 2 cm].
Compound 10 (0.6 mg), which was isolated as a yellow gum, had a molecular formula of C32H30O14 as deduced by HRESIMS, 1H, 13C and edited-HSQC NMR data (Figure S20), indicating an index of hydrogen deficiency of 18. Analysis of the NMR data indicated 10 as an asymmetric heterodimer with structural similarity to 9. Key differences between 9 and 10 were chemical shift values and splitting patterns of H-5, H-6, and H2-7 (Table 4, Figures S17 and S20). A proton singlet in 9 (δH 4.12), corresponding to H-5, was replaced by a doublet of doublets (δH 3.93, dd, J = 11.2, 2.7) in 10, suggesting a pseudodiaxial trans orientation of H-5/H-6 in 10. These data indicated that the secalonic acid moiety in 10 was similar to the monomeric units of 1, while the 11-deoxyblennolide D moiety was preserved, as in 9. A 2,6′-linkage in 10, also similar to 9, was confirmed by diagnostic HMBC correlations of H-3 with C-6′ and H-7′ with C-2, respectively, establishing the planar structure of 10 (Figure 4). As for 9, the absolute configuration of 10 was deduced based on the monomeric constituents and ECD calculations as (5S,6R,10aS,2′S,9′S,10′S) (Figures 9, S18 and S21). The trivial name blennolide J was ascribed to compound 10.
In addition to compounds 1–10, the biogenetically related benzophenone, nidulalin B (15, 1.7 mg) was isolated and identified by comparison of its spectroscopic and spectrometric data with those reported in the literature (Figure S26).[20]
Resorcylic acid lactones
Compounds 11–13 were identified as the resorcylic acid lactones, aigialomycin A (4.2 mg),[21] hypothemycin (19.6 mg),[22] and dihydrohypothemycin (3.7 mg),[21] respectively (Figures S22–S24), while compound 14 was the biogenetically related pyrenochaetic acid C (2.7 mg) (Figure S25).[23] In all cases, the spectroscopic and spectrometric data were in agreement with those reported in the literature.
Compound 16 (0.8 mg) was isolated as a colorless oil with a molecular formula of C12H14O5 as revealed by HRESIMS, 1H, 13C and edited-HSQC NMR data (Figure S27), indicating an index of hydrogen deficiency of six. UV absorption maxima of 301, 270, and 233 nm were indicative of an aromatic carbonyl compound.[24] Inspection of the NMR data showed signals characteristic of six aromatic carbons, two of which were oxygenated, two doublet aromatic protons with coupling constant values of 2.3 Hz, and two phenolic protons (Table 5, Figure S27). These data suggested a 1,2,3,5-tetrasubstituted benzene ring with two aromatic protons meta to each other (Figure S27, Table 5). Benzene ring substituents were confirmed by HMBC correlations (Figure 4). COSY data identified a 1,2,3-trisubtituted pentane moiety (H2-4/H-3/H-11/H-12/H-13) with C-3 and C-11 being oxygenated and C-4 attached to the benzene ring (Figure 4). HMBC correlations from H2-4 to C-6 and C-10 confirmed C-5 as the attachment point of the aliphatic side chain with the aromatic ring. Chemical shift values (δH/δC 4.43/81.1) for H-3/C-3 indicated esterification of C-3 to form a six membered ring with the ester carbonyl (δC 170.2) that was attached to the benzene ring at C-2. Further examination of the NMR data yielded the planar dihydroisocoumarin structure of 16, which was ascribed the trivial name pyrenomycin. The absolute configuration of 16 was assigned via a modified Mosher’s ester method,[25] establishing the configuration as 3R and 11S (Figure 10). Similar dihydroisocoumarin compounds were isolated previously from marine sponges, plants, fungi, and insects, including hiburipyranone,[26] 3-(2-hydroxypropyl)-8-hydroxy-3,4-dihydroisocoumarin,[27] and (3S,1′R)-3-(1-hydroxyethyl)-6,8-dihydroxy-7-methyl-3,4-dihydroisocoumarin.[28]
Table 5.
NMR data for 16 (700 MHz for 1H, 175 MHz for 13C; chemical shifts in δ, coupling constants in Hz, Methanol-d4)
| Position | δC | δH mult (J in Hz) |
|---|---|---|
| 1 | 170.2 | |
| 3 | 81.1 | 4.43, dt (12.7, 3.3) |
| 4 | 29.0 | 2.75, dd (16.4, 3.3) |
| 3.11, dd (16.4, 12.7) | ||
| 5 | 142.2 | |
| 6 | 107.2 | 6.21, brd (2.3) |
| 7 | 164.2 | |
| 8 | 100.9 | 6.15, d (2.3) |
| 9 | 168.8 | |
| 10 | 99.6 | |
| 11 | 73.1 | 3.58, dt (8.6, 3.3) |
| 12 | 25.1 | 1.67, m |
| 13 | 9.2 | 1.03, t (7.5) |
| 9-OH | 8.55, s | |
| 11-OH | 1.28, brs | |
| 7-OH | 4.57, brs |
Figure 10.
