

Abstract
Immunotherapies that augment antitumor T cells have had recent success for treating patients with cancer. Here we examined whether tumor-specific CD4+ T cells enhance CD8+ T-cell adoptive immunotherapy in a lymphopenic environment. Our model employed physiological doses of tyrosinase-related protein 1-specific CD4+ transgenic T cells-CD4+ T cells and pmel-CD8+ T cells that when transferred individually were subtherapeutic; however, when transferred together provided significant (p ≤ 0.001) therapeutic efficacy. Therapeutic efficacy correlated with increased numbers of effector and memory CD8+ T cells with tumor-specific cytokine expression. When combined with CD4+ T cells, transfer of total (naïve and effector) or effector CD8+ T cells were highly effective, suggesting CD4+ T cells can help mediate therapeutic effects by maintaining function of activated CD8+ T cells. In addition, CD4+ T cells had a pronounced effect in the early posttransfer period, as their elimination within the first 3 days significantly (p < 0.001) reduced therapeutic efficacy. The CD8+ T cells recovered from mice treated with both CD8+ and CD4+ T cells had decreased expression of PD-1 and PD-1-blockade enhanced the therapeutic efficacy of pmel-CD8 alone, suggesting that CD4+ T cells help reduce CD8+ T-cell exhaustion. These data support combining immunotherapies that elicit both tumor-specific CD4+ and CD8+ T cells for treatment of patients with cancer.
Keywords: Cancer immunotherapy, CD4+ T-cell help, Metastatic melanoma, PD-1, T cell
Introduction
Metastatic melanoma is a devastating disease with a poor overall survival rate. Two immunotherapies, IL-2 and anti-CTLA-4, provide objective clinical responses in approximately 15% of patients and are approved drugs 1,2. Recently, the combination of anti-CTLA-4 and anti-PD-1 has shown to provide profound and rapid anticancer activity in approximately half of the patients receiving treatment 3. Preclinical models have identified T-cell-dependent mechanisms as being the primary mediators of these antitumor effects 4. Adoptive T-cell immunotherapy in patients made lymphopenic by nonmyeloablative chemotherapy has also provided significant success against this disease 5,6. Many of these studies utilize T cells expanded from tumor-infiltrating lymphocyte (TIL) cultures; however, the use of human T cells transduced to express tumor-reactive TCRs or chimeric antigen receptors provides a practical alternative with the potential to be broadly applied to patients with almost any type of cancer 7,8. Furthermore, studies with chimeric antigen receptors have shown long-term tumor regression and tumor-specific T-cell persistence for over 6 months 7.
Both clinical trials and murine models, studying adoptive immunotherapy using tumor-specific TILs or TCR transgenic (Tg) T cells have shown that immunotherapy is more effective in a lymphopenic than in a lymphoreplete environment 5,6. A lymphopenic environment can be established in a number of ways including chemotherapy (cyclophosphamide), radiation, a combination of both or by using mice that lack endogenous T and B cells (RAG−/− deficient mice) 9,10. Each lymphopenia inducing method provides a different combination of mechanisms that enhances immunotherapy. In addition to the direct killing of tumor cells, by radiation or chemotherapy, mechanisms that augment T-cell expansion and effector function include elimination or absence of suppressive cells as well as cells that serve as cytokine sinks, increased access to antigen-presenting cells (APCs), and access to proinflammatory signals that may activate APCs or aid in overcoming T-cell exhaustion 11. Additionally, combination adoptive immunotherapy that included induction of lymphopenia prior to adoptive transfer and IL-2 administration, has resulted in persistence of memory and circulating CD8+ T cells and correlated with successful clinical outcome 12–14. This supports the concept that CD8+ T cells play a dominant role in tumor elimination by directly killing tumor cells or by the secretion of cytokine or chemokines that have antitumor effects and recruit other adaptive or innate immune components. Thus combinations that maintain or increase tumor-specific CD8+ T cells would be expected to improve therapeutic efficacy 15.
The importance of CD4+ T-cell help for both priming and maintenance of memory CD8+ T-cell immunity has long been appreciated 16,17. Our lab and others have shown that immunotherapy with tumor-specific CD8+ T cells in CD4-deficient MHC class II−/− mice resulted in regression of pulmonary metastases, but did not result in long-term antitumor immunity and tumors eventually recurred 18,19. In contrast, multiple studies have shown that partial or transient CD4-depletion can enhance antitumor responses, but since these models do not eliminate CD4+ T cells completely, there may be a small population of CD4+ T cells programming or maintaining CD8+ T-cell function 20–22. Moreover, induction of lymphopenia is thought to abrogate the need for CD4+ help since it increases CD8+ T-cell exposure to homeostatic cytokines, IL-7 and IL-15, driving memory T-cell formation and enhancing antitumor immune responses 23,24.
Although it has not been directly examined, it is possible that low objective response rates in some studies are due to the transfer of tumor-specific CD8+ T cells in the absence of tumor-specific CD4+ T cells. Two studies utilizing MART-1 and/or gp100-specific HLA class I restricted TCR gene transfer for treatment of metastatic melanoma resulted in objective clinical response rates of 13% (2/15) and 30% (6/20), which were lower than the response rates achieved using bulk CD4+ and CD8+ TILs (51–71%). While these are small studies, one possible explanation for the low response rate is the absence of tumor-specific CD4+ T cells. However, attempts to identify tumor-specific CD4+ T cells in the peripheral blood of patients with cancer who experienced an objective clinical response following nonmyeloablative chemotherapy and adoptive immunotherapy with TILs have been difficult 25. Furthermore, CD4+ T cells alone have been expanded and adoptively transferred with high-dose IL-2 and have resulted in few clinical responses 26. Given the prior studies underscoring the significant contribution, CD4+ T cells play in supporting the therapeutic efficacy of tumor-specific CD8+ T cells and a number of clinical studies suggesting that neither CD8+ or CD4+ T cells are effective alone, in this paper, we investigated the importance of tumor-specific CD4+ T-cell help in augmenting antitumor immunity of CD8+ T cells under conditions of lymphopenia-driven homeostasis.
