

Abstract
Objectives
We investigated the safety, tolerability, pharmacokinetics and pharmacodynamics of evacetrapib.
Methods
Healthy volunteers received multiple daily doses of evacetrapib (10–600 mg) administered for up to 15 days in a placebo-controlled study.
Key findings
Mean peak plasma concentrations of evacetrapib occurred at 4–6 h and terminal half-life ranged 24–44 h. Steady state was achieved at approximately 10 days; all subjects had undetectable levels of evacetrapib 3 weeks after their last dose. The trough inhibition of cholesteryl ester transfer protein (CETP) activity was 65 and 84% at 100 and 300 mg, respectively. At the highest dose (600 mg), evacetrapib significantly inhibited CETP activity (91%), increased HDL-C (87%) and apo AI (42%), and decreased LDL-C (29%) and apo B (26%) relative to placebo. For the highest dose tested, levels of evacetrapib, CETP activity, CETP mass, HDL-C and LDL-C returned to levels at or near baseline after a 2-week washout period. Evacetrapib at the highest dose tested did not produce any significant effect on 24-h ambulatory systolic or diastolic blood pressure.
Conclusions
Multiple doses of evacetrapib potently inhibited CETP activity, leading to substantial elevations in HDL-C and lowering of LDL-C. Evacetrapib was devoid of clinically relevant effects on blood pressure and mineralocorticoid levels.
Keywords: cholesteryl ester transfer protein, evacetrapib, high-density lipoprotein, pharmacodynamic, pharmacokinetic
Introduction
Although aggressive lowering of low-density lipoprotein cholesterol (LDL-C) has demonstrated 20–35% relative reduction in cardiovascular (CV) events, additional therapies targeting other lipid-related risk factors are needed to address the residual cardiovascular disease (CVD). Low levels of high-density lipoprotein-cholesterol (HDL-C) were identified as an important risk factor for coronary heart disease over 3 decades ago.[1] HDL-C levels are inversely correlated with CVD risk, even in subjects with low LDL-C levels.[2] This epidemiological data suggest that agents that elevate HDL-C levels may have important clinical benefits in atherosclerosis management.
Current pharmacotherapeutic options available for increasing HDL-C, including statins, fibrates and niacin, produce only modest elevations in HDL-C or have limited tolerability.[3] Although strong pharmacologic inhibition of cholesteryl ester transfer protein (CETP) can increase HDL-C levels by more than 100%,[4] the CV benefit of CETP inhibition has yet to be demonstrated in prospective, randomized CV outcome trials.
Two CETP inhibitors, torcetrapib and dalcetrapib, have failed in late-stage clinical trials. Torcetrapib, the first drug in this class, was terminated after an excess risk of mortality and CV events, and increases in blood pressure and aldosterone levels were observed in the Phase 3 ILLUMINATE trial.[5] Increases in blood pressure and overall mineralocorticoid activity with torcetrapib have also been reported in vitro and in animal models,6–8 Dalcetrapib, a lower potency CETP inhibitor, was stopped based on interim results of the Phase 3 dal-OUTCOMES trial, which demonstrated futility in achieving the targeted outcomes with continued treatment.[9] In this trial, the mean systolic blood pressure was significantly higher (approximately 0.6 mmHg) in the dalcetrapib group than in the placebo group, even though there were no significant between-group differences in diastolic blood pressure or levels of plasma aldosterone, potassium or bicarbonate.
To date, anacetrapib and evacetrapib have not shown effects on either blood pressure or mineralcorticoid activity in preclinical and clinical studies.[4],[10] In the DEFINE Phase 3 safety study, anacetrapib had robust effects on LDL-C and HDL-C, while no changes were noted in blood pressure or electrolyte or aldosterone levels through 76 weeks.[4] In a Phase 2 study with nearly 400 dyslipidemic patients, evacetrapib showed significant dose-dependent inhibition of CETP activity, increased HDL-C levels by up to 129% and decreased LDL-C by up to 36%, without having effects on blood pressure or mineralocorticoid activity.[10]
In the current manuscript, we report further safety, tolerability, pharmacokinetic (PK) and pharmacodynamic (PD) results from a multiple ascending dose study of evacetrapib administered to healthy volunteers for up to 15 days.
