

Abstract
Nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatis (NASH) are becoming the dominant liver diseases in the US and Western World. Extensive work is being done to diagnose, understand, and explore the pathogenesis of these multivariable complex diseases. Recently a new avenue of biologic regulation is being explored. MicroRNAs are non-coding RNAs that modulate the expression of multiple genes and have been implicated in multiple diseases. Recently there is a growing body of evidence supporting a significant role of microRNAs in NAFLD pathogenesis and progression to NASH, and hinting at their use as targets, biomarkers and potential therapeutic tools. This review is designed to highlight some of the recent work on a few of the key microRNAs involved in the pathogenesis of NAFLD and NASH.
Keywords: Nonalcoholic fatty liver disease, NAFLD, nonalcoholic steatohepatis, biologic regulation, biomarkers, therapeutic tools, pathogenesis, micro-RNA, miR-122, miR-33a, miR-34a, miR-24
Introduction
Nonalcoholic fatty liver disease (NAFLD) encompasses a spectrum of disease that spans from isolated fatty liver (IFL) to nonalcoholic steatohepatitis (NASH) to cirrhosis and hepatocellular cancer (HCC). NASH can be defined by necro-inflammation with hepatocyte injury and ballooning, along with a variable amount of fibrosis and potential for progression to cirrhosis. NALFD is considered by most to be the hepatic manifestation of the metabolic syndrome, which is defined by the presence of three or more of the following: visceral obesity, elevated fasting plasma glucose, hypertension, hypertriglyceridemia, or low high-density lipoprotein levels [1].
Global NAFLD prevalence rates ranges from 2.8% to 46% [2-3]. With time, NAFLD has become the most common cause of chronic liver disease in the United States and other western countries, and its prevalence has mirrored the rising obesity and diabetes mellitus (DM) epidemics. Recent global analysis estimates 1.6 billion adults are overweight with a body mass index (BMI) of 25 kg/m2 or greater, and 500 million adults are obese with a BMI of 30 kg/m2 or greater [4]. In the United States, approximately 33.8% of the population is obese and 10.6% have type 2 DM [5].
The National Health and Nutrition Examination Surveys (NHANES) performed in the United States from 1988 to 2008 showed stable prevalence rates for particular chronic liver diseases, such as hepatitis B, hepatitis C, and alcoholic liver disease. Recent advances in the treatment of HCV will likely lessen its prevalence and impact on public health [6]. However, the prevalence of NAFLD dramatically increased from 5.5% to 11%, in the same time period. Overall, NAFLD represented approximately 47% of all chronic liver disease in 1988, rising to 75% by 2008 [7].
Estimating the true incidence and prevalence of NAFLD, specifically NASH, remains a challenge due both to the asymptomatic presentation of the disease as well as the lack of accurate and noninvasive diagnostic measurements. The prevalence of NAFLD was first estimated to be 36% in lean patients and 72% in obese patients in 1990 via autopsy. The prevalence of NASH in this same cohort was found to be 2.7% in lean patients and 18.5% in obese patients [8]. The evaluation of patients undergoing bariatric surgery confirmed the high prevalence rates among obese populations in which the prevalence of NAFLD and NASH was found to be 91% and 37%, respectively [9]. Of note, although NAFLD is strongly associated with obesity and other metabolic syndrome components, it can occur in patients without obesity and obesity does not portend the presence of NAFLD.
What is especially concerning, NASH may progress to cirrhosis from any stage [10]. After the development of cirrhosis the patient is at risk for development of hepatocellular carcinoma (HCC) [11] and liver-related mortality [12]. Even without the development of NASH obesity alone is associated to the risk of developing HCC. Large meta-analysis of cohort studies investigating non-viral causes of HCC, show a 90% increased risk of HCC in obese patients [13, 14]. This has been suggested as a major cause of the growing rates of HCC in developed countries. In particular, the annual incidence of HCC over the last two decades in the United States has increased 80% [15-17], The association between NASH and HCC represents a growing area of study and concern as metabolic syndrome and obesity rates continue to rise.
The pathogenesis of NASH may be inherently involved with the metabolic syndrome and its associated insulin resistance, both of which are thought to be proinflammatory processes [18]. For example, obesity is frequently linked to type-II diabetes and consequently, to hyperinsulinemia, which leads to increased insulin-like factor 1 (IGF-1) and elevated production of sex steroids and cytokines. In 2010, it was reported that obesity is associated with a chronic inflammatory state characterized by the release of IL-6 and TNFα, two well-known inflammation promoting cytokines [19]. However, the cause(s) of NAFLD and the progression of NAFLD to NASH have yet to be defined.
Further hindering the study of NAFLD and NASH is the difficulty in determining the presence of NASH by non-invasive means. To that end multiple avenues investigating different markers and pathways associated with the development of NASH are being investigated. One of the promising possible mechanisms in the development of NASH is altered expression of micro ribonucleic acids (microRNAs). This review will discuss the current breakthroughs in the investigation of microRNAs and their relation to the development of NASH. After covering the background of how microRNAs function we will discuss new data relating to miR-122, miR-33, miR-34a and miR-24.