Results
Immunotherapy with both pmel and TRP-1 augments therapeutic efficacy
Since adoptive transfer of large numbers (5 × 104–2 × 105) of tyrosinase-related protein 1-specific CD4+ transgenic T cells (TRP-1) cytotoxic CD4+ Tg T cells alone can induce regression of established B16-F10 melanoma 27,28 we sought, to generate a model where therapeutic efficacy was dependent on both CD8+ and CD4+ T cells. Thus, we needed to identify a dose of TRP-1-CD4+ Tg T cells that was not therapeutic on its own. To do this, 3-day experimental pulmonary metastases were established by i.v. injection of the poorly immunogenic subclone, B16BL6-D5 melanoma (D5) in tyrosinase-related protein 1 (tyrp-1)bwRAG1−/− (RAG1−/− tyrp-1 protein deficient 29) female mice, which lack endogenous T and B cells. Mice received adoptive transfer of CD4-enriched TRP-1 Tg cells at doses ranging from 1 × 106 to 50 cells. TRP-1-CD4+ T cells failed to fully eliminate metastases regardless of their number, but animals treated with 5000 or less CD4+ T cells had greater than 50 metastases (Supporting Information Fig. 1). Consequently, a dose of 1000 CD4-enriched TRP-1 cells, a physiologically relevant number, was used for subsequent experiments. For all experiments, gp100-specific CD8+ Tg T cells (pmel) CD8+ T cells were activated by αCD3/IL-2 expansion. RAG1−/− tyrp1 protein-deficient lymphopenic mice with 3-day D5 experimental pulmonary metastases were treated with 106 αCD3/IL-2-expanded pmel and 1000 naïve CD4-enriched TRP-1 (Pmel + TRP-1), 106 αCD3/IL-2-expanded pmel alone, 1000 TRP-1 alone or no treatment (Fig. 1A). All mice received 90 000 IU IL-2 i.p. daily for 3 days. Ten and 20 days after adoptive transfer mice treated with both pmel and TRP-1 cells had significantly less tumor burden than mice treated with either pmel or TRP-1 alone (Fig. 1B). Mice treated with both pmel and TRP-1 showed no evidence of tumor after 40 days, while pmel or TRP-1 alone groups succumbed to tumor burden before 27 days (Fig. 1C).
Figure 1.
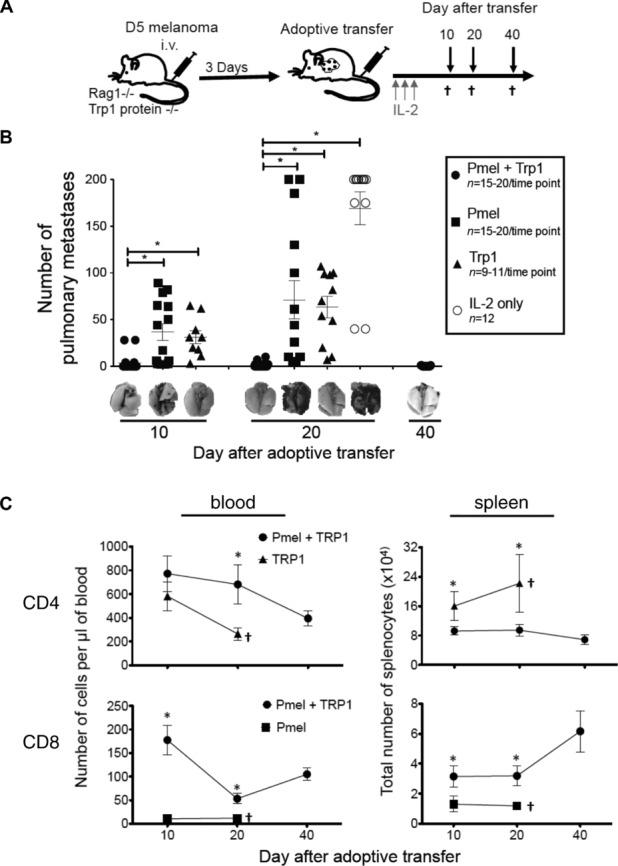
Treatment with tumor-specific pmel and TRP-1 T cells of lymphopenic tumor-bearing RAG1−/− mice eliminates tumor and increases pmel T cells in the blood and spleen. (A) Overview of RAG1−/− lymphopenic experimental pulmonary metastases model. RAG1−/− lymphopenic tyrp1 protein-deficient mice were treated with 2 × 105 D5 melanoma cells i.v. After 3 days, the same mice were treated with 1 × 106 αCD3/IL-2 stimulated pmel-CD8+ Tg cells and 1000 CD4-enriched TRP-1 Tg cells (Pmel + TRP-1), pmel alone (Pmel), TRP-1 alone (TRP-1) or no cells (IL-2 only). All mice also received 3 doses of 90 000 IU IL-2 i.p. daily. Mice were euthanized (†) for analysis 10, 20, and 40 days after adoptive immunotherapy. (B) Number of pulmonary metastases at each time point with representative resected lungs. (C) Total number of CD3+CD4+ or CD3+CD8+ cells in the blood or spleen at day 10, 20, and 40 following adoptive immunotherapy. (A and B) Data are shown as mean ± SEM of n = 9–20 mice per time point and are combined from four independent experiments. *p < 0.001; Students t-test.
Treatment with both pmel and TRP-1 significantly increased the number of CD8+ T cells in the blood and the spleen 10 and 20 days following adoptive transfer, compared to treatment with pmel alone (Fig. 1C). This is notable because persistence of antitumor CD8+ T cell has been correlated with long-term tumor control 2,30,31. Treatment with pmel and TRP-1 also resulted in increased CD4+ T cells in the blood compared to TRP-1 treatment alone; however, there were decreased CD4+ T cells in the spleen (Fig. 1C).
The addition of TRP-1 T cells enhances the number and function of pmel-CD8+ T cells
CD4+ T cells are known to be important for maintenance of effector (TEff) and memory (Tmemory) CD8+ T cells 5, therefore, we determined whether adoptive transfer of both pmel and TRP-1 T cells increased the number and frequency of Tmemory and TEff pmel-CD8+ T cells compared to adoptive transfer with pmel alone. Mice receiving combined therapy exhibited an increased total number of TEff (CD44+CD62LloCD127lo) pmel-CD8+ T cells in the blood and spleen (Fig. 2A). There was also an increase in total Tmemory (CD44+CD127+) pmel-CD8+ T cells in the blood and effector memory (TEM) (CD44+CD127+CCR7lo) pmel-CD8+ T cells in the spleen.
Figure 2.