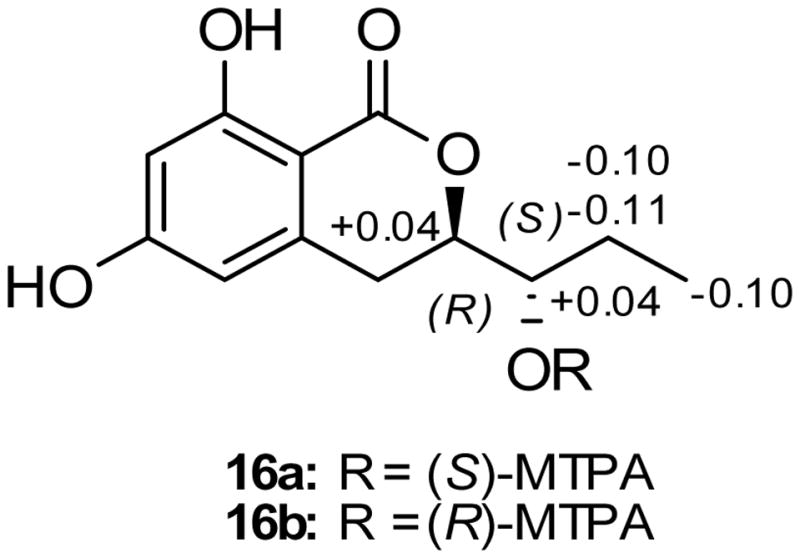
ΔδH values [Δδ (in ppm) = δS − δR] obtained for (S)- and (R)-MTPA esters (16a and 16b, respectively) of pyrenomycin (16) in pyridine-d5.
Cytotoxicity and antimicrobial evaluation of isolated compounds
Compounds (1–15) were tested for cytotoxicity in the MDA-MB-435 and SW-620 cancer cell lines. Of the tetrahydroxanthone derivatives, 1 and 4 were the most potent with IC50 values less than 0.50 μM in both cell lines (Table 6). Based on the cytotoxicity data of related analogues, the importance of the configuration of C-5/C-5′ became evident (Table 6). Compounds 1 and 4, with 5S,5′S configuration, were the most potent, with a 2,2′- and a 2,4′-linkages for 1 and 4, respectively. A 5S,5′R configuration, as in 3, was approximately 20 times less active than 1, while a 5R,5′R configuration rendered 2 inactive; both had a 2,2′-linkage of the monomeric units. For those with a 2,4′-linkage, compound 5 with 5R,5′R was approximately 30 times less active than 4. Heterodimeric 10, which was composed of the monomeric units of 1 and compound 7, was approximately 25 times less active than 1. Compound 9, which was composed of the monomeric units of 2 and compound 7 was inactive. The difference in the potencies of 10 vs. 9 further supported the importance of the 5S,5′S configuration, and all three monomeric compounds (6–8) were inactive. Consistent with the cytotoxicity data obtained in this study, 1 and its chemically rearranged 2,4′-dimer 4 were reported to have equipotent activity against cultured mouse leukemia L1210 cells.[2d] Moreover, 1 has been reported to have a protecting effect of the dopaminergic neurons from 1-methyl-4-phenylpyridinium (MPP+)-induced cell death[29] and to attenuate colchicine-induced apoptosis of the cortical neurons.[30]
Table 6.
Cytotoxicity of compounds 1–15 against two human tumor cell lines.
| Compound[a] | IC50 values in μM[b] | |
|---|---|---|
| MDA-MB-435[c] | SW-620[c] | |
| 1 | 0.16 | 0.41 |
| 2 | NA | 19.12 |
| 3 | 3.27 | 3.67 |
| 4 | 0.18 | 0.21 |
| 5 | 5.20 | 5.55 |
| 10 | 4.06 | 6.14 |
| 12 | 0.58 | 2.14 |
Compounds 6–9, 11, and 13–15 were inactive with IC50 values >20 μM.
IC50 values were determined as the concentration required to inhibit growth to 50% of control with 72 h incubation.
Positive control was vinblastine tested at concentration of 1 nM in MDA-MB-435 cells and 10 nM in SW620 cells, which had 23% and 76% viable cells, respectively.
In agreement with the literature,[5] cytotoxicity data of the structurally related RALs (11–13) indicated the importance of the (Z)-enone for activity, as in hypothemycin (12) (IC50 value of 0.58 μM, MDA-MB-435). Alternatively, aigialomycin A (11), with an (E)-enone, and dihydrohypothemycin (13), with a reduced enone, were both inactive (IC50 value >10 μM). RALs containing a (Z)-enone have been reported as potent inhibitors of several ATPases and kinases, including TAK1.[31]
The antimicrobial activity of the isolated compounds (1–15) was evaluated against a panel of bacteria and fungi (Tables 7 and S5). Compounds 3 and 5 showed promising activity against the Gram-positive bacterium Micrococcus luteus with MIC values of 5 and 15 μg/mL, respectively. Secalonic acid A (1) was reported to have activity against Bacillus subtilis and Piriculaia oryzae[2b] and a phlogistic activity,[32] while penicillixanthone A (4) was reported to have medium antibacterial activity against M. luteus, Pseudoalteromonas nigrifaciens, and B. subtilis.[33] Interestingly, compounds 3 and 5, which were both inactive in cytotoxicity assays, were the most promising in the antimicrobial assays. This finding demonstrates the importance of testing isolated natural compounds in a variety of assays.
Table 7.