The Significance of microRNA in Human disease
Investigations of the human transcriptome following completion of the Human Genome Project in 2003 revealed that our genome encodes non-coding RNAs. First described in 1993[20], microRNAs play an important role in posttranscriptional gene-expression regulation [21].
MicroRNAs are thought to fine-tune entire intracellular molecular cascades such as intracellular signaling [22]. MicroRNA networks are differentially expressed: microRNA expression is organ/tissue specific [23] and is differentially altered during disease states. Thus, induction or inhibition of microRNA expression may provide unique therapeutic tools [24, 25]. Aberrant expression of microRNAs in liver tissue has been implicated in the progression of liver fibrosis, and hepatocarcinogenesis [26-28].
MicroRNAs are small non-coding RNAs 21-25 nucleotides in size that play an important role in the regulation of gene expression. They are produced as a pre-microRNA and exported from the nucleus to the cytoplasm where they are cleaved by the endoribonuclease called Dicer. During this process the cleaved portion(s) are unwound to single strands and loaded into the RNA-induced silencing complex (RISC). This microRNA-RISC complex can interact with the 3′-end untranslated region (3′UTR) of the target gene's messenger-RNA resulting in the suppression of mRNA translation or direct degradation of target mRNA [29]. It has been found that a single microRNA can have several target genes, allowing it to play a broad role in gene regulation.
MicroRNAs and mRNA can be released from cells and carried by exosomes produced from the endoplasmic reticulum [30]. Recent work has demonstrated that intercellular signaling performed by the exosome is through the action of microRNA. From the donor cell, microRNA-containing exosomes are released through ceramide-dependent secretory machinery and transferred to a receiving target cell [31]. The transfer can occur through actively secreted micro vesicles, as demonstrated in human blood and cell cultures [32]. Virsuses such as EBV may produce microRNAs themselves that are then brought to target cells by exosomes where then modulate the immune response [33]. In the setting of liver disease, HCC has been demonstrated to use microRNA exchange via exosomes in intercellular communication [34].
Since their discovery microRNAs have been investigated for possible connections in human disease. Alterations in their expression patterns have been seen in multiple disease states compared to normal tissues and serum. In particular changes in microRNA expression in heart disease, sepsis, malignancies, and autoimmune diseases (reviewed in [35]), have suggested a new avenue of research and targets for treating human disease. This promising new field has propelled research and opened the possibility that stable microRNAs which are detectable in serum and plasma may serve as biomarkers for early disease states or be non-invasive means of determining the severity of disease [36,37]. In particular, miR-122, miR-33a, miR-34a, and miR-24 have shown recent promise as markers for hepatic inflammation in the setting of NASH/NAFLD or as factors contributing to or involved in the development of NASH/NAFLD.
MiR-122
In 2008, expression profiling studies of patients identified miR-122 as differentially expressed after comparing normal patients and NASH patients' microRNA profiles, indicating a possible role in the pathogenesis of NASH [38]. MiR-122 has received considerable attention as a biomarker of liver disease because it comprises approximately 70% of microRNAs in the liver [39,40]. In fact compared to the rest of the body miR-122 has been found to be the most abundant in the liver, and is released into circulation during hepatocyte damage [41, 42].
In the liver, increased expression of miR-122 is modulated by multiple transcription factors involved in hepatocyte development and regulation such as HNF1A, HNF3A and HNF3B [43]. Within liver tissue miR-122, has been found to be a key regulator of cholesterol and fatty-acid metabolism[44]. Mir-122 has also been found to be increased in multiple disease states including hepatitis B and C infections, drug-induced liver injury, and NAFLD [38,44-50]. Due to its increased expression in a broad range of diseases with necro-inflammatory activity, miR-122 has been suggested as a serum or tissue marker of general hepatic inflammation. Unfortunately, the circulating level of mirR-122 dose not correlate with fibrosis stage and functional capacity of the liver and cannot serve as a non-invasive means of staging chronic liver disease [46].
In the setting of acute inflammation miR-122 shows more promise. It has been found that circulating levels of miR-122 correlated with the age of patients with HBV acute-on-chronic liver failure [50,51]. Two independent studies addressing acetaminophen (APAP) toxicity, a common toxin precipitating DILI, in experimental murine models identified miR-122 and miR-192 as potential circulating biomarkers in this setting of acute toxicity [52-53].
Most recently, a study demonstrated miR-122 serum levels were robustly increased early on in a diet-induced model of NAFLD and correlated with ALT and AST levels. Though most work has supported miR-122 as a broad marker for liver inflammation, this work supports the notion that miR-122 correlates to and can indicate inflammation in the setting of NASH [54].
One function of miR-122 is post-transcriptional regulation of CYP7A1, the rate-limiting enzyme controlling bile acid synthesis in human hepatocytes [55]. Recent studies showed that miR-122 knockout mice accumulate triacylglycerol in the liver, due to an up regulation of enzymes responsible for triacylglycerol synthesis and storage [56].