Treatment of tumor-bearing RAG1−/− mice with tumor-specific pmel and TRP-1 T cells increases survival and function of TEff and Tmemory pmel T cells. (A) TEff (CD44+CD62LloCD127lo) and Tmemory (CD44+CD127+) CD8+ or CD4+ T cells in the blood and TEff and TEM (CD44+CD62LloCD127+CCR7lo) in the spleen 10 and 20 days after adoptive immunotherapy with both pmel and TRP-1 compared to treatment with pmel alone or TRP-1 alone. (B) Distribution of Tnaïve, TEff, TEM (EM), and TCM (CM) phenotype CD8+ or CD4+ T cells in the spleen at the day of transfer (day 0), 10 or 20 days after adoptive transfer. (C) Percent of tumor-specific IFN-γ expressing CD8+ or CD4+ T cells stimulated with specific D5 CIITA (D5II), nonspecific syngeneic MCA-310 CIITA (MCAII) or unstimulated (no stim) 20 days after adoptive immunotherapy. (A–C) Data are shown as mean ± SEM of n = 9–20 mice per time point and are combined from four independent experiments. *p < 0.001, Student’s t-test. (D) ICS of CD8+-gated cells using splenocytes from pmel + TRP-1 or pmel-treated mice stimulated with αCD3 or unstimulated. Representative pies from one experiment out of four performed.
The composition of naïve (CD62L+CD44lo), TEff (CD44+CD127loCCR7lo), TEM (CD44+CD127+CCR7lo), and central memory (TCM) (CD44+CD127+CCR7+) pmel-CD8+ T cells 10 and 20 days after adoptive transfer indicates a much smaller proportion of TEM phenotype pmel-CD8+ T cells in mice treated with pmel T cells alone (Fig. 2B). The presence of tumor-specific TRP-1-CD4+ T cells increased the number and frequency of pmel TEff CD8+ T cells that eliminated the tumor and also increased the number of long-lived memory T cells (Fig. 2A and B). Analysis of CD8+ T-cell function by intracellular cytokine staining 10 days after transfer, showed that pmel-CD8+ T cells from pmel and TRP-1-treated mice had a significantly (p ≤ 0.001) higher frequency of D5-specific IFN-γ-expressing cells compared to stimulation with the syngeneic but unrelated MCA-310 sarcoma. However, CD8+ T cells from pmel-only-treated mice also exhibited an increased percentage of IFN-γ-positive pmel-CD8+ T cells, but this difference did not reach statistical significance (Fig 2C). Pmel and TRP-1-treated mice also had a higher frequency of CD8+ T cells exhibiting polyfunctional cytokine expression (TNF-α, IFN-γ, Granzyme B, and IL-2), which has been associated with long-lived T cells (Fig 2D) 8,32–34. We analyzed the phenotype of TRP-1-CD4+ T cells in blood and spleen and found increased numbers of TEff and TEM in TRP-1-only treated mice compared to pmel and TRP-1-treated mice (Fig 2A). There were significantly more TEff TRP-1-CD4+ T cells in the blood of pmel- and TRP-1-treated mice, however, this was only at the day 20 time point and did not translate to a proportional difference in the entire population (naive, TEff, TEM, and TCM) as we observed in the CD8+ T cells (Fig 2A and B). Mice treated with either pmel and TRP-1 or TRP-1 only had an equal percentage of tumor-specific IFN-γ-producing CD4+ T cells (Fig 2C). Since previous studies using the D5 experimental metastases model documented that CD8+ T cells were the dominant mechanism for eliminating tumor when endogenous tumor vaccine-primed T cells were used for adoptive immunotherapy, we focused on the effect CD4+ T cells had on the CD8+ T cells 7,35.
TRP-1 T cells help maintain pmel-CD8+ T cells
We found that following adoptive transfer of tumor-specific Tg CD4+ and CD8+ T cells, tumor had not recurred by 40 days and most animals were apparently cured of their disease (Fig 1B and data not shown). We hypothesized that CD4+ T cells could be helping to prime a small number of Tnaïve CD8+ T cells that still remain after αCD3/IL-2 expansion. Therefore, we phenotyped αCD3/IL-2-expanded pmel at the time of adoptive immuno-therapy (day 0). This analysis revealed a large population (15–20%) of phenotypically “naïve” (CD44loCD62L+) CD8+ T cells (referred to as αCD3/IL-2 expanded CD44loCD62L+) (Fig 2B). This observation surprised us, so we examined whether CD4+ T cells needed this αCD3/IL-2-expanded CD44loCD62L+ population to help prime CD8+ T cells or whether they were maintaining activated TEff phenotype CD8+ T cells. To test this, we eliminated the CD44loCD62L+ CD8+ T cells from the αCD3/IL-2 expanded population by sorting on TEff phenotype pmel (CD44+CD62Llo). We then compared treatment with 5 × 105 sorted effector CD44+CD62Llo pmel and 1000 TRP-1 T cells (sort Pmel+ TRP-1), 5 × 105 sorted TEff CD44+CD62Llo pmel alone (sort Pmel), 5 × 105 total αCD3/IL-2-expanded pmel and TRP-1 (total Pmel + TRP-1) or 5 × 105 total pmel alone (total Pmel) (Supporting Information Fig. 2B).
Immunotherapy with sorted TEff pmel or total pmel, combined with TRP-1 T cells had significantly less tumor growth at 10 and 20 days following treatment compared to mice treated with either pmel population alone (Fig 3A). The majority of mice treated with both pmel and TRP-1, either sorted or total, survived longer than 40 days with no symptoms of tumor progression (Fig 3B). Furthermore, while mice treated with sorted pmel and TRP-1 had fewer splenic CD8+ T cells than mice receiving total pmel and TRP-1, their numbers were still increased compared to mice treated with only total or sorted pmel T cells 10 days after transfer (Fig 3C). Since elimination of the αCD3/IL-2 expanded CD44loCD62L+ did not diminish the antitumor effects in vivo, it suggests that tumor-specific TRP-1-CD4+ T cells are able to maintain activated pmel-CD8+ T cells.
Figure 3.
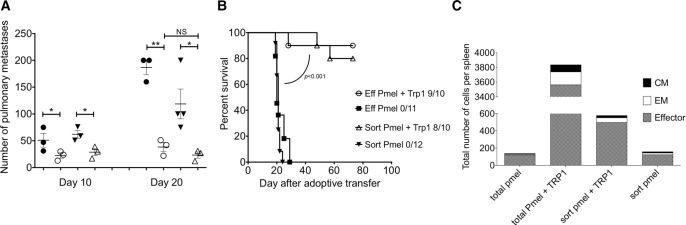
Tumor-specific TRP-1 T cells are important for maintaining activated pmel in the lymphopenic environment. (A and B) Mice were treated with 5 × 105 αCD3/IL-2-expanded sorted TEff (CD44+CD62Llo) pmel and 1000 TRP-1 cells (sort pmel + TRP-1), 5 × 105 total CD3/IL-2-expanded pmel and 1000 TRP-1 (total pmel + TRP-1), sort pmel alone or total pmel alone. (A) Number of pulmonary metastases 10 and 20 days after adoptive transfer. Data are shown as mean ± SEM of n = 3 mice per group and are representative of one out of two independent experiments. *p < 0.05, **p < 0.01; Student’s t-test (B) Numbers are mice that are tumor free of total. Data shown are from five to six mice and combined from two independent experiments. (C) Total number of CD3+CD8+ T cells with distribution of TEff, TEM, and TCM phenotypes in the spleen 10 days after immunotherapy. Data shown are one representative of two independent experiments.
