

Abstract
Sophora moorcroftiana, a Leguminosae shrub species that is restricted to the arid and semi-arid regions of the Qinghai-Tibet Plateau, is an ecologically important foundation species and exhibits substantial drought tolerance in the Plateau. There are no functional genomics resources in public databases for understanding the molecular mechanism underlying the drought tolerance of S. moorcroftiana. Therefore, we performed a large-scale transcriptome sequencing of this species under drought stress using the Illumina sequencing technology. A total of 62,348,602 clean reads were obtained. The assembly of the clean reads resulted in 146,943 transcripts, including 66,026 unigenes. In the assembled sequences, 1534 transcription factors were identified and classified into 23 different common families, and 9040 SSR loci, from di- to hexa-nucleotides, whose repeat number is greater than five, were presented. In addition, we performed a gene expression profiling analysis upon dehydration treatment. The results indicated significant differences in the gene expression profiles among the control, mild stress and severe stress. In total, 4687, 5648 and 5735 genes were identified from the comparison of mild versus control, severe versus control and severe versus mild stress, respectively. Based on the differentially expressed genes, a Gene Ontology annotation analysis indicated many dehydration-relevant categories, including ‘response to water ‘stimulus’ and ‘response to water deprivation’. Meanwhile, the Kyoto Encyclopedia of Genes and Genomes pathway analysis uncovered some important pathways, such as ‘metabolic pathways’ and ‘plant hormone signal transduction’. In addition, the expression patterns of 25 putative genes that are involved in drought tolerance resulting from quantitative real-time PCR were consistent with their transcript abundance changes as identified by RNA-seq. The globally sequenced genes covered a considerable proportion of the S. moorcroftiana transcriptome, and the expression results may be useful to further extend the knowledge on the drought tolerance of this plant species that survives under Plateau conditions.
Introduction
The Qinghai-Tibet Plateau is generally called “the roof of the world” because of its extremely high altitude and extreme environment. The Plateau plays an important role in determining the formation and variation of regional weather and climate in East and South Asia, as well as the Northern Hemisphere atmospheric circulation in general [1]. It was recently discovered that the dry and warming climate and other factors have caused desertification expansion in some portions of the Plateau [2], [3].
Sophora moorcroftiana is an endemic Leguminosae shrub species that is restricted to the arid and semi-arid regions of the Plateau. S. moorcroftiana mainly grows in the middle and upper reaches of the dry valley region of the Yarlung Tsangpo River at high altitudes ranging from 2,800 m to 4,400 m. This species is a unique Sophora characterized in this area by strong drought resistance after long-term adaptation to the local environment and climate. This species is currently the preferred drought-resistant afforestation tree species in the sand land in the Plateau.
With the development of molecular technologies and ‘omics’ tools, studies of the transcriptome and Differential Expression Genes (DEGs) under certain condition have become powerful strategies for the global analysis of plant genes. Using transcriptome and DEG data, the abiotic stress response of Arabidopsis and other non-model plants has been widely studied [4]–[9]. However, until now, no genes have been identified, and no molecular research of this species has been reported, despite the importance of the genus. Considering the large genome size of the plant, the whole genome sequencing of S. moorcroftiana is difficult; therefore, the construction of large EST collections of this species is the most promising approach for providing functional-genomics-level information in S. moorcroftiana.
In this study, in order to identify drought-tolerant genes, potential dehydration-responsive genes were first identified based on Illumina tag-sequencing, and then, the DEGs were screened and further validated by qRT-PCR. This study will be helpful for elucidating the molecular responsive mechanism of S. moorcroftiana to drought stress and for further improving drought resistance by genetic modification in the future.
Results and Discussion
Leaf water potential during drought stress
To evaluate the drought stress conditions, three trees were selected for each treatment, and three biological replicates were set. Water stress was imposed by withholding water until a desired stress condition was reached. The standard used for differentiating the mild drought- stressed and severe drought- stressed trees was the leaf water potential. The potential of control trees was between -0.9 and -1.1 MPa, (2) while the mild drought-stressed wasbetween-1.9 and -2.1 MPa, and (3) trees the severe drought-stressed was between 3.0 and 3.2 MPa. The lower leaves of the severely stressed trees were slightly wilted, while the leaves of the mildly stressed and the control trees experienced normal growth.
Transcriptome sequencing and assembly
Transcriptome sequences are valuable resources, especially for species without a sequenced genome, such as the non-model plant species S. moorcroftiana. These sequences accelerate gene discovery, permitting expression analysis evolutionary genome dynamics studies. In this study, Next-Generation Sequencing enabled the generation of large numbers of sequence reads in a rapid and cost-effective manner. The Illumina sequencing data of S. moorcroftiana were deposited into the NCBI SRA database under the accession number SRP041237. A total of 63,855,218 raw reads were generated. After removing the low-quality reads and trimming off the adapter sequences, 62,348,602 clean reads were obtained. The assembly of the clean reads resulted in 146,943 transcripts, including 66,026 unigenes that ranged from 201 to 17064 bp with an N50 length of 1438 bp (Table 1). The length statistics of the assembled unigenes were displayed (Figure 1). To our knowledge, this is the first report of S. moorcroftiana transcriptome data.
Table 1. Summary of sequences analysis.
| Description | Number |
| Before trimming | |
| Raw reads | 63,855,218 |
| After trimming | |
| Clean reads | 62,348,602 |
| Clean bases (Gb) | 6.24 |
| GC ontent (%) | 40 |
| Q20 percentage (%) | 98.53 |
| After assembly | |
| Unigenes | 66,026 |
| Min length (bp) | 201 |
| Max length (bp) | 17,064 |
| Average length (bp) | 773 |
| N50 (bp) | 1438 |
| N90 (bp) | 291 |
Figure 1. Length distribution of the assembled unigenes.

Blast analysis
To predict and analyze the function of the assembled transcripts, non-redundant sequences were submitted to a BLASTx search against the following databases: Nr (NCBI non-redundant protein sequences), Nt (NCBI non-redundant nucleotide sequences), Pfam (Protein family), Swiss-Prot (a manually annotated and reviewed protein sequence database), GO (Gene Ontology), KOG (eukaryotic orthologous groups) and KEGG (Kyoto Encyclopedia of Genes and Genomes). The unigenes were subjected to public databases for similarity searching. Among these unigenes, 31,117 (47.12%), 20,646 (31.26%) and 21,493 (32.55%) unigenes showed identity with sequences in the NCBI Nr, Nt and SwissProt databases, respectively, with an e value <1e-5.