Within this same miR-122 knock out model, the microRNA was also found to regulate fibrogenic factors, such as Kruppel-like factor 6 that targets transforming growth factor β1. When miR-122 is increased there is an activation of hepatic stellate cells and fibrogenic processes. Also coordinated role for miR-122 in liver health is supported the development of steatosis and abnormal levels of very-low density lipoproteins and high-density lipoproteins, in miR-122 knockout mice [56].
MiR-33a
MiR-33a is another microRNA that has been found to be involved in bile acid regulation, in addition to fatty acid oxidation and cholesterol homeostasis [57, 58]. When cellular cholesterol levels decrease, miR-33a expression is co-induced with its host gene sterol regulatory element-binding protein 2 (SREBP2) mRNA. MiR-33a silencing studies demonstrated regression of atherosclerosis in mice, which suggests that miR-33a acts in synergy with SREBP2 to regulate cholesterol homeostasis [59]. Unfortunately, this decrease in atherosclerosis comes with the price of increased intrahepatic cholesterol. It was found when SREBP2 and miR-33a are activated; hepatic efflux transporters and bile acid synthesis are down regulated [60]. This was confirmed in work with hepatic stellate cells exposed to free cholesterol, which resulted in enhanced SREBP2 and miR-33a signaling with suppression of peroxisome proliferator-activated receptor γ signaling. This altered signaling results increased free cholesterol in the hepatic stellate cells, leading TGFβ-induced activation and exaggerated liver fibrosis in NASH [61]. The picture is complex however; miR-33a knockout mice develop obesity and liver steatosis, possibly due to expression of SREBP1 which was enhanced in the absence of miR-33a [62]. MiR-33a and -33b have been also described to inhibit genes involved in fatty acid metabolism and insulin signaling in hepatocytes [63], further implying their pathogenic role during NAFLD.
MiR-34a
As previously discussed, expression profiling studies have identified several differentially expressed microRNAs, including miR-21, miR-34a, miR-122, and miR-155, in human [37] and mouse NASH, [64-66] and that the altered expression of these microRNAs suggest a significant role in the pathogenesis of NASH. However, despite some recent success in the investigation and clinical management of NASH, major gaps remain in our understanding. This includes our limited understanding of the biochemical etiology, the broad spectrum of its presentation, and the basic process of the diseases progression from NAFLD to NASH [67-68]. In particular whether there is different susceptibility of certain patients or tissues to progress to NASH, and could this susceptibility be associated with an altered expression of microRNAs.
MiR-34a has been implicated as a central factor in the development of NASH. Past work has shown miR-34a involvement in cell maintenance through activity and activation of senescence, cell cycle arrest, and apoptosis [69]. MiR-34a induction of apoptosis has been theorized to involve interactions with target proteins in the proper biological setting. The best characterized target of miR-34a is Sirtuin 1 (SIRT1), a NAD-dependent deacetylase that modulates apoptosis in the context of oxidative stress and genomic damage [70]. Specifically, SIRT1 decreases p53 activity. When upregulated, miR-34a suppresses SIRT1, thereby increasing p53 acetylation and transcription and in turn leads to the induction of pro-apoptotic genes such as PUMA. This effect on apoptosis may be partially due to miR-34a suppression of KLF4 (Kruppel-like factor 4) [71], which is transcriptionally activated by p53 following DNA damage [72] and causes cell cycle arrest at both the G1/S and G2/M boundaries [73,74].
This miR-34a/SIRT1/p53 pro-apoptotic pathway in NAFLD represents a positive feedback loop whereby miR-34 and similar microRNAs are direct transcriptional targets upregulated by p53 [75]. Other work has observed that the inflammation in human liver that occurs with NAFLD and NASH is paralleled with increases in p53 [76]. This has also been supported by p53 activation of mitochondrial pathways of apoptosis in an animal model of NASH [77]. Together these data suggests a key role of miR-34a in hepatocyte apoptosis in the pathogenesis of NAFLD and NASH. Inhibition of miR-34a and p53 activity, or preserving SIRT1 function are logical therapeutic approaches that may benefit patients who have NAFLD and NASH.
One such advance has been made in modulating the miR-34a/SIRT1/p53pro-apoptotic pathway through treatment with ursodeoxycholic acid (UDCA) [78]. Rui et. al. treated rat hepatocytes with UDCA resulting in increased SIRT1 expression and subsequent decreased acetylation of p53. As expected by the reduced p53 activity, apoptosis was also reduced. This protective activity of UDCA was effective even in a model of miR-34a overexpression. Their work indicates that the mechanism of UDCA and its inhibition of miR-34a is partly due to the prevention of p53 transactivation, and thus preventing the positive feedback loop. In turn, the decrease of miR-34a (by reducing p53 positive feedback) leads to increased SIRT1.
Recent results have also demonstrated that miR-34a expression in both human liver and human serum is increased in patients with NAFLD. Importantly a correlation was made of the liver miR-34a level and the severity of NAFLD [79]. Due to both of these factors, miR-34a may not only represent a therapeutic target in the treatment of NAFLD progression, but may also be useful as a non-invasive biomarker to assess disease progression. However, an association of increased circulating miR-34a with type 2 diabetes may complicate the use of this marker [80].