Antimicrobial activities of compounds 1–15
| Compound[a] | Antimicrobial Activity MIC (μg/mL) | |
|---|---|---|
| M. luteus | S. aureus | |
| 1 | 38 | 75 |
| 2 | 36 | NA[b] |
| 3 | 5 | 39 |
| 4 | 46 | 93 |
| 5 | 15 | 59 |
| 10 | 43 | 43 |
| Vancomycin[c] | -- | 0.25 |
See table S5 for compounds 6–9, 11, and 12–15.
NA: not active with MIC >145 μg/mL.
Positive control..
Conclusions
A total of 16 polyketides (1–16), of which six were new, were isolated from a fungal culture of S. terrestris (MSX45109). Cytotoxicity assays suggested compounds 1, 4, and 12 as the most potent. When evaluated for antimicrobial activity, compounds 3 and 5 showed promising M. luteus activity. This fungus was a prolific producer of polyketides, affording the opportunity to examine many analogues in a series of bioassays concomitantly. Moreover, while a few of the known compounds were first described decades ago, this study also served to update the literature with spectroscopic and spectrometric data derived from modern instruments.
Experimental Section
General Procedures
UV and ECD spectra were obtained using a Varian Cary 100 Bio UV-vis spectrophotometer (Varian Inc., Walnut Creek, CA, USA) and an Olis DSM 17 ECD spectrophotometer (Olis, Inc. Bogart, GA, USA), respectively. NMR data were collected using either a JEOL ECA-500 NMR spectrometer operating at 500 MHz for 1H and 125 MHz for 13C, or a JEOL ECS-400 NMR spectrometer operating at 400 MHz for 1H and 100 MHz for 13C and equipped with a high sensitivity JEOL Royal probe and a 24-slot autosampler (both from JEOL Ltd., Tokyo, Japan), or an Agilent 700 MHz NMR spectrometer (Agilent Technologies, Inc., Santa Clara, CA, USA), equipped with a cryoprope, operating at 700 MHz for 1H and 175 MHz for 13C. Residual solvent signals were utilized for referencing. High resolution mass spectra (HRMS) were obtained using a Thermo LTQ Orbitrap XL mass spectrometer equipped with an electrospray ionization source (Thermo Fisher Scientific, San Jose, CA, USA). A Waters Acquity UPLC system (Waters Corp., Milford, MA, USA) utilizing a Waters BEH C18 column (1.7 μm; 50 × 2.1 mm) was used to check the purity of the isolated compounds with data collected and analyzed using Empower software. Phenomenex Gemini-NX C18 analytical (5 μm; 250 × 4.6 mm), preparative (5 μm; 250 × 21.2 mm), and semipreparative (5 μm; 250 × 10.0 mm) columns (all from Phenomenex, Torrance, CA, USA) were used on a Varian Prostar HPLC system equipped with ProStar 210 pumps and a Prostar 335 photodiode array detector (PDA), with data collected and analyzed using Galaxie Chromatography Workstation software (version 1.9.3.2, Varian Inc.). Flash chromatography was performed on a Teledyne ISCO CombiFlash Rf 200 using Silica Gold columns (both from Teledyne Isco, Lincoln, NE, USA) and monitored by UV and evaporative light-scattering detectors.
Fungal Strain Isolation and Identification
Mycosynthetix fungal strain MSX45109 was isolated from leaf litter collected in a mangrove habitat in 1989. Molecular techniques were used to identify MSX45109 by sequencing the internal transcribed spacer regions 1 & 2 and 5.8S nrDNA (ITS).[34] DNA extraction, PCR amplification, sequencing, and phylogenetic analyses were performed as described previously.[1b, 35] BLAST search in GenBank and the curated BOLD systems (http://www.boldsystems.org/index.php/IDS_OpenIdEngine) database, as well as Q-bank using ITS rDNA sequences suggested that MSX45109 shared high sequence similarity with several Centraalbureau voor Schimmelcultures (CBS) strains of Setophoma terrestris (Pleosporales, Ascomycota) with ≥80% query coverage and 99–100% sequence identity. The top BLAST matches were downloaded using Seaview v4.4.2. and Maximum Likelihood analysis was performed using methods outlined earlier.[36] Based on the results of the BLAST search and Maximum Likelihood phylogeny of the ITS region (Figure S28), the strain MSX45109 was identified as Setophoma terrestris (H. N. Hansen) Gorenz J.C. Currently, three species of Setophoma have been described with S. terrestris as the type species.[37] The ITS sequences of four isolates of MSX45109 were deposited in the GenBank (accession numbers KM203887, KM203888, KM203889, and KM203890).