Functional annotation and pathway assignment
Gene Ontology (GO) assignments were used to classify the functions of the predicted S. moorcroftiana genes; 23,310 (35.3%) unigenes were classified into three major functional categories (Biological Process, Cellular Component and Molecular Function) and 47 subcategories (Figure 2). In terms of Biological Processes, ‘metabolic processes’ and ‘cellular processes’ were the top two GO terms, indicating that the leaves were undergoing extensive metabolic activities, which is consistent with the results from the leaves of another Leguminosae genus Prosopis alba in Argentina [10]. In terms of Molecular Function, the top three GO terms were related to the following categories: ‘binding’, ‘catalytic activity’ and ‘transporters activity’. A detailed analysis of the Cellular Component showed that the most representative categories were ‘cell part’, ‘organelle’ and ‘macromolecular region part’. These results are similar with the GO assignments of transcriptome data from abiotic-tolerant Reaumuria trigyna, Prosopis alba, Anthurium, and Chorispora bungeana [9]–[12]. To classify the orthologous gene products, 11,801 (17.87%) unigenes were subdivided into 26 eukaryotic Orthologous Groups (KOG) classifications (Figure 3). Among these classifications, the cluster of ‘general function prediction only’ represented the largest group, followed by ‘post-translational modification’, ‘protein turnover’, ‘chaperon’, ‘translation’ and ‘signal transduction’. The two categories involving ‘cell motility’ and ‘unnamed protein’ represented the smallest KOG classifications (Figure 3). To identify the biological pathways in the annotated sequences using the Kyoto Encyclopedia of Genes and Genomes (KEGG), the assembled unigenes were assigned to five specific pathways, including Cellular Processes, Environmental Information Processing, Genetic Information Processing, Metabolism, and Organism Systems (Figure 4). A summary of the unigenes annotation is given in Table S1.
Figure 2. Gene categorization of the assembled unigenes.

The unigenes with the best BLAST hits were aligned. All 23,310 unigenes were classified into three major functional categories and 47 sub-categories. The right Y-axis represents the number of genes in a category; the left Y-axis indicates the percentage of a specific category of genes in each main category.
Figure 3. KOG classification of the putative proteins.
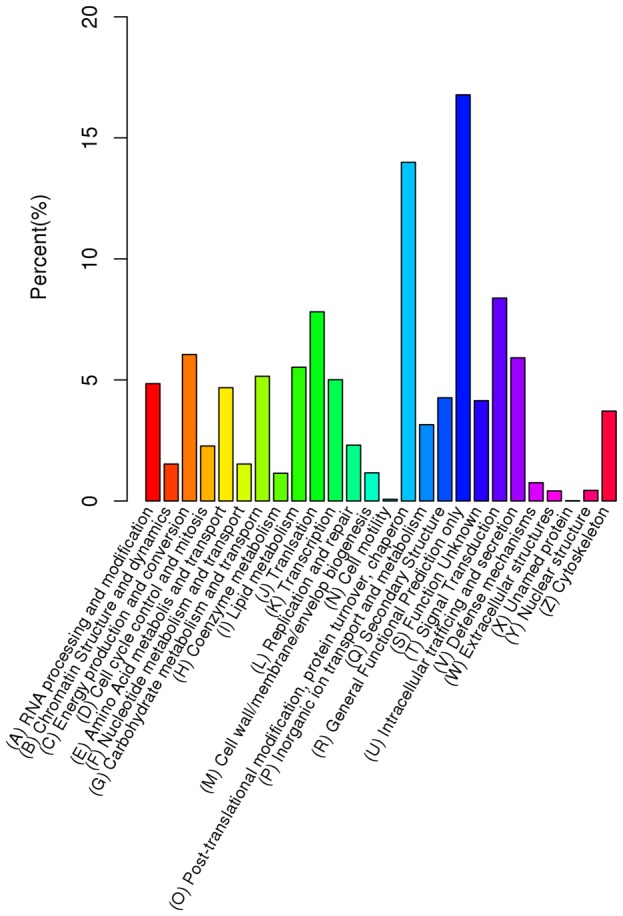
All 11,801 unigenes were subdivided into 26 eukaryotic Orthologous Group (KOG) classifications. The Y-axis indicates the number of unigenes in a specific functional cluster.
Figure 4. Histogram presentation of the KEGG classification of the annotated transcripts.

The left Y-axis indicates the KEGG pathway. The right Y-axis indicates the sub-branches. A, cellular processes; B, environmental information processing; C, genetic information processing; D, metabolism; E, organismal systems. The X-axis indicates the percentage of unigenes that were assigned to a specific pathway.
Transcription factors
Transcription factors (TFs) are important upstream regulatory proteins and play significant roles in plant responses to abiotic and biotic stresses. In this study, 1534 TFs were identified and classified into 23 different common families by searching from unique transcripts (Figure 5). The largest group of TFs was the bZIP family (160, 10.43%), followed by MYB (115, 7.5%), bHLH (107, 6.98%), zinc finger (103, 6.71%), and WRKY (103, 6.71%). This result is similar to the results from the Arachis hypogaea transcriptome, whose largest group is bZIP, followed by MYB, NAC, bHLH, AP2-EREB and WRKY [13], and similar to the results from the Chrysanthemum morifolium transcriptome, whose largest group is MYB, followed by C3H, AP2, C2H2, bHLH and the WRKY big groups [14]. These results further suggest that bZIP, MYB, bLHL and WRKY TFs are superfamilies in plants, and expression of most numbers are affected by abiotic stress [4], [8], [14].
Figure 5. Number of unique transcripts that were annotated as transcription factors.

SSR identification
SSRs have much higher levels of polymorphisms than do most other marker systems due to their codominance, hypervariability, high reproducibility and abundance in eukaryotic genomes. The utility of SSRs in genetic studies is well established [15], [16]. In this study, the EST-SSRs in the transcriptome of S. moorcroftiana were discovered based on an analysis of the assembled contig templates. A total of 12,086 distinct SSR loci were identified. Among these loci, SSR loci from di- to hexa-nucleotide, whose repeat number is greater than five, accounted for 9040. Most of these satellites are di- or tri-nucleotide motifs, being 2182 and 2199, respectively (Table 2). The AG/CT was the most frequent di-nucleotide SSR repeat and accounted for 1326, and the AAG/CTT was the most frequent tri-nucleotide SSR repeat and accounted for 568. Details of the identified SSRs are listed in Table S2. Likewise, AG/CT and AAG/CTT were the most frequent di- and tri-nucleotide SSR repeats in Ammopiptanthus mongolicus [17], which might be due to the fact that both S. moorcroftiana and A. mongolicus belong to legume plants and may contain similar SSR characteristics in their genomes.