MiR-24
One group has started to investigate miR-24 in the development of NASH stemming from hepatocyte-specific microRNA profiling studies in mouse livers. They showed that miR-24 is highly and specifically expressed in hepatocytes [81]. Ng et. al. performed this study by microRNA profiling of livers from hepatocyte specific Dicer1 knockout and wild-type mice. The microRNAs whose expression was at least 1.7 fold higher in WT than Dicer1 knockout mice were defined as hepatocyte-specific microRNAs. This experiment also confirmed the importance of pro-inflammatory miR-122 and miR-192, which were also enriched in this study.
They then confirmed the importance of miR-24 in NASH through high fat diet (HFD) treatment of C57Bl/6 mice, which demonstrated up regulation of miR-24. MiR-24 up regulation in the setting of fatty acid exposure was then confirmed with Oleic acid treatment of primary human hepatocyte and HepG2 cell cultures. Both cell cultures also demonstrated increased miR-24 expression.
Using the PubMed GEO Databse, five normal and eight human NAFLD/NASH liver samples were compared to identify possible targets of miR-24. With this analysis 411 down-regulated genes were identified in human fatty livers. The 3′ UTR of mRNA of these potential targets were then compared with the conserved seed region of miR-24. This led the group to identify 48 potential target genes that have binding motifs for miR-24. These 48 targets were cross-referenced with the 411 down-regulated genes found in the control fatty liver array comparison, identifying 3 genes Insig1, KFL6 (Kruppel-like factor 6) and CXADR (Coxsackie virus and adenovirus receptor).
Insig1 is a polytopic membrane protein of the endoplasmic reticulum (ER) that regulates lipid synthesis by retaining sterol regulatory element–binding proteins (SREBPs) in the ER and preventing their proteolytic activation in the Golgi apparatus [82-84]. The movement of SREBPs from ER to the Golgi complex is a central event in lipid homeostasis in animal cells [85-87]. Significantly, 18-20 SREBPs have been identified as membrane-bound transcription factors that activate genes encoding enzymes required for synthesis of cholesterol and triglycerides [86-87].
The three SREBP isoforms, SREBP1a, SREBP1c and SREBP2, have different roles in lipid synthesis. SREBP1c is involved in fatty acid and triglyceride synthesis, whereas SREBP2 is relatively specific to cholesterol synthesis [87]. Immediately after their synthesis, SREBPs bind to SCAP (SREBP cleavage-activating protein). When Insig1 protein levels are low, SCAPs escort SREBPs to the Golgi where they are processed and released into the cytosol, and can then enter the nucleus and activate transcription of lipogenic genes [86].
Next, they assessed the functional contribution of increased Insig1 expression to the development of fatty liver by reducing miR-24 expression in obese mice. They synthesized LNA (locked nucleic acid) anti-miR-24 ASO specifically targeting miR-24. To confirm that the phenotype observed in miR-24-ASO-injected mice was due to specific microRNA deficiency, and not toxicity caused by the ASO, they generated miR-24- mis-matched-anti-sense oligo (miR-24-MM-ASO), a control ASO that differs from microRNAs in 4 mismatched base pairs. C57Bl/6 mice, which had been on a HFD for 8 weeks, were injected with either miR-24-ASO or miR-24-MM-ASO for 4 weeks. They observed an 88% reduction of hepatic miR-24 expression in mice that received miR-24-ASO compared to miR-24-MM-ASO, and a three-fold increase of Insig1, as revealed by qRT-PCR and immunostaining [81].
MiR-24-ASO treatment had no effect on body and liver weight. However, antagonizing miR-24 significantly reduced high levels of triglycerides and cholesterol in liver and triglycerides in plasma in HFD animals treated with miR-24-ASO, in contrast to plasma total cholesterol levels. Nile-Red and H&E staining further confirmed that miR-24 knockdown reduced hepatic lipid accumulation in livers of HFD-treated mice. These findings suggest that the crosstalk between miR-24 and Insig1 plays an important role in NAFLD and hypertriglyceridemia, and miR-24-ASO may be a potential therapeutic target for these disorders. This is supported by past work where the overexpression of Insig1 in liver inhibits lipogenesis and knockout of Insig1 leads to increased total content of both liver and plasma triglycerides [88-89], suggesting that the relationship between miR-24 and Insig1 may play an important role in the development of NAFLD and hyperlipidemia.
MiR-24 has been shown to be up-regulated in NAFLD/NASH and hepatocyte cell culture models of fatty acid exposure. Up regulation of miR-24 leads to deceased Insig1, increased nuclear SREBP1c andSREBP2, and subsequently elevated lipid accumulation in primary human hepatocytes and HepG2 cells. In contrast, antagonizing miR-24 leads to the opposite and more potentially therapeutic effects. These data suggest miR-24 may have a significant role in the development of NAFLD/NASH and so far shows promise as a therapeutic target.