Fermentation, Extraction and Isolation
Storage, fermentation, and extraction procedures of fungal strain MSX45109 were as described previously.[1b, 1d, 1h] In brief, a seed culture of strain MSX45109 grown in YESD medium was used to inoculate a 2.8-L Fernbach flask (Corning, Inc., Corning, NY, USA) containing 150 g rice and 300 mL H2O. The solid culture was incubated at r.t. for 14 days and then extracted by addition of a 500 mL mixture of 1:1 MeOH/CHCl3. The culture was chopped into small pieces and left to shake at 125 rpm at r.t., followed by vacuum filtration. The solid phase was washed with 100 mL of 1:1 MeOH/CHCl3. To the filtrate, 900 mL CHCl3 and 1500 mL H2O were added so that the final ratio of CHCl3/MeOH/H2O was 4:1:5. The mixture was stirred for ½ h and then transferred into a separatory funnel. The organic bottom layer was drawn off and evaporated to dryness and then re-constituted in 100 mL of 1:1 MeOH/CH3CN and 100 mL of hexanes. The biphasic solution was stirred for 15 min and then transferred to a separatory funnel. The MeOH/CH3CN layer was drawn off and evaporated to dryness under vacuum. The defatted material (1.5 g) was dissolved in a mixture of CHCl3/MeOH, adsorbed onto Celite 545, and fractionated via flash chromatography using a gradient solvent system of hexane/CHCl3/MeOH at a 40 mL/min flow rate and 53.3 column volumes over 63.9 min to afford five fractions. Fraction 3 (627 mg) was subjected to preparative reversed phase HPLC over a Phenomenex Gemini-NX C18 preparative column using a gradient system of 40:60 to 60:40 over 30 min of CH3CN/H2O (acidified with 0.1% formic acid) at a flow rate of 21.24 mL/min to yield thirteen sub-fractions. Sub-fractions 4, 5, 10, and 12 yielded compounds 11 (4.2 mg), 12 (19.6 mg), 1 (35.1), and 2 (48.4 mg), which eluted at 7.8, 8.7, 25.1, and 29.0 min, respectively. The other sub-fractions were subjected to further purifications as follows: sub-fraction 1 (2.3 mg) was subjected to semi-preparative HPLC purification over a Phenomenex Gemini-NX C18 column using a gradient system of 50:50 to 70:30 of MeOH/H2O (0.1% formic acid) over 15 min at a flow rate of 4.72 mL/min to yield compound 16 (0.8 mg), which eluted at 13.1 min. Sub-fraction 6 (8.5 mg) was subjected to preparative HPLC purification using a Phenomenex Gemini-NX C18 and a gradient system of 50:50 to 70:30 of MeOH/H2O (0.1% formic acid) over 15 min at a flow rate of 21.24 mL/min to yield compounds 15 (1.7 mg) and 13 (3.7 mg), which eluted at 8.0 and 18.9 min, respectively. Sub-fraction 7 (1.5 mg) was subjected to semipreparative HPLC purification using a Phenomenex Gemini-NX C18 column and a gradient system of 50:50 to 70:30 of MeOH/H2O (0.1% formic acid) over 15 min at a flow rate of 4.72 mL/min to yield compound 6 (0.8 mg), which eluted at 17.5 min. Sub-fraction 9 (18.2 mg) was subjected to preparative HPLC purification using a Phenomenex Gemini- NX C18 and a gradient system of 50:50 to 70:30 of MeOH/H2O (0.1% formic acid) over 15 min at a flow rate of 21.24 mL/min to yield compounds 7 (5.4 mg), 8 (1.0 mg), and 14 (2.7 mg), which eluted at 11.6, 13.5 and 19.5 min, respectively. An aliquot of sub-fraction 10 (20 mg) was further purified using preparative HPLC over a Phenomenex Gemini-NX C18 utilizing a gradient solvent system of 70:30 to 90:10 of MeOH/H2O (0.1% formic acid) over 15 min at a flow rate of 21.24 mL/min to yield compounds 10 (0.6 mg) and 1 (3.1 mg), which eluted at 9.5 and 14.8 min, respectively. An aliquot of sub-fraction 11 (20 mg) was subjected to semi-preparative HPLC purification over a Phenomenex Gemini-NX C18 column using a gradient system of 70:30 to 90:10 of MeOH/H2O (0.1% formic acid) over 15 min at a flow rate of 4.72 mL/min to yield compound 3 (9.5 mg), which eluted at 12.3 min, and compound 9 (1.1 mg), which was obtained by yet another round of similar purification. Sub-fraction 13 (21.5 mg) was subjected to preparative HPLC purification using a Phenomenex Gemini-NX C18 and a gradient system of 70:30 to 90:10 of MeOH/H2O (0.1% formic acid) over 15 min at a flow rate of 21.24 mL/min to yield compounds 2 (2.9 mg) and 4 (5.4 mg), which eluted at 10.5 and 19.9 min, respectively, and compound 5 (2.1 mg), which was obtained by another round of HPLC purification.
Penicillixanthone B (5)
Yellow powder; (c = 0.03 in acetone); UV (MeOH) λmax (log ε) 349 (3.64), 336 (3.63), 250 (3.58) nm; CD (c 6.26 × 10−5 M, CHCl3) λ (Δε) 225 (−55.3) nm, 243 (+48.4) nm, 332 (−18.9) nm; 347 (+2.6) nm, 376 (−8.8) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Table 1 and Supplementary Figure S9; HRESIMS m/z 639.1691 [M + H]+ (calcd for C32H31O14 639.1708).
Blennolide H (6)
Yellow gum; (c = 0.01 in chloroform); UV (MeOH) λmax (log ε) 283 (3.48), 193 (3.68) nm; CD (c 4.27 × 10−4 M, MeOH) λ (Δε) 231 (+11.9) nm, 269 (−20.9) nm, 298 (−19.8) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 175 MHz), see Table 2 and Supplementary Figure S11; HRESIMS m/z 305.1012 [M + H]+ (calcd for C16H17O6 305.1020).