Table 2. Summary of EST-SSR detected from transcriptome data.
| Motif length | Repeat numbers | Total(%) | |||||||
| 5 | 6 | 7 | 8 | 9 | 10 | >10 | |||
| Di- | 762 | 453 | 354 | 267 | 246 | 92 | 8 | 2182 | 48.27% |
| Tri- | 1335 | 565 | 264 | 33 | - | - | 2 | 2199 | 48.65% |
| Tetra- | 96 | 19 | - | - | - | 2 | 1 | 118 | 0.38% |
| Penta- | 17 | - | - | - | - | - | - | 17 | 0.38% |
| Hexa- | - | 1 | 1 | - | 1 | - | 1 | 4 | 0.09% |
| Total | 2210 | 1038 | 619 | 300 | 247 | 94 | 12 | 4520 | |
Comparison of the unigenes that are differentially expressed under drought stress
The expression of a high number of unigenes was affected in the drought-treated trees. Only the genes whose expression was identified as being significantly changed with a p value-adjusted (pdaj) <0.05 were retained. To analyze the similarities and differences among the drought-responsive transcriptome, a hierarchical clustering was prepared to represent the transcripts of all of the DEGs in the three replicates of the control, mild stress and severe stress trees. These results indicated significant differences in the gene expression profiles among the control, mild stress and severe stress (Fig 6A). When the DEGs were compared under the three conditions, we discovered 4687, 5648 and 5735 genes in the comparisons of mild versus control, severe versus control and severe versus mild stress, respectively. Notably, more genes were differentially expressed under severe stress compared to mild stress, suggesting that a severe stress treatment could affect more drought stress-related genes than mild stress. Moreover, a total of 601 genes overlapped with those of mild versus control, severe versus control and severe versus mild stress, indicating a linkage among three comparisons and a progressive biological process (Fig. 6B).
Figure 6. Cluster analysis of the DEGs across three comparisons.
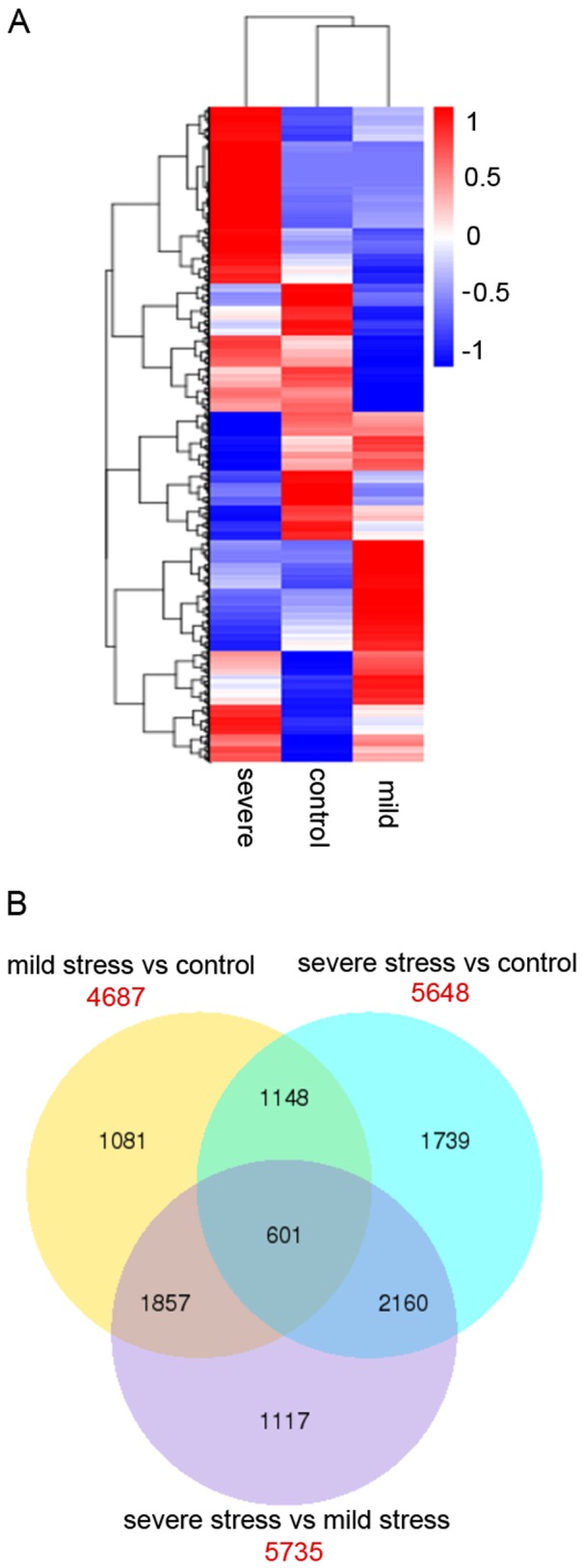
A. The differentially expression levels were log10 transformed and are shown with high expression represented by red and low expression represented by blue. B. Venn diagrams showing DEGs across three comparisons (mild stress versus control; severe stress versus control; and severe stress versus mild stress). The red values correspond to the total number of DEGs in each comparison; the overlapping values correspond to the number of differentially expressed genes in two/three comparisons.
Functional classification of the drought-stressed genes by Gene Ontology analysis
To identify the genes that are differentially expressed under drought stress, a functional categorization was carried out by GO analysis. By comparing mild stress versus control, 3068 DEGs, including 1223 down-regulated genes and 1845 up-regulated genes, revealed by DEG analysis were functionally assigned to the relevant terms in three categories (Biological Process, Cellular Component, and Molecular Function) of the GO database. The GO terms of the ‘oxidation-reduction process’ in Biological Process and ‘oxidoreductase activity’ in Molecular Function were significantly overrepresented (Figure 7A). By comparing severe stress with control, 3283 DEGs, including 1200 down-regulated genes and 2083 up-regulated genes, were functionally assigned to the relevant terms; ‘metabolic process’ in Biological Process was significantly overrepresented, followed by ‘oxidation-reduction process’ in Biological Process and ‘oxidoreductase activity’ in Molecular Function (Figure 7B). By comparing severe stress with mild stress, 3579 DEGs, including 1811 down-regulated genes and 1768 up-regulated genes, were functionally assigned to the relevant terms. Similar to the above two comparisons, the ‘oxidation-reduction process’ in Biological Process and ‘oxidoreductase activity’ in Molecular Function were significantly overrepresented (Figure 7C). These data, overall, suggest that ‘oxidation-reduction’ and ‘oxidoreductase activity’ were strongly affected may due to that the drought stress-induced generation of active oxygen at the cellular level is tightly controlled at both the production and consumption levels in vivo through increased antioxidative systems to avoid drought injury under drought conditions [18].