Furthermore, comparison of proliferation and function of ex vivo αCD3/IL-2-expanded CD44loCD62L+, αCD3/IL-2-expanded TEff CD44+CD62Llo cells, and unstimulated pmel splenocytes showed that αCD3/IL-2 expanded CD44loCD62L+ and TEff CD44+CD62Llo pmel cells proliferated (>5×) and showed comparable functionality (IFN-γ+TNFα+) whereas unstimulated pmel splenocytes did not (Supporting Information Fig. 3). These data support that tumor-specific CD4+ T cells can act in conjunction with TEM CD8+ T cells to increase their numbers, tumor-specific function and efficacy, even in the absence of naïve T-cell priming.
TRP-1 help occurs early after adoptive immunotherapy
We attempted to determine when, in relation to adoptive transfer, tumor-specific CD4+ T cells were needed to maintain antitumor immunity. According to the model described in Figure 1A, CD4+ cells were depleted 1 day prior, 3 and 10 days following adoptive immunotherapy with pmel and TRP-1 cells. Anti-CD4 antibody was administered 1 day prior to adoptive transfer even though RAG1−/− mice have no T cells to ensure CD4+ cells were immediately eliminated upon transfer. We expected this to replicate adoptive transfer with pmel alone. Indeed, depletion of CD4+ cells 1 day prior to adoptive immunotherapy resulted in a large tumor burden that was similar to pmel treatment alone. CD4-depletion 10 days after transfer resulted in less tumor burden, similar to undepleted mice, depletion at day 3 gave results that were intermediate (Fig 4A). This suggests that in this model tumor-specific CD4+ T cells exert their effects during the first 10 days following adoptive transfer. We examined whether CD4 depletion changed the number of CD8+ T cells in the blood and spleen 17/18 days after immunotherapy, CD4-depleted groups showed a substantial decrease in the number of pmel-CD8+ T cells (Fig 4B and data not shown). We also looked at expression of the exhaustion marker PD-1 (J43 antibody) on splenocytes 17/18 days after adoptive transfer. PD-1 expression on pmel-CD8+ T cells was significantly higher among CD8+ cells in all nontherapeutic CD4-depleted groups (Day 1, 3 and pmel alone) than in CD8+ cells from undepleted mice (Fig 4C). Depletion of CD4+ cells 1 day before or 3 days after adoptive transfer also correlated with decreased expression of IFN-γ, IL-2, TNF-α, and Granzyme B measured at day 17/18, compared to deletion at day 10 or undepleted mice (Fig 4D). However, long-term antitumor immunity was still compromised in mice depleted of CD4+ T cells 10 days after transfer compared to undepleted mice (Fig 4E). Interestingly, mice depleted of CD4+ cells 10 days after transfer often developed tumors at metastatic sites, such as the skin (Fig 4E). This suggests that CD4+ T cells continue to maintain pmel-CD8+ T cells or potentially act to support trafficking of CD8+ T cells in order to control tumor metastases 36.
Figure 4.
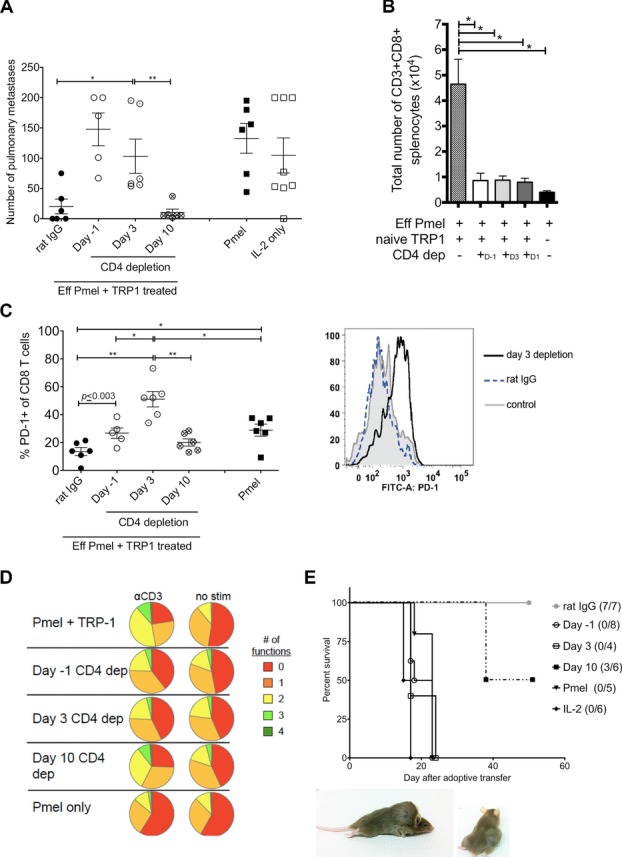
TRP-1 T cells are important early to reduce tumor burden. Mice treated with pmel and TRP-1 were depleted of CD4 cells one day prior, 3 or 10 days following adoptive transfer and compared to pmel alone or no treatment. All analysis was done 17/18 days following immunotherapy. (A) Total number of pulmonary metastases 17/18 days after adoptive transfer and survival. (B) Total number of CD3+CD8+ T cells in the spleen. (C) Frequency of PD-1 expressing CD3+CD8+ T cells 17/18 days after transfer. Histogram shows representative PD-1 expression on CD3+CD8+ cells comparing treatment with rat IgG treatment or CD4-depletion 3 days after transfer. (D) Summary of the number of cytokines expressed by CD8+ T cells (IL-2, TNF-α, IFN-γ, and Granzyme B) with 24 h αCD3 or no stimulation. (E) Survival curve and representative of distant skin metastases that occurred 38 days after transfer in mice depleted of CD4+ T cells 10 days after transfer. (A–C) Data are shown as mean ± SEM of n = 3–4 mice per group and are two combined experiments performed with all groups out of three independent experiments *p < 0.05, **p < 0.01; one-way ANOVA. (D and E) Data are one representative experiment of three independent experiments.