11-Deoxyblennolide D (7)
Colorless oil; (c = 0.03 in chloroform); UV (MeOH) λmax (log ε) 349 (3.21), 270 (3.45), 224 (3.37) nm; CD (c 1.56 × 10−4 M, CHCl3) λ (Δε) 235 (+6.2) nm, 267 (−53.8) nm, 307 (−16.4) nm; 346 (+0.5) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Table 3 and Supplementary Figure S13; HRESIMS m/z 321.0963 [M + H]+ (calcd for C16H17O7 321.0969).
Paecilin B (8)
Colorless oil; (c = 0.09 in chloroform); UV (MeOH) λmax (log ε) 346 (3.20), 271 (3.49), 193 (3.70) nm; CD (c 5.62 × 10−4 M, CHCl3) λ (Δε) 235 (−44.7) nm, 267 (+35.2) nm, 307 (+17.0) nm; 346 (−8.7) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Table 3 and Supplementary Figure S14; HRESIMS m/z 321.0961 [M + H]+ (calcd for C16H17O7 321.0969).
Blennolide I (9)
Yellow powder; (c = 0.09 in chloroform); UV (MeOH) λmax (log ε) 349 (2.81), 258 (3.69), 224 (3.69) nm; CD (c 6.26 × 10−5 M, CHCl3) λ (Δε) 241 (+13.9), 275 (−6.3) nm, 334 (−8.7) nm; 1H NMR (CDCl3, 700 MHz) and 13C NMR (CDCl3, 175 MHz), see Table 4 and Supplementary Figure S17; HRESIMS m/z 639.1682 [M + H]+ (calcd for C32H31O14 639.1708).
Blennolide J (10)
Yellow powder; (c = 0.08 in chloroform); UV (MeOH) λmax (log ε) 349 (3.37), 336 (3.37), 237 (3.4) nm; CD (c 1.72 × 10−4 M, CHCl3) λ (Δε) 245 (+38.9), 262 (−14.9) nm, 337 (−13.9) nm, 377 (−16.7) nm; 1H NMR (CDCl3, 700 MHz) and 13C NMR (CDCl3, 175 MHz), see Table 4 and Supplementary Figure S20; HRESIMS m/z 639.1683 [M + H]+ (calcd for C32H31O14 639.1708).
Pyrenomycin (16)
Coreless oil; (c = 0.05 in chloroform); UV (MeOH) λmax (log ε) 301 (3.09), 270 (3.12), 233 (3.07) nm; 1H NMR (Methnol-d4, 700 MHz) and 13C NMR (Methnol-d4, 175 MHz), see Table 5 and Supplementary Figure S27; HRESIMS m/z 239.0909 [M + H]+ (calcd for C12H16O5 239.0914).
Preparation of The (R)- And (S)-MTPA Ester Derivatives of Pyrenomycin (16)
To 0.1 mg of compound 16 were added 400 μL of pyridine-d5, and the solution was transferred into an NMR tube. To initiate the reaction, 20 μL of S-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl (MTPA) chloride were added with careful shaking and then monitored immediately by 1H NMR at the following time points: 0, 5, 10, 15, 30, 60, and 120 min. The reaction was found to be complete in 2 h, yielding the mono (R)-MTPA ester derivative (16b) of 16. 1H NMR data of 16b (500 MHz, pyridine-d5): δH 1.85 (3H, d, J = 7.5, H3-13), 1.81 (1H, m, H-12a), 1.90 (1H, m, H-12b), 4.81 (1H, m, H-11), 5.61 (1H, m, H-3). In an analogues manner, 0.1 mg of compound 16 dissolved in 400 μL pyridine-d5 were reacted in a second NMR tube with 20 μL (R)-(−)-α-MTPA chloride for 1 h, to afford the mono (S)-MTPA ester (16a). 1H NMR data of 16a (500 MHz, pyridine-d5): δH 1.75 (3H, d, J = 6.9, H3-13), 1.70 (1H, m, H-12a), 1.80 (1H, m, H-12b), 4.85 (1H, m, H-11), 5.65 (1H, m, H-3).
Molecular Modeling Calculations
Theoretical calculations of ECD spectra for compounds 4, 5 and 7–10, and their corresponding enantiomers or epimers, were performed with the Gaussian 09 (Gaussian Inc., Wallingford CT, USA) program package as described previously[13e] (for detail see Supplementary Information).[38]
Cytotoxicity assay
The cytotoxicity of compounds (1–15) were tested against the MDA-MB-435[39] human melanoma (HTB-129, ATCC) and the SW-620[40] human colorectal adenocarcinoma (CCL-227, ATCC) cell lines as described previously.[41]
Antimicrobial assay
Minimal inhibitory concentrations (MICs) of compounds (1–15) were measured against a panel of bacteria and fungi as described previously.[41a] All measurements were made in duplicate.
Supplementary Material
Acknowledgments
This research was supported by program project grant P01 CA125066 from the National Cancer Institute/National Institutes of Health, Bethesda, MD, USA. The high resolution mass spectrometry data were acquired at the Triad Mass Spectrometry Laboratory at the University of North Carolina at Greensboro. We are indebted to Dirección General de Cómputo y de Tecnologías de Información y Comunicación (DGTIC), UNAM, for providing the resources to carry out computational calculations through the KanBalam system.