Figure 7. GO classifications of DEGs across three comparisons.

The right Y-axis represents the number of DEGs in a category; the left Y-axis indicates the percentage of a specific category of DEGs in each main category. A, mild stress versus control; B, severe stress versus control; C, severe stress versus mild stress.
In addition, as expected, the GO terms ‘response to water stimulus’, ‘response to water deprivation’, ‘hyperosmotic response’, and ‘response to stress’ were highly enriched in the DEGs, further confirming the efficiency of the drought treatments and the reliability of the gene expression data. At the same time, other terms that were related to the response to various other types of abiotic and biotic stresses, such as ‘response to abiotic stimulus’ ‘hyperosmotic salinity response’, ‘response to temperature stimulus’, and ‘immune response’, were also highly enriched in the DEGs, indicating the crosstalk of different stress responses in S. moorcroftiana, similar to the previously reported plant species [9], [14]. Furthermore, a general induction and enhancing of gene expression occurred under drought stress. Moreover, the DEGs were classified into additional GO terms of Molecular Function, which might due to more genes being differentially expressed in severe stress compared to mild stress (Figure 6B). However, for the Cellular Component category, dozens of DEGs were identified by comparing mild stress with control, while several DEGs were identified by comparing severe stress with control, and no DEG was identified by comparing severe stress with mild stress, suggesting that the expression of the unigenes that are involved in Cellular Component were not obviously changed under drought stress despite the degree of drought stress.
KEGG pathway analysis of the drought-responsive genes
To determine whether the drought stress-responsive genes engaged in specific pathways, the DEGs were used as objects to search against the KEGG pathway database. The top 20 obviously enriched pathways are shown in Fig. 8. By comparing mild stress with control, the DEGs were enriched in ‘metabolic pathways’, and 165 DEGs were related to ‘biosynthesis of secondary metabolites’ (Fig. 8A). However, this is only approximately 12% and 15% of the total genes that are involved in ‘metabolic pathways’ and ‘biosynthesis of secondary metabolites’ (Table S3), respectively. Although, only 52 and 58 genes that are related to the pathways ‘photosynthesis’ and ‘porphyrin and chlorophyll metabolism’, more than 30% DEGs were enriched (Table S3), suggesting that mild drought affected plant photosynthesis, which also occurs in other plant species under drought, including C. morifolium [14], B. nivea [4], and A. mongolicus [17]. By comparing severe stress with control, among the genes that are related to the pathways ‘ribosome’ and ‘plant hormone signal transduction’ (Fig. 8B), approximately 25% and 22% of the DEGs were enriched, respectively (Table S4). By comparing severe stress with mild stress, similar to the results of comparing mild stress with control, more DEGs were enriched in the pathways ‘metabolic pathways’ and ‘biosynthesis of secondary metabolites’ (Fig. 8C). However, the enrichment was only approximately 15% and 17%, respectively. In addition, among the genes that are related to the pathways ‘ribosome’, ‘plant hormone signal transduction’, ‘ascorbate and aldarate metabolism’, and ‘carbon fixation in photosynthetic organisms’, approximately 24%, 26%, 24%, and 22% of the DEGs were enriched, respectively (Table S5). These results are consistent with drought-stressed B. nivea, in which the ‘ribosome’ pathway enriched the most DEGs, followed by ‘ascorbate and aldarate metabolism’ and ‘carbon fixation in photosynthetic organisms’ and other pathways [4]. In general, these data indicate that severe stress strongly affects ‘ribosome’, ‘plant hormone signal transduction’, ‘ascorbate and aldarate metabolism’, and ‘carbon fixation in photosynthetic organisms’ in S. moorcroftiana, possibly due to the drought stress decreasing the CO2 assimilation rates that because of reduction of stomatal conductance, decreases the contents and activities of photosynthetic carbon reduction cycle enzymes, and induces plant hormone signal transduction [18], [19]. In addition, severe drought may affect the transcription of genes by changing the expression of ‘ribosome’-related genes.
Figure 8. KEGG enrichments of the annotated DEGs across three comparisons.
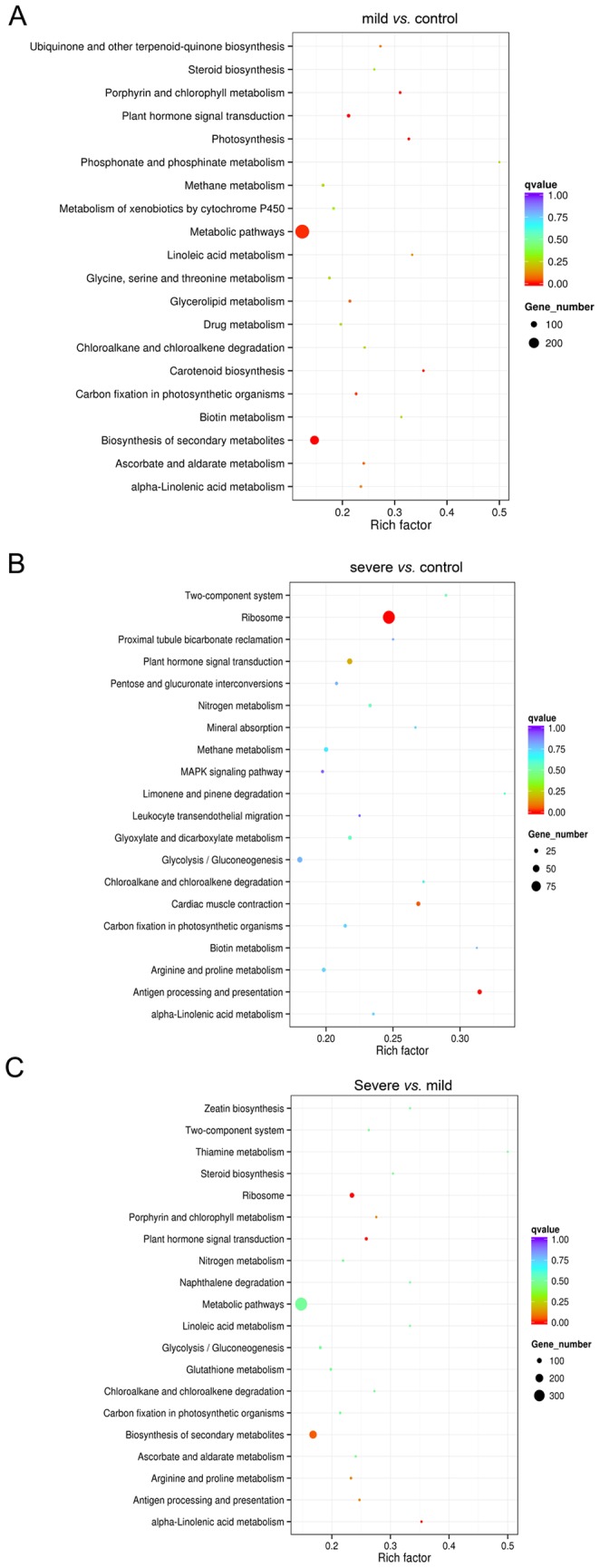
The left Y-axis indicates the KEGG pathway. The X-axis indicates the Rich factor. A high q value is represented by blue, and a low q value is represented by red. A, mild stress versus control; B, severe stress versus control; C, severe stress versus mild stress.