A summary of the discussed micro-RNAs can be found in Table 1. Figure 1 is a representation of the expression and activity of the discussed micro-RNAs.
Table 1. Selected microRNA Activities.
| MicroRNA | Human disease | Activity In NAFLD/NASH | References |
|---|---|---|---|
| miR-24 | NASH | Insig1 down regulation | Ng et al, 2014 |
| miR-33 | NASH | Bile acid regulation | Allen et al, 2012 |
| miR-34a | Elevated in HCV, NAFLD and NASH | Senescence, cell cycle arrest, and apoptosis-by suppressing SIRT1 | Panasiuk et al, 2006 Najafi-Shoushtari et al, 2010 |
| miR-122 | Elevated in HBV, HCV and NAFLD | Correlates with necro-inflammatory activity. Regulate CYP7A1-bile acid synthesis. Also loss of miR-122 may lead to increased fibrosis through Klf6. | Bihrer et al, 2011 Xu et al, 2011 Cermelli et al, 2011 |
Figure 1.
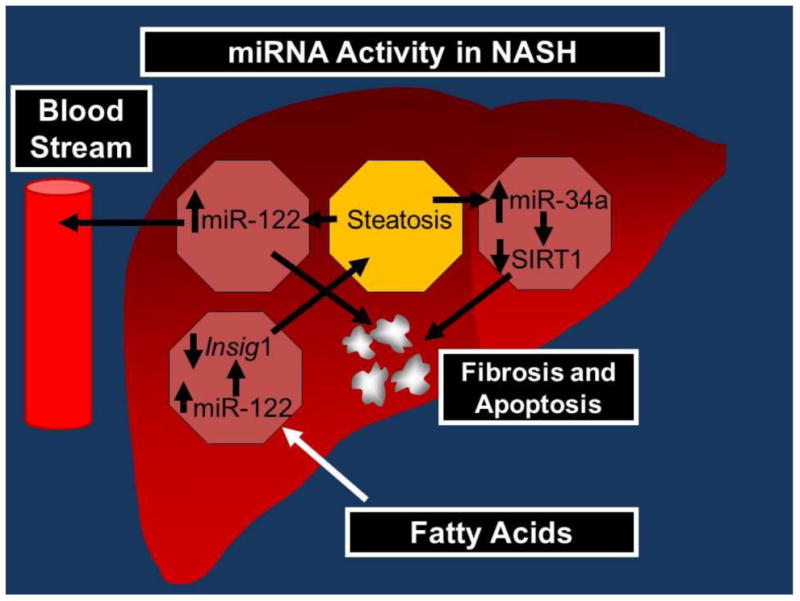
MiR-122 increases in expression with steatosis which leads to Fibrosis, while being detected in the patient's serum. MiR-24 increases with hapatic exposure to Fatty acids, resulting in increased Insig1 expression which may increase steatosis. Steatosis may also increase miR-34a expression which then decreases SIRT1, leading to increased fibrosis.
Conclusion
The incidence of NAFLD and NASH is increasing dramatically in the world population with the increase of metabolic syndrome across the globe. As the prevalence of more western high fat diets spread, both NASH and metabolic syndrome will continue to become a major concern in human health. Furthermore, with the development of more effective HBV and HCV treatments, NASH and NAFLD will become the most dominant cause of liver disease throughout the world. These developments underscore the critical need to be able to detect and track the severity of NASH and NAFLD in patients, through non-invasive means. With the discovery and growing knowledge of microRNAs we may be able to find better non-invasive markers of the disease to detect it earlier and to investigate therapeutic interventions. Furthermore, microRNAs show promise as possible targets for those interventions.
Acknowledgments
This work was supported, in whole or in part, by NIH Grants R01 DK081450-03 and T32 07150-37 to AJS
Footnotes
Table included with summary of the discussed microRNA originally produced Image provided originally drawn and produced.
Compliance with Ethics Guidelines
Conflict of interest: Robert Vincent and Arun Sanyal have no conflict of interest.
Human and Animal Rights and Informed Consent: Subjects approved by the institutional review board and with informed consent.