Footnotes
Supporting Information: Molecular modeling calculations, UPLC chromatograms of compounds 1–16, 1H and 13C NMR spectra for compounds 1–16, key COSY, HMBC, and NOESY correlations of 4, comparison of the experimental ECD spectra of penicillixanthone A (4) and penicillixanthone B (5), experimental and calculated ECD spectra for blennolide H (6), comparison of the experimental ECD spectra of blennolide I (9) and blennolide J (10), DFT B3LYP/DGDZVP global minimum energy models, relative Gibbs free energies (ΔGrel)a and equilibrium population (p)b values for the most relevant conformations of compounds 4, 5, and 7–10, and their correponding enantiomers 4a, 5a, and 7a–10a, phylogram of the most likely tree; ECD and optical rotation data, experimental vs. literature, of compounds 1–4, 6, 8–9, and blennolide G, 1H NMR data for 1–3, NMR data for 4, and NMR data for 8 and paecilin B.
References
- 1.a) El-Elimat T, Figueroa M, Ehrmann BM, Cech NB, Pearce CJ, Oberlies NH. J Nat Prod. 2013;76:1709–1716. doi: 10.1021/np4004307. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) El-Elimat T, Figueroa M, Raja HA, Graf TN, Adcock AF, Kroll DJ, Day CS, Wani MC, Pearce CJ, Oberlies NH. J Nat Prod. 2013;76:382–387. doi: 10.1021/np300749w. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) El-Elimat T, Zhang X, Jarjoura D, Moy FJ, Orjala J, Kinghorn AD, Pearce CJ, Oberlies NH. ACS Med Chem Lett. 2012;3:645–649. doi: 10.1021/ml300105s. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Figueroa M, Graf TN, Ayers S, Adcock AF, Kroll DJ, Yang J, Swanson SM, Munoz-Acuna U, Carcache de Blanco EJ, Agrawal R, Wani MC, Darveaux BA, Pearce CJ, Oberlies NH. J Antibiot. 2012;65:559–564. doi: 10.1038/ja.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Sy-Cordero AA, Graf TN, Adcock AF, Kroll DJ, Shen Q, Swanson SM, Wani MC, Pearce CJ, Oberlies NH. J Nat Prod. 2011;74:2137–2142. doi: 10.1021/np2004243. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Sy-Cordero AA, Pearce CJ, Oberlies NH. J Antibiot. 2012;65:541–549. doi: 10.1038/ja.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Ayers S, Ehrmann BM, Adcock AF, Kroll DJ, Wani MC, Pearce CJ, Oberlies NH. Tetrahedron Lett. 2011;52:5733–5735. doi: 10.1016/j.tetlet.2011.08.125. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Ayers S, Graf TN, Adcock AF, Kroll DJ, Matthew S, Carcache de Blanco EJ, Shen Q, Swanson SM, Wani MC, Pearce CJ, Oberlies NH. J Nat Prod. 2011;74:1126–1131. doi: 10.1021/np200062x. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Ayers S, Graf TN, Adcock AF, Kroll DJ, Shen Q, Swanson SM, Matthew S, Carcache de Blanco EJ, Wani MC, Darveaux BA, Pearce CJ, Oberlies NH. J Antibiot. 2012;65:3–8. doi: 10.1038/ja.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Ayers S, Graf TN, Adcock AF, Kroll DJ, Shen Q, Swanson SM, Wani MC, Darveaux BA, Pearce CJ, Oberlies NH. Tetrahedron Lett. 2011;52:5128–5230. doi: 10.1016/j.tetlet.2011.07.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Braäse S, Encinas A, Keck J, Nising CF. Chem Rev. 2009;109:3903–3990. doi: 10.1021/cr050001f. [DOI] [PubMed] [Google Scholar]; b) Masters KS, Bräse S. Chem Rev. 2012;112:3717–3776. doi: 10.1021/cr100446h. [DOI] [PubMed] [Google Scholar]; c) Stoll A, Renz J, Brack A. Helv Chim Acta. 1952;35:2022–2034. [Google Scholar]; d) Kurobane I, Iwahashi S, Fukuda A. Drugs Exp Clin Res. 1987;13:339–344. [PubMed] [Google Scholar]; e) Liao G, Zhou J, Wang H, Mao Z, Xiao W, Wang H, She Z, Zhu Y. Oncol Rep. 2010;23:387–395. [PubMed] [Google Scholar]; f) McPhee F, Caldera PS, Bemis GW, McDonagh AF, Kuntz ID, Craik CS. Biochem J. 1996;320(Pt 2):681–686. doi: 10.1042/bj3200681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Ito T, Masubuchi M. J Antibiot. 2014;67:353–360. doi: 10.1038/ja.2014.12. [DOI] [PubMed] [Google Scholar]; b) Yang JY, Sanchez LM, Rath CM, Liu X, Boudreau PD, Bruns N, Glukhov E, Wodtke A, de Felicio R, Fenner A, Wong WR, Linington RG, Zhang L, Debonsi HM, Gerwick WH, Dorrestein PC. J Nat Prod. 2013;76:1686–1699. doi: 10.1021/np400413s. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Klitgaard A, Iversen A, Andersen MR, Larsen TO, Frisvad JC, Nielsen KF. Anal Bioanal Chem. 2014;406:1933–1943. doi: 10.1007/s00216-013-7582-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang W, Krohn K, Ullah Z, Florke U, Pescitelli G, Di Bari L, Antus S, Kurtan T, Rheinheimer J, Draeger S, Schulz B. Chem - Eur J. 2008;14:4913–4923. doi: 10.1002/chem.200800035. [DOI] [PubMed] [Google Scholar]