Expression analysis of the genes that are potentially involved in the drought response
To identify drought-responsive genes, 25 unigenes were selected. The expression of 25 unigenes was significantly up-regulated or down-regulated under drought treatment.
Transcription factors. Transcription factors are widely involved in drought stress, especially DREB TFs. In this study, 1534 TFs were identified from the transcriptome data, many of which were differentially expressed among the control and drought-stressed leaves. Eleven TFs were selected for further expression analysis; all seven were drought-responsive TFs (Fig. 9). Among these TFs, nine genes encoding DREB, Zinc-Finger Protein (ZFP), Zinc-Finger Protein Kinase (ZFPK), MYB, NAC, and WRKY were induced, while an ERF was repressed by drought; these results indicate that TFs are widely involved in plant response to drought stress, and most of these TFs fall into the AP2/ERF, NAC, MYB, Zinc-Finger and WRKY superfamilies [20], [21]. Consistently, 12 TFs from B. nivea, including AP2, NAC, MYB, Zinc-Finger, and WRKY, were drought stress-responsive TFs [4]. Eight DREB genes were responsive to dehydration in chrysanthemum [14], and two genes encoding NAC and ERF were up-regulated in A. mongolicus [17]. In addition, expression of AP2/ERF, NAC, MYB, Zinc-Finger, WRKY and other TF genes were induced in drought-tolerant rice under drought [22]. It is worth noting that a recent study reported a novel TF, named Far-Red impaired response 1 (FAR1), which is derived from ancient mutator-like transposases and belongs to the FRS gene family in Arabidopsis, that was remarkably up-regulated by drought treatment [23]; our results also demonstrate that the FAR gene was induced by drought in S. moorcroftiana, suggesting that FAR1 may be involved in additional processes in plants.
Figure 9. Verification of 25 putative genes that are involved in the drought response by qRT-PCR.

The left Y-axis indicates the normalized expression pattern of the DEGs under drought. The normalized expression patterns were log2 transformed. Both Actin and GAPDH were used as internal controls.
Regulating plant hormones
Extensive overlaps exist between the drought response and several plant hormone responses, including auxin and ABA in Arabidopsis [24]. The promoters of many drought-inducible genes contain the ABA-responsive element, including some TFs; 67% of drought-regulated genes are significantly regulated by ABA [25]. In this study, the expression of an ABA-Responsive (ABR) gene was induced by drought, suggesting that the drought response may be linked to ABA signaling. In contrast, the expression of a gene encoding Auxin-Induced Protein (AIP) in S. moorcroftiana was down-regulated under drought; our data agree with the observation that auxin-responsive genes were down-regulated by drought in Arabidopsis [25], suggesting that S. moorcroftiana drought tolerance also linked to the auxin signaling network.
Adjust the osmotic pressure
Water-channel proteins and sugar transporters are believed to function in the transport of water and sugars to adjust the osmotic pressure under stress [26]. The transcripts of most Aquaporins (AQPs) were elevated under drought stress in Malus species [27]. In addition, at least five AQPs were up-regulated in rice under drought [28]. These studies suggest that the expression of AQPs may be induced by drought; however, the transcripts some AQPs were down-regulated in rice, sugarcane, and Chinese cabbage under drought [28]–[30]. In this study, three selected AQPs were induced by drought (Fig. 9). These results indicating that the role of AQPs in drought stress is complex. Moreover, the transcript of a gene encoding Sugar Transporter (SUT) was induced by drought in this study (Fig. 9), which could be explained by additional sugar transportation adjusting the osmotic pressure under drought.
Stabilizing cell structures
Dehydrins (DHNs) are a class of hydrophilic, thermostable stress proteins with a high number of charged amino acids. DHNs function to protect cells from damage caused by stress-induced dehydration [31]. The expression of DHNs is usually induced by drought stress. For example, the transcripts of DHNs were strongly up-regulated by drought stress in peach and tobacco [32], [33]. Similarly, similar to previous reports, a selected DHN from S. moorcroftiana was also induced by drought in this study. However, this is not always the case, as some DHNs also remained unchanged under drought in grape and barley [34]–[36], possibly resulting from differences in the DHNs expression levels often being dependent on the duration of the stress.
Reducing the oxidative damage
The induction of Reactive Oxygen Species (ROS)-related factors suggests that drought stress is accompanied by the production of ROS in plant roots, and the induction of ROS-related enzymes may be involved in the protection of tissues from oxidative damage under stress conditions [37]. The response and dynamics of antioxidant enzyme transcripts, including SOD, POD, PRX, APX and GR et al., are commonly used to study plant stress responses. Of the antioxidative enzymes, Peroxidases (POD) play key roles in cellular ROS detoxification, Peroxiredoxins (PRX) are a recently discovered family of antioxidant enzymes that catalyze the reduction of peroxides. In the woody plant species Tamarix hispida, transcripts of 10 PODs were highly induced by drought in different organs [38]. In agreement with this observation, a POD was obviously induced under drought in this study. Moreover, the expression of a PRX of Xerophyta viscosa was up-regulated when exposed to dehydration [39]. However, the expression patterns of four PRXs showed temporal and organ specificity under PEG treatment in X. viscosa; all four PRXs were down-regulated in the leaves, and three of them were generally decreased in the roots, while three PRXs was up-regulated in the stems under PEG stress [40]. Similar with these results, expression of PRX was significantly increased in the leaves under drought. In addition, in Arabidopsis, deficiency of Glycerol-3-phosphate dehydrogenase (GPDH) leads to a higher cellular level of ROS, and a noticeable increase in the GPDH transcript was observed after exposing the trees to drought stress [41]. The expression pattern was also observed in this study, in which a GPDH was increased under drought in S. moorcroftiana. These results suggest that the POD, PRX and GPDH in S. moorcroftiana are drought-induced genes. Arabidopsis contains 244 Cytochrome 450 (CYP450) genes in its genome; transforming Arabidopsis AtCYP78A7 into rice increased the drought tolerance of the transgenic rice [42], whereas the disruption of CYP707A3 in Arabidopsis results in increased drought tolerance, and its over-expression results in an increased transpiration rate and reduced drought tolerance [43], suggesting that different plant CYPs could be positive or negative regulators of drought tolerance. In this study, drought stress increased the transcripts of a selected CYP gene, indicating that this CYP may be a positive regulator of drought tolerance in S. moorcroftiana.