PD-1-blockade augments the therapeutic efficacy of pmel-CD8+ T cells
We next examined whether blocking PD-1 could restore the therapeutic efficacy of pmel T cells alone by treating mice with a PD-1-blocking antibody. PD-1-blockade augmented therapeutic efficacy of pmel T cells alone, significantly (p < 0.01) reducing the number of metastases compared to pmel T cells alone (31 ± 5 versus 82 ± 16 mean ± SEM) (Fig 5A). Since PD-1 treatment did not result in complete elimination of metastases, we examined other mechanisms of CD4+ T-cell help including CD40-CD40L, increasing survival or decreasing apoptosis 37–39. There was no difference in overall survival or in the total number and function of T cells with CD40-CD40L blockade (Supporting Information Fig. 4) suggesting that in our model enhancement of CD8+ T cells was not dependent on CD40–CD40L interactions. We also examined expression of the survival factor Bcl-2 and the apoptotic factor TRAIL in CD8+ T cells from pmel and TRP-1 or pmel-alone-treated mice. There was no difference in Bcl-2 expression; however, there was a substantial decrease in TRAIL expression when mice were treated with both pmel and TRP-1 T cells (Fig 5B), implying the addition of tumor-specific CD4+ T cells increases CD8+ T-cell persistence by reducing exhaustion by the PD-1 pathway and decreasing TRAIL-induced apoptosis.
Figure 5.
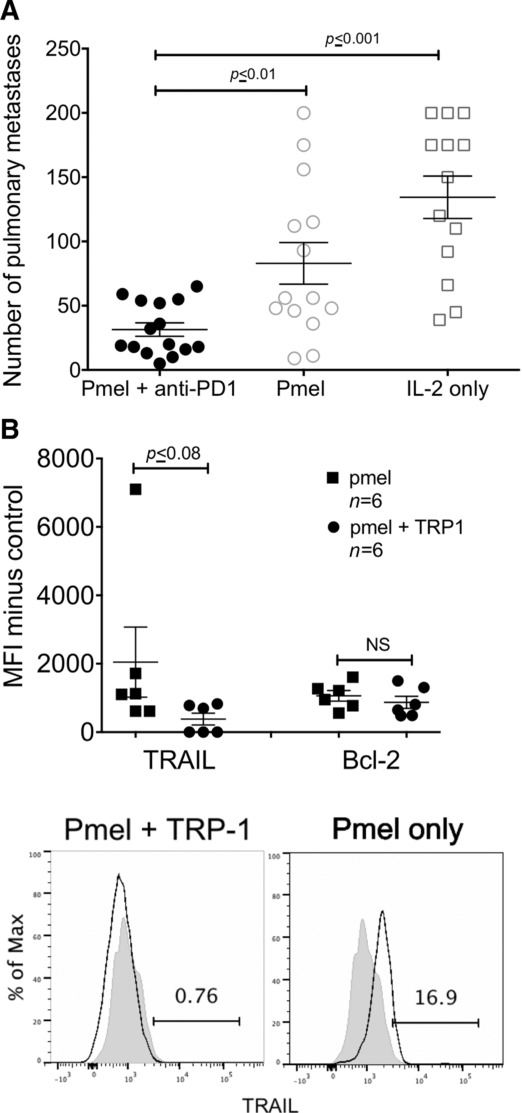
PD-1-blockade augments the therapeutic efficacy of pmel alone. (A) Number of pulmonary metastases from mice treated with pmel plus PD-1-blocking antibody (RMP1–14) or rat IgG. All mice received IL-2 (B). Mice treated with pmel and TRP-1 cells were compared to mice treated with pmel alone. Flow cytometry is shown for Live-CD3+CD8+ cells 18 days after adoptive transfer. MFI minus control (FMO) of Bcl-2 and TRAIL. Histograms representative TRAIL expression for pmel and TRP-1 or pmel-alone-treated mice. Black line is treatment, gray fill is FMO. Data are shown as mean ± SEM of (A) n = 3–6 or (B) n = 3 mice and are combined from (A) three or (B) two independent experiments. Statistics are (A) one-way ANOVA or (B) Students t-test.
Discussion
We recently found that tumor-vaccine-specific CD4+ T cells augmented therapeutic efficacy of immunotherapy with tumor-specific CD8+ T cells in the RAG1−/− lymphopenic environment (Friedman, manuscript in preparation). Here we take advantage of the tumor-specific TRP-1 MHC class II-restricted TCR Tg CD4+ T cells to examine the role of tumor-specific CD4+ T cells in the lymphopenic environment. Our results suggest that tumor-specific CD4+ T cells in combination with tumor-specific CD8+ T cells augment therapeutic efficacy, maintain long-term tumor control, and increase total survival and function of CD8+ T cells. Additionally, we show that physiological doses of tumor-specific CD4+ T cells could significantly (p < 0.001) augment therapeutic efficacy of immunotherapy with tumor-specific CD8+ T cells (Fig 1B). Treatment with both pmel and TRP-1 increased tumor-specific function, polyfunctionality, and total number of effector and memory CD8+ T cells compared to pmel alone (Fig 1C and2). This suggests tumor-specific CD4+ T cells enhance memory, survival, and effector function of tumor-specific CD8+ T cells. Adoptive immunotherapy with low doses of TRP-1-CD4+ cells used here did not result in significant therapeutic efficacy (Fig 1B). However, the TRP-1-only treated mice had significantly more CD4+ T cells in the spleen than pmel- and TRP-1-treated mice 10 and 20 days after transfer (Fig 1C and2A). This suggests that even though there are more CD4+ T cells in the spleen with TRP-1 treatment alone, the CD4+ T cells are less effective at eliminating tumor, possibly because they are trafficking to the spleen rather than the tumor.
Our data also suggest that tumor-specific CD4+ T cells can support therapeutic efficacy by maintaining effector CD8+ T cells even in the absence of phenotypically naïve CD8+ T cells (Fig 3 and Supporting Information Fig. 3). Ten days after transfer mice given TRP-1 and CD44+CD62L+ pmel had less total CD8+ T cells than those treated with TRP-1 and total pmel; however, this number was considerably more than either group treated with pmel alone (Fig 3C) and TRP-1 and CD44+CD62L+ pmel-treated mice did not develop tumor after more than 70 days (Fig 3A and B). This ability of CD4+ and CD8+ T cells to cure mice of systemic tumor burden in the absence of a source of naive CD8+ T cells is strong evidence that CD4+ T cells are maintaining CD8+ effector T cells in this model. Characterizing αCD3/IL-2 ex vivo expanded pmel showed that αCD3/IL-2-expanded CD44loCD62L+ cells were expressing some TNF-α and IFN-γ and proliferating similarly to the TEff phenotype cells (Supporting Information Fig. 3). One explanation for this apparent disconnect between phenotype and production of effector cytokines could be that CD62L and CD44 are being upregulated and downregulated during the αCD3/IL-2 stimulation causing them to display an atypical phenotype, including a population of CD44loCD62Llo cells that are neither a Tnaïve (CD44loCD62L+) or TEff (CD44+CD62Llo) phenotype (data not shown). If this is the case, it further supports that tumor-specific TRP-1 CD4+ T cells are acting to maintain activated CD8+ T cells in the lymphopenic environment.