References
Papers of particular interest, published recently, have been highlighted as:
* Of importance
** Of major importance
- 1.Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–5. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 2.Lazo M, Clark JM. The epidemiology of nonalcoholic fatty liver disease: a global perspective. Semin Liver Dis. 2008;28:339–50. doi: 10.1055/s-0028-1091978. [DOI] [PubMed] [Google Scholar]
- 3•.Lazo M, Hernandez R, Eberhardt MS, et al. Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutritional Examination Survey, 1988-1994. Am J Epidemiol. 2013;178:38–45. doi: 10.1093/aje/kws448. Recent epidemiology study of the NASH epidemic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finucane MM, Stevens GA, Cowan MJ, et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet. 2011;377(9765):557–67. doi: 10.1016/S0140-6736(10)62037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flegal KM, Carroll MD, Ogden CL, et al. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303:235–41. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 6.Liang TJ, Ghany MG. Therapy of Hepatitis C - Back to the Future. NEJM. 2014 May; doi: 10.1056/NEJMe1403619. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Younossi ZM, Stepanova M, Afendy M, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol. 2011;9(6):524–30. doi: 10.1016/j.cgh.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 8.Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatology. 1990;12:1106–10. doi: 10.1002/hep.1840120505. [DOI] [PubMed] [Google Scholar]
- 9.Machado M, Marques-Vidal P, Cortez-Pinto H. Hepatic histology in obese patients undergoing bariatric surgery. J Hepatol. 2006;45:600–6. doi: 10.1016/j.jhep.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 10.Malik SM, deVera ME, Fontes P, et al. Outcome after liver transplantation for NASH cirrhosis. Am J Transplant. 2009;7:782–93. doi: 10.1111/j.1600-6143.2009.02590.x. [DOI] [PubMed] [Google Scholar]
- 11.Siegel AB, Zhu AX. Metabolic syndrome and hepatocellular carcinoma. Cancer. 2009;115:5651–61. doi: 10.1002/cncr.24687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ong JP, Pitts A, Younossi ZM. Increased overall mortality and liver-related mortality in non-alcoholic fatty liver disease. J Hepatol. 2008;49:608–12. doi: 10.1016/j.jhep.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 13.Blonski W, Kotlyar DS, Forde KA. Non-viral causes of hepatocellular carcinoma. World J Gastroenterol. 2010;16(29):3603–3615. doi: 10.3748/wjg.v16.i29.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2006;40(Suppl 1):S5–S10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- 15.Bosch FX, Ribes J, Diaz M, Cléries R. Primary liver cancer: world-wide incidence and trends. Gastroenterology. 2004;127:S3–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 16.El-Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340(10):745–750. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- 17.Gomaa AI, Khan SA, Toledano MB, Waked I, Taylor-Robinson SD. Hepatocellular carcinoma: epidemiology, risk factors and pathogenesis. World J Gastroenterol. 2008;14(27):4300–4308. doi: 10.3748/wjg.14.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brunt E, Kleiner DE, Wilson LA, Belt P, Neuschwander Tetri BA. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology. 2011;53:810–820. doi: 10.1002/hep.24127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee RC, Feinbaum RL, Ambros V. The C elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 21.Lagos-quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 22.Berezikov E. Evolution of microRNA diversity and regulation in animals. Nat Rev Genet. 2011;12:846–860. doi: 10.1038/nrg3079. [DOI] [PubMed] [Google Scholar]
- 23.Lagos-quintana M, Rauhut R, et al. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 24.Davidson BL, Mccray PB., Jr Current prospects for RNA interference-based therapies. Nat Rev Genet. 2011;12:329–340. doi: 10.1038/nrg2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaucsa'r T, Ra'c ZZ, Hamar P. Posttranscriptional gene-expression regulation by micro RNA (miRNA) network in renal diseaseAdv. Drug Deliv Rev. 2010;30:1390–1401. doi: 10.1016/j.addr.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Murakami Y, Toyoda H, Tanaka M, Kuroda M, Harada Y, et al. The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS One. 2011;6:e16081. doi: 10.1371/journal.pone.0016081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, et al. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006;25:2537–2545. doi: 10.1038/sj.onc.1209283. [DOI] [PubMed] [Google Scholar]
- 28.Braconi C, Henry JC, Kogure T, Schmittgen T, Patel T. The role of microRNAs in human liver cancers. Semin Oncol. 2011;38:752–763. doi: 10.1053/j.seminoncol.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He L, Hannon G. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(7):522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 30.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, et al. Exosomemediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 31.Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, et al. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010;285:17442–17452. doi: 10.1074/jbc.M110.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Liu D, Chen X, Li J, Li L, et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol Cell. 2010;39:133–144. doi: 10.1016/j.molcel.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 33.Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A. 2010;107:6328–6333. doi: 10.1073/pnas.0914843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kogure T, Lin WL, Yan IK, Braconi C, Patel T. Intercellular nanovesiclemediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology. 2011;54:1237–1248. doi: 10.1002/hep.24504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kosaka N, Iguchi H, Ochiya T. Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010;101:2087–2092. doi: 10.1111/j.1349-7006.2010.01650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Ba Y, Ma L, Cai X, Yin Y, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18:997–1006. doi: 10.1038/cr.2008.282. [DOI] [PubMed] [Google Scholar]