- 5.Bräse S, Gläser F, Kramer CS. In: The Chemistry of Mycotoxins. Bräse S, Gläser F, Kramer C, Lindner S, Linsenmeier AM, Masters KS, Meister AC, Ruff BM, Zhong S, editors. Springer; Vienna: 2013. pp. 91–108. [PubMed] [Google Scholar]
- 6.Dictionary of Natural Products Online 23.1. Taylor & Francis Group; London: 2014. [Google Scholar]
- 7.a) Andersen R, Buchi G, Kobbe B, Demain AL. J Org Chem. 1977;42:352–353. doi: 10.1021/jo00422a042. [DOI] [PubMed] [Google Scholar]; b) Steyn PS. Tetrahedron. 1970;26:51–57. doi: 10.1016/0040-4020(70)85006-2. [DOI] [PubMed] [Google Scholar]
- 8.a) Steyn PS. Tetrahedron. 1970;26:51–57. doi: 10.1016/0040-4020(70)85006-2. [DOI] [PubMed] [Google Scholar]; b) Elsässer B, Krohn K, Flörke U, Root N, Aust HJ, Draeger S, Schulz B, Antus S, Kurtán T. Eur J Org Chem. 2005;2005:4563–4570. [Google Scholar]
- 9.Kurobane I, Vining LC, Mcinnes AG. J Antibiot. 1979;32:1256–1266. doi: 10.7164/antibiotics.32.1256. [DOI] [PubMed] [Google Scholar]
- 10.Franck B, Gottschalk EM, Ohnsorge U, Hüper F. Chem Ber. 1966;99:3842–3862. [Google Scholar]
- 11.Howard CC, Johnstone RAW, Entwistle ID. J Chem Soc, Chem Commun. 1973:464–464. [Google Scholar]
- 12.Kurobane I, Vining LC, McInnes AG. Tetrahedron Lett. 1978;19:4633–4636. [Google Scholar]
- 13.a) Bringmann G, Bruhn T, Maksimenka K, Hemberger Y. Eur J Org Chem. 2009;2009:2717–2727. [Google Scholar]; b) Stephens PJ, Harada N. Chirality. 2010;22:229–233. doi: 10.1002/chir.20733. [DOI] [PubMed] [Google Scholar]; c) Stephens PJ, Pan JJ, Devlin FJ, Urbanová M, Hájícček J. J Org Chem. 2007;72:2508–2524. doi: 10.1021/jo062567p. [DOI] [PubMed] [Google Scholar]; d) Acunña UM, Figueroa M, Kavalier A, Jancovski N, Basile MJ, Kennelly EJ. J Nat Prod. 2010;73:1775–1779. doi: 10.1021/np100322d. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) El-Elimat T, Raja HA, Figueroa M, Falkinham JO, III, Oberlies NH. Phytochemistry. 2014;104:114–120. doi: 10.1016/j.phytochem.2014.04.006. [DOI] [PubMed] [Google Scholar]; f) Ding Y, Li XC, Ferreira D. J Nat Prod. 2008;72:327–335. doi: 10.1021/np800146v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Godbout N, Salahub DR, Andzelm J, Wimmer E. Can J Chem. 1992;70:560–571. [Google Scholar]; b) Andzelm J, Wimmer E. The Journal of Chemical Physics. 1992;96:1280–1303. [Google Scholar]
- 15.Jiang T, Tian L, Guo AH, Fu HZ, Pei YH, Lin WH. Acta Pharm Sin. 2002;37:271–274. [PubMed] [Google Scholar]
- 16.Kurobane I, Vining LC, Mcinnes AG U. S. Patent, editor. Asahi Kasei Kogyo Kabushiki Kaisha; Osaka, Japan: 1984. pp. 1–9. [Google Scholar]
- 17.Guo ZY, She ZG, Shao CL, Wen L, Liu F, Zheng ZH, Lin YC. Magn Reson Chem. 2007;45:777–780. doi: 10.1002/mrc.2035. [DOI] [PubMed] [Google Scholar]
- 18.Arunpanichlert J, Rukachaisirikul V, Tadpetch K, Phongpaichit S, Hutadilok-Towatana N, Supaphon O, Sakayaroj J. Phytochem Lett. 2012;5:604–608. [Google Scholar]
- 19.Bose NK, Chaudhury DN. J Indian Chem Soc. 1966;43:411–415. [Google Scholar]
- 20.Kawahara N, Sekita S, Satake M, Udagawa S, Kawai K. Chem Pharm Bull. 1994;42:1720–1723. [Google Scholar]
- 21.Isaka M, Suyarnsestakorn C, Tanticharoen M, Kongsaeree P, Thebtaranonth Y. J Org Chem. 2002;67:1561–1566. doi: 10.1021/jo010930g. [DOI] [PubMed] [Google Scholar]
- 22.Agatsuma T, Takahashi A, Kabuto C, Nozoe S. Chem Pharm Bull. 1993;41:373–375. doi: 10.1248/cpb.41.1726. [DOI] [PubMed] [Google Scholar]
- 23.Sato H, Konoma K, Sakamura S. Agric Biol Chem. 1981;45:1675–1679. [Google Scholar]