Tumor-specific TRP-1-CD4+ T cells were particularly important early following adoptive transfer, as elimination within 3 days, but not 10 days (Fig 4A) resulted in partial loss of therapeutic efficacy and correlated with an increase in the exhaustion marker PD-1 on CD8+ T cells at day 18. The increase of PD-1 was most significant for the group depleted of CD4+ cells 3 days after transfer (Fig 4C), suggesting that the early time points may be particularly important for CD4+ help. These findings are consistent with reports showing that antigen-specific CD4+ T-cell help can decrease PD-1 expression on CD8+ T cells 20,40–42. Elimination of CD4+ cells 10 days after adoptive transfer did not reduce therapeutic efficacy, measured by enumeration of pulmonary metastases at day 18 or result in increased PD-1 expression on CD8+ T cells compared to undepleted mice. However, we did see a decrease in total number of pmel-CD8+ T cells in the day 10 depleted group. One possible explanation is that by day 10, when the CD4+ T cells are depleted, the majority of tumor has been eliminated and there is less antigen-driven proliferation of CD8+ T cells. Both undepleted mice and day 10 depleted mice also had polyfunctional (IFN-γ, TNF-α, Granzyme B, IL-2) CD8+ T cells, 17/18 days after adoptive transfer, which may be responsible for the enhanced antitumor efficacy at this time point (Fig 4D). Interestingly, eliminating CD4+ T cells 10 days after adoptive transfer resulted in late-onset distant metastases (skin, ovaries) 40 days after transfer (Fig 4E). This development of distant metastases is consistent with one previous study using adoptive immunotherapy with CD8+ T cells in MHC class II-deficient mice 18. Distant tumor metastases were not observed in mice that received both pmel and TRP-1 (undepleted), even 200 days after immunotherapy and might be explained by the role of CD4+ T-cell help in CD8+ T-cell trafficking, which has been observed in other models 36,43 or that CD4+ help is maintaining memory CD8+ T cells, which are important for tumor immune surveillance 44.
Investigating the mechanism of this CD4 support, we identified that PD-1 interactions play a role in limiting the antitumor effects of immunotherapy with nonhelped pmel-CD8+ T cells, as PD-1 blockade significantly (p < 0.01) improved therapeutic efficacy (Fig 5A). In contrast, blockade of CD40–CD40L interactions in animals treated with both tumor-specific CD4+ and CD8+ T cells did not decrease therapeutic efficacy, suggesting that these interactions are not essential (Supporting Information Fig. 4). Such an independence of CD40–CD40L interactions has been described in a number of other preclinical cancer immunotherapy models 35. Further, phenotypic characterization of T cells at times following adoptive transfer suggest tumor-specific TRP-1-CD4+ T cells maintain pmel-CD8+ T cells, in the lymphopenic environment, by decreasing TRAIL expression (Fig 5B). Together these data suggest that tumor-specific CD4+ T cells are important to reduce PD-1-mediated exhaustion and potentially TRAIL-mediated apoptosis during initial tumor elimination.
Approximately 33% of all pmel- and TRP-1-treated mice developed amelanotic tumors, 100–200 days after adoptive transfer, at the primary metastatic site in the pleural cavity (data not shown). This frequency of amelanotic tumor recurrence in mice treated with pmel and TRP-1 T cells is consistent with a recent report from Jensen et al. evaluating recurrence of experimental subcutaneous melanoma 45. In our study, mice that recurred with amelanotic tumor had a decreased total number and function of CD4+ and CD8+ T cells, and tumor cells had reduced expression of gp100 and TRP-1 protein (data not shown). This indicates antigen loss is a potential problem and antigen-specific CD4+ and CD8+ T cells may need additional combination therapy to prevent tumor recurrence or should consider targeting antigens that are critical to tumor’s survival. Furthermore lymphoreplete mice made lymphopenic by radiation and given immunotherapy with the same number of TRP-1 and pmel that were therapeutic in Rag1−/− mice, did not experience a significant reduction in tumor burden (Supporting Information Fig. 5). These findings underscore the complexities of the immune response and the preclinical model systems and identify opportunities for studies that may uncover novel mechanisms that limit antitumor immunity. Given our findings of the important role tumor-specific CD4 T cells play in augmenting therapy, combining CD8+ T-cell adoptive immunotherapy with vaccination that includes targets with CD4+ epitopes may be a good way to induce endogenous tumor-specific CD4+ helper T-cell responses. This approach has the theoretical advantage of developing a broad range of CD4+ T cells by targeting multiple tumor antigens or by eliciting epitope spreading of endogenous CD4+ T cells 46,47. This broad CD4+ T-cell repertoire might reduce the significance of tumor antigen-loss variants, which are seen in multiple preclinical models where a single antigen is targeted 47. Together our data strongly argues that tumor-specific CD4+ T cells play an important role in maintaining long-term systemic antitumor immunity and suggest that investigators designing clinical immunotherapy trials should consider a range of vaccine or adoptive immunotherapy options that include or promote both tumor-specific CD4+ and CD8+ T cells in order to optimize clinical outcome of patients on these trials.
Materials and methods
Tumor cell lines and metastases
We used the poorly immunogenic subclone, D5, isolated from the spontaneously arisen B16BL6 melanoma. This tumor cell line is defined as poorly immunogenic as vaccination with irradiated D5 fails to protect mice from a subsequent challenge with viable D5 48. MCA-310 or D5-G6, a D5 clone stability transduced to secrete GM-CSF were used to generate MCA-310 or D5-specific CD4+ T cells, as previously described 18. T-cell stimulation assays were done using D5 CIITA and the unrelated syngeneic sarcoma MCA-310 CIITA; both were modified to express CIITA 6. D5, D5-G6, D5 CIITA, and MCA-310 CIITA were propagated using 10% FBS RPMI 1640 supplemented with 2 mmol/L l-glutamine, 0.1 mmol/L nonessential amino acids, 1 mmol/L sodium pyruvate, 5 μg/mL gentamicin-sulfate (Lonza), and 50 μM/L β-mercaptoethanol (Sigma). All tumor cell lines were propagated for less than 6 weeks. Three-day established pulmonary metastases were generated by injecting 2 × 105 D5 cells i.v.