- 38.Cheung O, Puri P, Eicken C, et al. Nonalcoholic steatohepatitis is associated with altered hepatic microRNA expression. Hepatology. 2008;48:1810–1820. doi: 10.1002/hep.22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 40.Chang J, Nicolas E, Marks D, Sander C, Lerro A, et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106–113. doi: 10.4161/rna.1.2.1066. [DOI] [PubMed] [Google Scholar]
- 41.Szabo G, Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol. 2013 Sep;10(9):542–52. doi: 10.1038/nrgastro.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42•.Trebicka J, et al. Hepatic and serum levels of miR-122 after chronic HCV induced fibrosis. J Hepatol. 2013;58(2):234–9. doi: 10.1016/j.jhep.2012.10.015. Additional support of miR-122 as secreted marker of HCV inflammation. [DOI] [PubMed] [Google Scholar]
- 43.Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28:3526–3536. doi: 10.1038/onc.2009.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esau C, Davis S, Murray SF, Yu XX, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 45.Pogribny IP, Starlard-Davenport A, Tryndyak VP. Difference in expression of hepatic microRNAs miR-29c, miR-34a, miR-155, and miR-200b is associated with strain-specific susceptibility to dietary nonalcoholic steatohepatitis in mice. Lab Invest. 2010;90:1437–1446. doi: 10.1038/labinvest.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bihrer V, Friedrich-Rust M, Kronenberger B, et al. Serum miR-122 as a biomarker of necroinflammation in patients with chronic hepatitis C virus infection. Am J Gastroenterol. 2011;106:1663–1669. doi: 10.1038/ajg.2011.161. [DOI] [PubMed] [Google Scholar]
- 47.Cermelli S, Ruggieri A, Marrero JA, et al. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS One. 2011;6:e23937. doi: 10.1371/journal.pone.0023937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Starkey Lewis PJ, Merz M, et al. Serum microRNA biomarkers for drug-induced liver injury. Clin Pharmacol Ther. 2012;92:291–293. doi: 10.1038/clpt.2012.101. [DOI] [PubMed] [Google Scholar]
- 49.Tryndyak VP, Latendresse JR, Montgomery B, et al. Plasma microRNAs are sensitive indicators of interstrain differences in the severity of liver injury induced in mice by a choline- and folate-deficient diet. Toxicol Appl Pharmacol. 2012;262:52–59. doi: 10.1016/j.taap.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50•.Waidmann O, Bihrer V, Pleli T, et al. Serum microRNA-122 levels in different groups of patients with chronic hepatitis B virus infection. J Viral Hepat. 2012;19:e58–e65. doi: 10.1111/j.1365-2893.2011.01536.x. MiR-122 also as a marker of HCV infection and inflammation. [DOI] [PubMed] [Google Scholar]
- 51.Ji F, Yang B, Peng X, Ding H, You H, et al. Circulating microRNAs in hepatitis B virus-infected patients. J Viral Hepat. 2011;18:e242–251. doi: 10.1111/j.1365-2893.2011.01443.x. [DOI] [PubMed] [Google Scholar]
- 52.Wang K, et al. Circulating microRNAs, potential biomarkers for drug-induced liver injury. Proc Natl Acad Sci U S A. 2009;106(11):4402–7. 34. doi: 10.1073/pnas.0813371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su YW, et al. A panel of serum microRNAs as specific biomarkers for diagnosis of compound- and herb-induced liver injury in rats. PLoS One. 2012;7(5):e37395. doi: 10.1371/journal.pone.0037395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54•.Clarke JD, Sharapova T, Lake AD, et al. Circulating microRNA 122 in the methionine- and choline-deficient mouse model of non-alcoholic steatohepatitis. Appl Toxicol. 2013 Nov 12; doi: 10.1002/jat.2960. Recent mouse diet model of NASH which correlates with miR-122 expression in serum. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song KH, Li T, Owsley E, Chiang JY. A putative role of micro RNA in regulation of cholesterol 7alpha-hydroxylase expression in human hepatocytes. J Lipid Res. 2010;51:2223–33. doi: 10.1194/jlr.M004531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56••.Tsai WC, Hsu SD, Hsu CS, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122:2884–97. doi: 10.1172/JCI63455. The use of knock out 122 mice to investigate its role in liver disease. This knock out had increased fibrosis and tumor formation- partially attributed to the action of a miR-122a target, the Klf6 transcript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–9. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58••.Allen RM, Marquart TJ, Albert CJ, Suchy FJ, et al. miR-33 controls the expression of biliary transporters, and mediates statin- and diet-induced hepatotoxicity. EMBO Mol Med. 2012;4:882–95. doi: 10.1002/emmm.201201228. The first work connected miR-33 with bile and cholesterol metabolism in the liver and the connection to NASH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rayner KJ, Sheedy FJ, Esau CC, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–31. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60•.Li T, Francl JM, Boehme S, Chiang JY. Regulation of cholesterol and bile acid homeostasis by the CYP7A1/SREBP2/miR-33a axis. Hepatology. 2013;58:1111–21. doi: 10.1002/hep.26427. Additional support and detail into the mechanism of mir-33a and bile acid metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61••.Tomita K, Teratani T, Suzuki T, et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology. 2014;59:154–69. doi: 10.1002/hep.26604. Evidence of how miR-33 is involved in inflammation that is part of fibrosis in NASH. [DOI] [PubMed] [Google Scholar]