- 24.Pretsch E, Bühlmann P, Affolter C. Structure Determination of Organic Compounds: Tables of Spectral Data. Springer; 2000. [Google Scholar]
- 25.Hoye TR, Jeffrey CS, Shao F. Nat Protoc. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- 26.Fusetani N, Sugawara T, Matsunaga S, Hirota H. J Org Chem. 1991;56:4971–4974. [Google Scholar]
- 27.Gao G, Qi S, Zhang S, Yin H, Xiao Z, Li M, Li Q. Pharmazie. 2008;63:542–544. [PubMed] [Google Scholar]
- 28.Jiang HL, Luo XH, Wang XZ, Yang JL, Yao XJ, Crews P, Valeriote FA, Wu QX. Fitoterapia. 2012;83:1275–1280. doi: 10.1016/j.fitote.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Zhai A, Zhu X, Wang X, Chen R, Wang H. Eur J Pharmacol. 2013;713:58–67. doi: 10.1016/j.ejphar.2013.04.029. [DOI] [PubMed] [Google Scholar]
- 30.Zhai A, Zhang Y, Zhu X, Liang J, Wang X, Lin Y, Chen R. Neurochem Int. 2011;58:85–91. doi: 10.1016/j.neuint.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 31.a) Serba C, Winssinger N. Eur J Org Chem. 2013;2013:4195–4214. [Google Scholar]; b) Wu JQ, Powell F, Larsen NA, Lai ZW, Byth KF, Read J, Gu RF, Roth M, Toader D, Saeh JC, Chen HW. ACS Chem Biol. 2013;8:643–650. doi: 10.1021/cb3005897. [DOI] [PubMed] [Google Scholar]
- 32.Harada M, Yano S, Watanabe H, Yamazaki M, Miyaki K. Chem Pharm Bull. 1974;22:1600–1606. doi: 10.1248/cpb.22.1600. [DOI] [PubMed] [Google Scholar]
- 33.Bao J, Sun YL, Zhang XY, Han Z, Gao HC, He F, Qian PY, Qi SH. J Antibiot. 2013;66:219–223. doi: 10.1038/ja.2012.110. [DOI] [PubMed] [Google Scholar]
- 34.Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W Fungal Barcoding Consortium. Proc Natl Acad Sci USA. 2012;109:6241–6246. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) El-Elimat T, Figueroa M, Raja HA, Adcock AF, Kroll DJ, Swanson SM, Wani MC, Pearce CJ, Oberlies NH. Tetrahedron Lett. 2013;54:4300–4302. doi: 10.1016/j.tetlet.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Figueroa M, Raja H, Falkinham JO, Adcock AF, Kroll DJ, Wani MC, Pearce CJ, Oberlies NH. J Nat Prod. 2013;76:1007–1015. doi: 10.1021/np3008842. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Raja HA, Oberlies NH, El-Elimat T, Miller AN, Zelski SE, Shearer CA. Mycoscience. 2013;54:353–361. [Google Scholar]; d) Raja HA, Oberlies NH, Figueroa M, Tanaka K, Hirayama K, Hashimoto A, Miller AN, Zelski SE, Shearer CA. Mycologia. 2013;105:959–976. doi: 10.3852/12-313. [DOI] [PubMed] [Google Scholar]
- 36.a) El-Elimat T, Raja HA, Graf TN, Faeth SH, Cech NB, Oberlies NH. J Nat Prod. 2014;77:193–199. doi: 10.1021/np400955q. [DOI] [PubMed] [Google Scholar]; b) Figueroa M, Jarmusch AK, Raja HA, El-Elimat T, Kavanaugh JS, Horswill AR, Cooks RG, Cech NB, Oberlies NH. J Nat Prod. 2014;77:1351–1358. doi: 10.1021/np5000704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.a) de Gruyter J, Woudenberg JH, Aveskamp MM, Verkley GJ, Groenewald JZ, Crous PW. Mycologia. 2010;102:1066–1081. doi: 10.3852/09-240. [DOI] [PubMed] [Google Scholar]; b) Quaedvlieg W, Verkley GJ, Shin HD, Barreto RW, Alfenas AC, Swart WJ, Groenewald JZ, Crous PW. Stud Mycol. 2013;75:307–390. doi: 10.3114/sim0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frisch MJTGW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota KFR, Hasegawa J, Ishida MNT, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, ReVision B.02. Gaussian Inc; Pittsburgh, PA: 2003. [Google Scholar]
- 39.Rae J, Creighton C, Meck J, Haddad B, Johnson M. Breast Cancer Res Treat. 2007;104:13–19. doi: 10.1007/s10549-006-9392-8. [DOI] [PubMed] [Google Scholar]
- 40.Leibovitz A, Stinson JC, McCombs WB, McCoy CE, Mazur KC, Mabry ND. Cancer Res. 1976;36:4562–4569. [PubMed] [Google Scholar]
- 41.a) El-Elimat T, Figueroa M, Raja HA, Swanson SM, Falkinham JO, Lucas DM, Grever MR, Wani MC, Pearce CJ, Oberlies NH. J Antibiot. 2014 doi: 10.1038/ja.2014.1125. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) El-Elimat T, Raja HA, Falkinham JO, Day CS, Oberlies NH. J Nat Prod. 2014 doi: 10.1021/np500497r. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.