Mice and adoptive immunotherapy
TRP-1 TCR × tyrp-1bwRAG−/− (RAG1−/− tyrp1 protein-deficient MHC Class II-restricted TCR Tg) male mice were used to isolate tumor-specific TRP-1-CD4+ splenocytes (gift from Dr. Restifo and The Jackson Laboratory). TRP-1-CD4+ T cells are specific for the murine tyrosinase related protein 1 peptide (36). Tyrp-1 protein is expressed by the B16BL6-D5 and TRP-1-CD4+ T cells secrete IFN-γ in response to stimulation with class II+ D5 tumor cells. Female RAG1−/− tyrp1-protein-deficient littermates, which lack the TCR transgene or RAG1−/− were used as hosts. RAG1−/− pmel-1 (MHC class I-restricted TCR Tg) mice were generated by breeding RAG1−/− (The Jackson Laboratory) with pmel-1 mice (gift of Dr. Restifo), and used to isolate pmel tumor-specific CD8+ T cells from male mice.
Recognized principles of laboratory animal care were followed (Guide for the Care and Use of Laboratory Animals, National Research Council, 1996) and all animal protocols were approved by the EACRI animal care and use committee.
Single-cell suspensions of pmel splenocytes were incubated for 2 days on 5 μg/mL anti-CD3 (2C11) in a 24-well plate followed by 3 days with 60 IU/mL IL-2 (Chiron) in a lifecell tissue culture bag (Baxter) (referred to as αCD3-IL-2-expansion). TRP-1-CD4+ splenocyte suspensions were enriched using a pan T-cell isolation kit (Miltenyi). Intravenous injections used 106 αCD3/IL-2-expanded pmel and/or 1000 enriched TRP-1 cells, unless otherwise noted. Mice also received i.p. injections of 90,000 IU IL-2 (Chiron) given daily for 3 days. Mouse lungs were resected and stored in Feketes solution. Metastases were enumerated by counting black nodules on the lung surface. Maximum tumor burden was recorded as 200 metastases.
Flow cytometry
Spleens were disrupted using a 3-mL syringe in a 6-well plate and filtered to single-cell suspensions. Red blood cells were lysed using ACK buffer (Lonza). Cells were stained for phenotyping and sorting with combinations of the following antibodies CD4-Qdot605 (Invitrogen), CD8-PE-Cy7, CD3-Percp-eFluor710, CD62L-Pacific blue/eFluor450, CD127-PE, CD127-allophycocyanin-eFluor780, PD-1-FITC, CCR7-allophycocyanin, CD44-AF700, PD-1-PE, TRAIL-PE, Bcl-2-FITC, FOXP3-eFluor450 (eBioscience), CD4-allophycocyanin-Cy7, CD4-allophycocyanin-H7 (Becton Dickinson). PD-1 staining was done on splenocytes harvested 17/18 days after adoptive transfer using the anti-PD-1 (J43) antibody. Intracellular staining was performed using the eBioscience fix-perm kit. The gating strategy for memory T cells is shown in Supporting Information Fig. 2A.
Blood counts were calculated using Flow-Count Fluorospheres (Beckman Coulter). For intracellular cytokine staining, splenocytes were incubated 18–24 h adding 5 μg/mL Brefeldin A (Sigma) after 2 h. Cells were stained with LIVE/DEAD fixable yellow stain (Invitrogen-Molecular Probes), CD8-V500 and CD4-APC-H7 (Becton Dickinson). Cells were fixed and permeablized (Becton Dickinson) and then stained with IFN-γ-PE (Becton Dickinson), TNFα-FITC, Granzyme-B-PE-Cy7, and/or IL-2-eFluor450/Pacific Blue (eBioscience) or IL-2-Brilliant Violet 421 (Biolegend). ICS cells were gated on live-singlet lymphocytes negative for live-dead dye, followed by CD4+ or CD8+ and individual cytokines. Proliferation was detected using CFSE (Invitrogen-Molecular Probes) as previously reported 49. Gates were based on fluorescence minus one controls. All samples were run on a BD LSRII or BD Aria and analyzed using FlowJo (Treestar), Pestle, and SPICE (Courtesy of Mario Roederer).
Depletion and blocking antibodies
CD4- and CD8-depleting antibodies were made from 2.43 or GK1.5 hybridomas (ATCC), respectively by purifying ascites using Biosephra MEP Hypercel (Ciphergen) as described previously 35 or purchased (BioXcell). CD40L-blocking antibody (MR1), PD-1 (RMP1–14) and hamster IgG were purchased (BioXcell). Rat IgG control antibody was purchased (Sigma).
Statistics
Unpaired, paired Student’s t-tests, or one-way ANOVAs were done for analysis of cell numbers and phenotype using Prism (Graphpad). Mantel-Cox log rank tests were used to analyze survival curves (Prism, Graphpad). A p value of <0.05 was considered significant.
Acknowledgments
We would like to thank Dan Haley for assistance with initial set-up of flow panels and sorting experiments, Tacy Brotherton and Amanda Lyon for breeding and caring for the mice, Dr. Dubay for initial help setting up polychromatic flow analysis, and Michael Afentoulis for help with experiments. We would like to additionally thank Dr. Urba, Dr. van de Ven, Dr. Twitty, Dr. Parker, Dr. Davey, and Dr. Weinberg for reviewing this manuscript and for thoughtful discussions throughout this work. Funding sources: NCI CA 80564, NIH T32 AI007472.