Secondary metabolism
The transcripts of some key enzymes that are related to important secondary metabolisms were significantly affected by drought. Chalcone isomerase (CHI) and chalcone synthase (CHS) are two key enzymes in the biosynthesis of flavonoids in plants. In chrysanthemum, the transcripts of CHS and CHI were down-regulated by drought [14]. However, a rapid increase in the expressions of CHS and CHI was observed under drought in two wheat cultivars [44]. Interestingly, CHS and CHI were up-regulated following short-term dehydration stress in roots [45], whereas they were repressed during long-term drought in the roots of alfalfa [46]. In this study, one CHI was induced by drought, while another selected CHI and a CHS were repressed by drought in S. moorcroftiana. This discrepancy might reflect differences in the function of plant CHI and CHS in response to drought stress.
In summary, the expression patterns of 25 putative genes that are involved in drought tolerance were consistent with changes in their transcript abundance, mainly through regulating hormone signaling, reducing oxidative damage, adjusting the osmotic pressure, and regulating secondary metabolism. Meanwhile, transcription factors play important roles in drought tolerance in S. moorcroftiana.
Conclusions
The combination of RNA-seq and DEGs analyses based on Illumina sequencing technology provided comprehensive information on gene expression. The substantially assembled sequences represented a considerable portion of the transcriptome of S. moorcroftiana. Based on the assembled de novo transcriptome, 4687, 5648 and 5735 DEGs were identified from the comparison of mild versus control, severe versus control and severe versus mild stress, respectively. Kyoto Encyclopedia of Genes and Genomes pathway analysis uncovered the differentially expressed genes were involved in important pathways, such as ‘metabolic pathways’, and ‘plant hormone signal transduction’. In addition, expression patterns of 25 selected DEGs were further validated with qRT-PCR, which reflected significant alteration in major biological processes and metabolic pathways during drought stress. The information provided here can also further extend the knowledge of the drought tolerance of this plant species that survives in the arid and semi-arid regions of the Qinghai-Tibet Plateau.
Methods
Plant material and stress treatment
This study was approved by the National Key Station for Field Scientific Observation & Experiment. This field studies did not involve endangered species. The seeds of S. moorcroftiana were identified and collected by Yanhui Ye (Agricultural and Animal Husbandry College, Tibet University) in Oct. 2010 from Milin County (N 29o12′24.58″, E 94o12′2.29″, H 2936 m), Nyingchi, Tibet. To elucidate the drought tolerance mechanism under natural Plateau conditions, two-year-old S. moorcroftiana trees (approximately 60 cm in height) in pots were selected from germplasm nursery (field conditions) of the Agricultural and Animal Husbandry College, Tibet University. The trees were randomly assigned to one of three different treatments: (1) control trees maintained under well-watered conditions, (2) mildly drought-stressed trees and (3) severely drought-stressed trees. The water potential was assessed by repeated measurements of the predawn leaf water potential using the PSYPRO Water potential system with C52 psychrometer (Wescor, USA) following the manufacturer's protocol.
Total RNA isolation and RNA-seq library construction
For the samples for RNA-seq, the leaves from the middle position of the trees were collected from the control and stressed plants and then immediately placed in liquid nitrogen. The total RNA was isolated using EASYspin Plus Plant RNA isolation kit (Aidlab, Beijing, China) following the manufacturer's instructions. An equal quantity of RNA from all three of the treatments and three independent biological replicates was blended for cDNA library construction to obtain the transcriptome data. The sequencing libraries were generated using NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB, USA) following manufacturer's recommendations, and index codes were added to attribute sequences to each sample.
Clustering and sequencing
The index-coded samples were clustered on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer's instructions. After the cluster generation, the library preparations were sequenced on an Illumina HiSeq 2000 platform, and paired-end reads were generated.
Sequence read mapping, assembly and SSR detection
The raw data (raw reads) in fastq format were first processed through in-house Perl scripts. In this step, clean data (clean reads) were obtained by removing the reads containing adapters, reads containing ploy-N and low-quality reads from the raw data. At the same time, the Q20, Q30, GC-content and sequence duplication level of the clean data were calculated. All of the downstream analyses were based on high-quality clean data. The clean reads were assembled using Trinity software as described for de novo transcriptome assembly without a reference genome. The SSRs of the transcriptome were identified using MISA (http://pgrc.ipk-gatersleben.de/misa/misa.html).
Gene expression quantification and differential expression analysis
The libraries were sequenced, and 50-bp single-end reads were generated. The clean data were mapped back onto the assembled transcriptome, and the read count for each gene was obtained from the mapping results. The gene expression levels were measured using the reads per kilobase per million reads (RPKM) method using the formula that was previously described by Mortazavi [47]. A differential expression analysis of three conditions was performed using the DESeq R package (1.10.1). Three independent biological replicates for each treatment were analyzed. The p values were adjusted using the Benjamini & Hochberg method. The corrected p value of 0.05 was set as the threshold for significantly differential expression.
Functional annotation
The GO enrichment analysis of the DEGs was implemented by the GOseq R package-based Wallenius noncentral hypergeometric distribution [48], which can adjust for gene length bias in DEGs. The KEGG pathway enrichment analysis of the DEGs was performed using KOBAS [49].
Validation of the DEGs by quantitative real-time PCR
For the quantitative RT-PCR of the mRNAs, 1 µg of total RNA was used to synthesize the cDNA using the PrimeScript RT reagent Kit (Takara, Japan). Real-time PCR was performed using SYBR premix Ex Taq (Takara, Japan) according to the manufacturer's instruction. The PCR amplification was performed under the following conditions: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s and 60°C for 10 s. The products were verified by melting curve analysis. Quantification was achieved by normalizing the number of target transcripts copies to both Actin and GAPDH genes using the normalized expression method. Three independent biological replicates for each sample and three technical replicates for each biological replicate were analyzed. All of the primers that were used are listed in Table S6.