- 62•.Horie T, Nishino T, Baba O, et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat Commun. 2013;4:2883. doi: 10.1038/ncomms3883. Communication on miR-33 and an additional SREBP it modulates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davalos A, Goedeke L, Smibert P, et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci U S A. 2011;108:9232–7. doi: 10.1073/pnas.1102281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang B, Majumder S, Nuovo G, et al. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino-defined diet in C57BL/6 mice. Hepatology. 2009;50:1152–1161. doi: 10.1002/hep.23100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dolganiuc A, Petrasek J, Kodys K, et al. MicroRNA expression profile in Lieber-DeCarli diet-induced alcoholic and methionine choline deficient diet-induced nonalcoholic steatohepatitis models in mice. Alcohol Clin Exp Res. 2009;33:1704–1710. doi: 10.1111/j.1530-0277.2009.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hill-Baskin AE, Markiewski MM, Buchner DA, et al. Diet-induced hepatocellular carcinoma in genetically predisposed mice. Hum Mol Genet. 2009;18:2975–2988. doi: 10.1093/hmg/ddp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marra F, Gastaldelli A, Svegliati Baroni G, et al. Molecular basis and mechanisms of progression of nonalcoholic steatohepatitis. Trends Mol Med. 2008;14:72–81. doi: 10.1016/j.molmed.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 68.Siegel AB, Zhu AX. Metabolic syndrome and hepatocellular carcinoma: two growing epidemics with a potential link. Cancer. 2009;115:5651–5661. doi: 10.1002/cncr.24687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193–199. doi: 10.1038/cdd.2009.56. [DOI] [PubMed] [Google Scholar]
- 70.Yamakuchi M, Ferlito M, Lowenstein CJ. MiR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71••.Chen Q, Lei Li, et al. MiR-34a regulates apoptosis in liver cells by targeting the KLF4 gene. Cellular & Molecular Biology Letters. 2014;19:52–64. doi: 10.2478/s11658-013-0115-y. The research linking miR-34a to apoptosis through KLF4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang W, Shields JM, Sogawa K, et al. The gut-enriched Kruppel-like factor suppresses the activity of the CYP1A1 promoter in a Spl-dependent fashion. J Biol Chem. 1998;273:17917–17925. doi: 10.1074/jbc.273.28.17917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yoon HS, Chen X, Yang VW. Kruppel-like factor 4 mediates p53- dependent G1/S cell cycle arrest in response to DNA damage. J Biol Chem. 2003;278:2101–2105. doi: 10.1074/jbc.M211027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yoon HS, Yang VW. Requirement of Kruppel-like factor 4 in preventing entry into mitosis following DNA damage. J Biol Chem. 2004;279:5035–5041. doi: 10.1074/jbc.M307631200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang TC, Wentzel EA, Kent OA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Panasiuk A, Dzieciol J, Panasiuk B, Prokopowicz D. Expression of p53, Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World J Gastroenterol. 2006;12:6198–6202. doi: 10.3748/wjg.v12.i38.6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Farrell GC, Larter CZ, Hou JY, et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J Gastroenterol Hepatol. 2009;24:443–452. doi: 10.1111/j.1440-1746.2009.05785.x. [DOI] [PubMed] [Google Scholar]
- 78••.Rui E, et al. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. Journal of Hepatology. 2013;58:119–125. doi: 10.1016/j.jhep.2012.08.008. The use of urosodeoxycholic as a way to modulate the miR-34a/SIRT1/p53 apoptotic pathway in NASH. [DOI] [PubMed] [Google Scholar]
- 79.Cermelli S, Ruggieri A, Marrero JA, et al. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS One. 2011;6:e23937. doi: 10.1371/journal.pone.0023937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kong L, Zhu J, et al. Significance of serum microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: a clinical study. Acta Diabetol. 2011 Mar 48;(1):61–9. doi: 10.1007/s00592-010-0226-0. [DOI] [PubMed] [Google Scholar]
- 81••.Ng R, Wu H, Xiao H, et al. Inhibition of miR-24 expression in liver prevents hepatic lipid accumulation and hyperlipidemia. Hepatology. 2014 Mar 28; doi: 10.1002/hep.27153. The primary study of miR-24 demonstrating its connection to NASH but also possible therapeutic benefit when inhibited. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Nat Acad Sci U S A. 2002;99:12753–12758. doi: 10.1073/pnas.162488899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gong Y, Lee JN, Lee PC, et al. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006;3:15–24. doi: 10.1016/j.cmet.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 84.Yang T, Espenshade PJ, Wright ME, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 85.Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Nat Acad Sci U S A. 1999;96:11041–11048. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Edwards PA, Tabor D, Kast HR, Venkateswaran A. Regulation of gene expression by SREBP and SCAP. Biochim Biophys Acta. 2000;1529:103–113. doi: 10.1016/s1388-1981(00)00140-2. [DOI] [PubMed] [Google Scholar]
- 87.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Engelking LJ, Kuriyama H, Hammer RE, et al. Overexpression of Insig-1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin-stimulated lipogenesis. J Clin Invest. 2004;113:1168–1175. doi: 10.1172/JCI20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Engelking LJ, Liang G, Hammer RE, et al. Schoenheimer effect explained–feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J Clin Invest. 2005;115:2489–2498. doi: 10.1172/JCI25614. [DOI] [PMC free article] [PubMed] [Google Scholar]