Glossary
- D5
poorly immunogenic subclone, B16BL6-D5 melanoma
- pmel
gp100-specific CD8+ transgenic T cell
- Tg
transgenic
- TIL
tumor-infiltrating lymphocyte
- TRP-1
tyrosinase-related protein 1-specific CD4+ transgenic T cell
- tyrp-1
tyrosinase-related protein 1
Conflict of interest
The authors declare no commercial or financial conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher’s web-site
References
- 1.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 2.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mulé JJ, Ettinghausen SE, Spiess PJ, Shu S, Rosenberg SA. Antitumor efficacy of lymphokine-activated killer cells and recombinant interleukin-2 in vivo: survival benefit and mechanisms of tumor escape in mice undergoing immunotherapy. Cancer Res. 1986;46:676–683. [PubMed] [Google Scholar]
- 5.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu H-M, Poehlein CH, Urba WJ, Fox BA. Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res. 2002;62:3914–3919. [PubMed] [Google Scholar]
- 7.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 10.North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J. Exp. Med. 1982;155:1063–1074. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, Klebanoff CA, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117:808–814. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, Borman ZA, et al. Determinants of successful CD8+ T cell adoptive immunotherapy for large established tumors in mice. Clin. Cancer Res. 2011;17:5343–5352. doi: 10.1158/1078-0432.CCR-11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barth RJ, Mulé JJ, Spiess PJ, Rosenberg SA. Interferon gamma and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J. Exp. Med. 1991;173:647–658. doi: 10.1084/jem.173.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 17.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat. Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu HM, Winter H, Urba WJ, Fox BA. Divergent roles for CD4+ T cells in the priming and effector/memory phases of adoptive immunotherapy. J. Immunol. 2000;165:4246–4253. doi: 10.4049/jimmunol.165.8.4246. [DOI] [PubMed] [Google Scholar]
- 19.de Goër de Herve M-G, Cariou A, Simonetta F, Taoufik Y. Heterospecific CD4 help to rescue CD8 T cell killers. J. Immunol. 2008;181:5974–5980. doi: 10.4049/jimmunol.181.9.5974. [DOI] [PubMed] [Google Scholar]
- 20.Kmieciak M, Worschech A, Nikizad H, Gowda M, Habibi M, Depcrynski A, Wang E, et al. CD4+ T cells inhibit the neu-specific CD8+ T-cell exhaustion during the priming phase of immune responses against breast cancer. Breast Cancer Res. Treat. 2011;126:385–394. doi: 10.1007/s10549-010-0942-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LaCelle MG, Jensen SM, Fox BA. Partial CD4 depletion reduces regulatory T cells induced by multiple vaccinations and restores therapeutic efficacy. Clin. Cancer Res. 2009;15:6881–6890. doi: 10.1158/1078-0432.CCR-09-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Côté AL, Byrne KT, Steinberg SM, Zhang P, Turk MJ. Protective CD8 memory T cell responses to mouse melanoma are generated in the absence of CD4 T cell help. PLoS ONE. 2011;6:e26491. doi: 10.1371/journal.pone.0026491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, Surh CD. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J. Exp. Med. 2002;195:1523–1532. doi: 10.1084/jem.20020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J. Exp. Med. 2000;192:557–564. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen PA, Kim H, Fowler DH, Gress RE, Jakobsen MK, Alexander RB, Mulé JJ, et al. Use of interleukin-7, interleukin-2, and interferon-gamma to propagate CD4+ T cells in culture with maintained antigen specificity. J. Immunother. Emphasis Tumor Immunol. 1993;14:242–252. doi: 10.1097/00002371-199310000-00012. [DOI] [PubMed] [Google Scholar]
- 26.Curti BD, Ochoa AC, Powers GC, Kopp WC, Alvord WG, Janik JE, Gause BL, et al. Phase I trial of anti-CD3-stimulated CD4+ T cells, infusional interleukin-2, and cyclophosphamide in patients with advanced cancer. J. Clin. Oncol. 1998;16:2752–2760. doi: 10.1200/JCO.1998.16.8.2752. [DOI] [PubMed] [Google Scholar]
- 27.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, et al. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010;207:637–650. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EKM, Muranski P, et al. Naive tumor-specific CD4+ T cells differentiated in vivo eradicate established melanoma. J. Exp. Med. 2010;207:651–667. doi: 10.1084/jem.20091921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011;364:2119–2127. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Precopio ML, Betts MR, Parrino J, Price DA, Gostick E, Ambrozak DR, Asher TE, et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J. Exp. Med. 2007;204:1405–1416. doi: 10.1084/jem.20062363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frankel TL, Burns WR, Peng PD, Yu Z, Chinnasamy D, Wargo JA, Zheng Z, et al. Both CD4 and CD8 T cells mediate equally effective in vivo tumor treatment when engineered with a highly avid TCR targeting tyrosinase. J. Immunol. 2010;184:5988–5998. doi: 10.4049/jimmunol.1000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Almeida JR, Price DA, Papagno L, Arkoub ZA, Sauce D, Bornstein E, Asher TE, et al. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J. Exp. Med. 2007;204:2473–2485. doi: 10.1084/jem.20070784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu H-M, Winter H, Ma J, Croft M, Urba WJ, Fox BA. CD28, TNF receptor, and IL-12 are critical for CD4-independent cross-priming of therapeutic antitumor CD8+ T cells. J. Immunol. 2002;169:4897–4904. doi: 10.4049/jimmunol.169.9.4897. [DOI] [PubMed] [Google Scholar]
- 36.Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010;70:8368–8377. doi: 10.1158/0008-5472.CAN-10-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 38.Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, et al. CD4 +T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 39.Rapetti L, Meunier S, Pontoux C, Tanchot C. CD4 help regulates expression of crucial genes involved in CD8 T cell memory and sensitivity to regulatory elements. J. Immunol. 2008;181:299–308. doi: 10.4049/jimmunol.181.1.299. [DOI] [PubMed] [Google Scholar]
- 40.Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha S-J, Barber DL, Ye L, et al. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc. Natl. Acad. Sci. USA. 2011;108:21182–21187. doi: 10.1073/pnas.1118450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fuse S, Tsai CY, Molloy MJ, Allie SR, Zhang W, Yagita H, Usherwood EJ. Recall responses by helpless memory CD8+ T cells are restricted by the up-regulation of PD-1. J. Immunol. 2009;182:4244–4254. doi: 10.4049/jimmunol.0802041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, Yagita H, et al. PD-1 blockade enhances T cell migration to tumors by elevating IFN-γ inducible chemokines. Cancer Res. 2012;72:5209–5218. doi: 10.1158/0008-5472.CAN-12-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8+ T lymphocyte mobilization to virus-infected tissue requires CD4+ T-cell help. Nature. 2009;462:510–513. doi: 10.1038/nature08511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 45.Jensen SM, Twitty CG, Maston LD, Antony PA, Lim M, Hu H-M, Petrausch U, et al. Increased frequency of suppressive regulatory T cells and T cell-mediated antigen loss results in murine melanoma recurrence. J. Immunol. 2012;89:767–776. doi: 10.4049/jimmunol.1103822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol. Immunother. 2005;54:721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butterfield LH, Ribas A, Dissette VB, Amarnani SN, Vu HT, Oseguera D, Wang H-J, et al. Determinant spreading associated with clinical response in dendritic cell-based immunotherapy for malignant melanoma. Clin. Cancer Res. 2003;9:998–1008. [PubMed] [Google Scholar]
- 48.Hu HM, Urba WJ, Fox BA. Gene-modified tumor vaccine with therapeutic potential shifts tumor-specific T cell response from a type 2 to a type 1 cytokine profile. J. Immunol. 1998;161:3033–3041. [PubMed] [Google Scholar]
- 49.Quah BJC, Warren HS, Parish CR. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protoc. 2007;2:2049–2056. doi: 10.1038/nprot.2007.296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher’s web-site