Supporting Information
Summary of the Unigenes annotation.
(DOCX)
Detailed frequencies of the EST-SSR repeat motifs.
(DOCX)
KEGG pathway enrichment results by DEGs from mild stress versus control.
(XLS)
KEGG pathway enrichment results by DEGs from severe stress versus control.
(XLS)
KEGG pathway enrichment results by DEGs from severe stress versus mild stress.
(XLS)
Primers for the qRT-PCR analysis.
(DOC)
Acknowledgments
We would like to thank Dr. Hao Sun (Institute of Applied Ecology, Chinese Academy of Science) for the help of designing this project. And we also would like to thank the editor Binying Fu Ph.D and two anonymous reviewers, for their valuable comments and suggestions.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are available from the NCBI SRA database (accession number SRP041237) and within Supporting Information files.
Funding Statement
This research was supported by the National Natural Science Foundation of China (31260189) and Collaborative Innovation Center Construction of Research and development on Tibetan characteristic agricultural and animal husbandry resources----plateau ecology. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cui X, Graf HF (2009) Recent land cover changes on the Tibetan Plateau: a review. Climatic Change 94: 47–61. [Google Scholar]
- 2. Xue X, Guo J, Han B, Sun Q, Liu L (2009) The effect of climate warming and permafrost thaw on desertification in the Qinghai-Tibetan Plateau. Geomorphology 108: 182–190. [Google Scholar]
- 3. Yang M, Wang S, Yao T, Gou X, Lu A, et al. (2004) Desertification and its relationship with permafrost degradation in Qinghai-Xizang (Tibet) plateau. Cold Reg Sci Technol 39: 47–53. [Google Scholar]
- 4. Liu T, Zhu S, Tang Q, Yu Y, Tang S (2013) Identification of drought stress-responsive transcription factors in ramie (Boehmeria nivea L. Gaud). Bmc Plant Biol 13: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. An D, Yang J, Zhang P (2012) Transcriptome profiling of low temperature-treated cassava apical shoots showed dynamic responses of tropical plant to cold stress. Bmc Genomics 13: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dugas DV, Monaco MK, Olson A, Klein RR, Kumari S, et al. (2011) Functional annotation of the transcriptome of Sorghum bicolor in response to osmotic stress and abscisic acid. Bmc Genomics 12: 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nishiyama R, Le DT, Watanabe Y, Matsui A, Tanaka M, et al. (2012) Transcriptome analyses of a salt-tolerant cytokinin-deficient mutant reveal differential regulation of salt stress response by cytokinin deficiency. Plos One 7: e32124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang XC, Zhao QY, Ma CL, Zhang ZH, Cao HL, et al. (2013) Global transcriptome profiles of Camellia sinensis during cold acclimation. Bmc Genomics 14: 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dang Z, Zheng L, Wang J, Gao Z, Wu S, et al. (2013) Transcriptomic profiling of the salt-stress response in the wild recretohalophyte Reaumuria trigyna . Bmc Genomics 14: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Torales SL, Rivarola M, Pomponio MF, Gonzalez S, Acuña CV, et al. (2013) De novo assembly and characterization of leaf transcriptome for the development of functional molecular markers of the extremophile multipurpose tree species Prosopis alba . Bmc Genomics 14: 705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tian DQ, Pan XY, Yu YM, Wang WY, Zhang F, et al. (2013) De novo characterization of the Anthurium transcriptome and analysis of its digital gene expression under cold stress. Bmc Genomics 14: 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao Z, Tan L, Dang C, Zhang H, Wu Q, et al. (2012) Deep-sequencing transcriptome analysis of chilling tolerance mechanisms of a subnival alpine plant, Chorispora bungeana . Bmc Plant Biol 12: 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guimarães PM, Brasileiro AC, Morgante CV, Martins AC, Pappas G, et al. (2012) Global transcriptome analysis of two wild relatives of peanut under drought and fungi infection. Bmc Genomics 13: 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu Y, Gao S, Yang Y, Huang M, Cheng L, et al. (2013) Transcriptome sequencing and whole genome expression profiling of chrysanthemum under dehydration stress. Bmc Genomics 14: 662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kantartzi SK (2013) Microsatellites: Methods and Protocols. Edited by Stella K. Kantartzi. Totowa, NJ: Humana Press 197 p. [Google Scholar]
- 16. Silva PI, Martins AM, Gouvea EG, Pessoa-Filho M, Ferreira ME (2013) Development and validation of microsatellite markers for Brachiaria ruziziensis obtained by partial genome assembly of Illumina single-end reads. Bmc Genomics 14: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu M, Shi J, Lu C (2013) Identification of stress-responsive genes in Ammopiptanthus mongolicus using ESTs generated from cold-and drought-stressed seedlings. Bmc Plant Biol 13: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reddy AR, Chaitanya KV, Vivekanandan M (2004) Drought-induced responses of photosynthesis and antioxidant metabolism in higher plants. J Plant Physiol 161: 1189–1202. [DOI] [PubMed] [Google Scholar]
- 19.Farooq M, Wahid A, Kobayashi N, Fujita D, Basra S (2009) Plant drought stress: effects, mechanisms and management. In: Sustainable Agriculture. Springer 153–188.
- 20. Nakashima K, Ito Y, Yamaguchi-Shinozaki K (2009) Transcriptional regulatory networks in response to abiotic stresses in Arabidopsis and grasses. Plant Physiol 149: 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fujita Y, Fujita M, Shinozaki K, Yamaguchi-Shinozaki K (2011) ABA-mediated transcriptional regulation in response to osmotic stress in plants. J Plant Res 124: 509–525. [DOI] [PubMed] [Google Scholar]
- 22. Moumeni A, Satoh K, Kondoh H, Asano T, Hosaka A, et al. (2011) Comparative analysis of root transcriptome profiles of two pairs of drought-tolerant and susceptible rice near-isogenic lines under different drought stress. Bmc Plant Biol 11: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tang W, Ji Q, Huang Y, Jiang Z, Bao M, et al. (2013) FHY3 and FAR1 transcription factors integrate light and abscisic acid signaling in Arabidopsis . Plant Physiol 163: 857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seo PJ, Xiang F, Qiao M, Park JY, Lee YN, et al. (2009) The MYB96 transcription factor mediates abscisic acid signaling during drought stress response in Arabidopsis . Plant Physiol 151: 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang D, Wu W, Abrams SR, Cutler AJ (2008) The relationship of drought-related gene expression in Arabidopsis thaliana to hormonal and environmental factors. J Exp Bot 59: 2991–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, et al. (2002) Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J 31: 279–292. [DOI] [PubMed] [Google Scholar]
- 27. Liu C, Li C, Liang D, Ma F, Wang S, et al. (2013) Aquaporin expression in response to water-deficit stress in two Malus species: relationship with physiological status and drought tolerance. Plant Growth Regul 70: 187–197. [Google Scholar]
- 28. Hadiarto T, Tran L-SP (2011) Progress studies of drought-responsive genes in rice. Plant Cell Rep 30: 297–310. [DOI] [PubMed] [Google Scholar]
- 29. da Silva MD, RLdO Silva, Neto Costa Ferreira JR, Guimarães ACR, Veiga DT, et al. (2013) Expression analysis of sugarcane aquaporin genes under water deficit. Journal of Nucleic Acids 2013: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu S, Zhang F, Yu Y, Zhang D, Zhao X, et al. (2012) Transcriptome profiling of dehydration stress in the Chinese cabbage (Brassica rapa L. ssp. pekinensis) by tag sequencing. Plant Mol Biol Rep 30: 17–28. [Google Scholar]
- 31.Eriksson SK, Harryson PD (2011) Molecular biology, structure and function. In Plant Desiccation Tolerance. Edited by Luttge U, Beck E, Bartels D. Berlin, Springer 289–305. [Google Scholar]
- 32. Dobrá J, Vanková R, Havlová M, Burman AJ, Libus J, et al. (2011) Tobacco leaves and roots differ in the expression of proline metabolism-related genes in the course of drought stress and subsequent recovery. J Plant Physiol 168: 1588–1597. [DOI] [PubMed] [Google Scholar]
- 33. Wisniewski ME, Bassett CL, Renaut J, Farrell R, Tworkoski T, et al. (2006) Differential regulation of two dehydrin genes from peach (Prunus persica) by photoperiod, low temperature and water deficit. Tree Physiol 26: 575–584. [DOI] [PubMed] [Google Scholar]
- 34. Suprunova T, Krugman T, Fahima T, Chen G, Shams I, et al. (2004) Differential expression of dehydrin genes in wild barley, Hordeum spontaneum, associated with resistance to water deficit. Plant, cell & environment 27: 1297–1308. [Google Scholar]
- 35. Qian G, Liu Y, Ao D, Yang F, Yu M (2008) Differential expression of dehydrin genes in hull-less barley (Hordeum vulgare ssp. vulgare) depending on duration of dehydration stress. Can J Plant Sci 88: 899–906. [Google Scholar]
- 36. Yang Y, He M, Zhu Z, Li S, Xu Y, et al. (2012) Identification of the dehydrin gene family from grapevine species and analysis of their responsiveness to various forms of abiotic and biotic stress. Bmc Plant Biol 12: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoshimura K, Masuda A, Kuwano M, Yokota A, Akashi K (2008) Programmed proteome response for drought avoidance/tolerance in the root of a C3 xerophyte (wild watermelon) under water deficits. Plant Cell Physiol 49: 226–241. [DOI] [PubMed] [Google Scholar]
- 38. Gao C, Wang Y, Liu G, Wang C, Jiang J, et al. (2010) Cloning of ten peroxidase (POD) genes from Tamarix hispida and characterization of their responses to abiotic stress. Plant Mol Biol Rep 28: 77–89. [Google Scholar]
- 39. Mowla SB, Thomson JA, Farrant JM, Mundree SG (2002) A novel stress-inducible antioxidant enzyme identified from the resurrection plant Xerophyta viscosa Baker. Planta 215: 716–726. [DOI] [PubMed] [Google Scholar]
- 40. Gao C, Zhang K, Yang G, Wang Y (2012) Expression analysis of four peroxiredoxin genes from Tamarix hispida in response to different abiotic stresses and exogenous abscisic acid (ABA). International journal of molecular sciences 13: 3751–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen W, Wei Y, Dauk M, Tan Y, Taylor DC, et al. (2006) Involvement of a glycerol-3-phosphate dehydrogenase in modulating the NADH/NAD+ ratio provides evidence of a mitochondrial glycerol-3-phosphate shuttle in Arabidopsis . The Plant Cell Online 18: 422–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nam KH, Nam KJ, An JH, Jeong SC, Park KW, et al. (2013) Comparative analysis of key nutrient composition between drought-tolerant transgenic rice and its non-transgenic counterpart. Food Science and Biotechnology 22: 1–7. [Google Scholar]
- 43.Bak S, Beisson F, Bishop G, Hamberger B, Höfer R, et al. (2011) Cytochromes P450. The Arabidopsis Book Volume 9. Washington, DC: BioOne/The American society of plant biologists: e0144. [DOI] [PMC free article] [PubMed]
- 44. Ma D, Sun D, Wang C, Li Y, Guo T (2014) Expression of flavonoid biosynthesis genes and accumulation of flavonoid in wheat leaves in response to drought stress. Plant Physiol Biochem 80: 60–66. [DOI] [PubMed] [Google Scholar]
- 45. Chen D, Liang MX, DeWald D, Weimer B, Peel MD, et al. (2008) Identification of dehydration responsive genes from two non-nodulated alfalfa cultivars using Medicago truncatula microarrays. Acta Physiol Plant 30: 183–199. [Google Scholar]
- 46. Kang Y, Han Y, Torres-Jerez I, Wang M, Tang Y, et al. (2011) System responses to long-term drought and re-watering of two contrasting alfalfa varieties. Plant J 68: 871–889. [DOI] [PubMed] [Google Scholar]
- 47. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- 48. Young MD, Wakefield MJ, Smyth GK, Oshlack A (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biology 11: R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mao X, Cai T, Olyarchuk JG, Wei L (2005) Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21: 3787–3793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of the Unigenes annotation.
(DOCX)
Detailed frequencies of the EST-SSR repeat motifs.
(DOCX)
KEGG pathway enrichment results by DEGs from mild stress versus control.
(XLS)
KEGG pathway enrichment results by DEGs from severe stress versus control.
(XLS)
KEGG pathway enrichment results by DEGs from severe stress versus mild stress.
(XLS)
Primers for the qRT-PCR analysis.
(DOC)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are available from the NCBI SRA database (accession number SRP041237) and within Supporting Information files.