Materials and Methods
Participants
We performed a single-centre, double-blind, placebo-controlled study that examined the safety, tolerability, PK and PD profiles of evacetrapib administered as multiple daily doses for up to 15 days in healthy male and female adult subjects. Day 14 data were used for the primary PK and PD analyses. Subjects had to have normal laboratory and heart rate measurements as determined by the investigator to be eligible for the study. Both supine and standing blood pressure had to be within the following limits: systolic blood pressure <140 mmHg and diastolic blood pressure and ≤90 mmHg. Body mass index had to be between 18.5 and 32 kg/m2, and fasting triglyceride and cholesterol levels had to be in the normal range. Within 14 days before dosing, subjects had to be willing to follow dietary restrictions throughout the study, maintaining relative consistency of sodium and potassium intake and avoidance of a low-sodium or high-potassium diet. Also, use of the following was excluded: herbal or dietary supplements, grapefruit and grapefruit products, and prescription and over-the-counter drugs known to inhibit cytochrome P450 (CYP) 3A activity. Medications for dyslipidemia were excluded for 30 days before dosing. Consumption of licorice products was excluded because licorice inhibits 11-β-hydroxysteroid dehydrogenase, with the potential to produce hyperaldosteronism-like clinical symptoms and signs.[11] Additional major exclusion criteria were: (1) regular use of drugs known to inhibit or induce CYP2C9, CYP2C8, CYP3A and CYP2D6 that may mediate drug–drug interactions and (2) smoking more than 10 cigarettes/day.
The Clinical Research Unit was located in Baltimore, Maryland and operated by Parexel. The study protocol was reviewed by Chesapeake Research Review, Inc. (Columbia, MD, USA), which has maintained full accreditation with the Association for the Accreditation of Hurman Research Protection Programs since 2004. All subjects provided written informed consent before participation in study procedures.
Study design
The study had a double-blind, randomized, parallel-group design. Adult subjects were randomized to receive placebo or evacetrapib in escalating doses of 10, 100, 300 and 600 mg administered as capsules once daily with a low-fat meal. Study drug was administered for 14 days in the 10 and 600-mg cohorts and for 15 days in the 100 and 300-mg cohorts, to accommodate additional PK/PD sampling for the drug–drug interaction analyses to be published elsewhere. Dose escalations did not occur until a minimum of five subjects received evacetrapib and tolerated the dose for at least 7 days of safety monitoring. An additional cohort was added as an amendment to the protocol to evaluate the effect of evacetrapib on blood pressure and the potential for skin rashes (in response to data from initial cohorts). This cohort consisted of subjects (n = 15) who were randomized to receive either placebo or evacetrapib 600 mg/day for 14 days, then crossed over to the other treatment after a 21-day washout period.
Bioanalysis
Plasma concentrations of evacetrapib were determined in acidified samples via use of a validated liquid chromatography with tandem mass spectrometry (LC/MS/MS) method. Briefly, evacetrapib was extracted from K2EDTA human plasma by solid phase extraction (SPE) with an anion exchange sorbent at basic pH using an Oasis MAX, 96-well plate (Cienytech, Santiago de Compostela, Spain). Before the extraction, isotope-labelled drug (evacetrapib – (13CD3)) was added as an internal standard. The SPE plate was washed with two separate methanol : water SPE washes before elution. Following the extraction, reconstituted extracts were injected into LC-MS/MS system (AB Sciex 4000 mass spectrometer, Analyst Version 1.4.1. equipped with a turbo ion spray interface, positive ionization mode; Framingham, MA, USA), utilizing a Zorbax Eclipse XDB-C18 column (4.6 × 75 mm, 3.5 μm; Agilent Technologies, Santa Clara, CA, USA) with a 250-micromolar ammonium formate buffer/H2O/methanol (4.0/9.0/87, v/v/v) mobile phase and flow rate of 1.0 ml/min. Selected reaction monitoring was used to detect evacetrapib and the internal standard with nominal mass transitions of m/z 639 → 314 and m/z 643.1 → 314, respectively. The lower limit of quantification was 1 ng/ml and the upper limit of quantification was 1000 ng/ml. Out-of-range samples were diluted and reanalyzed to yield results within the calibrated range. The interassay accuracy (% relative error) during validation ranged from −6.0 to 6.2%. The interassay precision (% relative standard deviation) during validation ranged 2.1–7.8%. All results were within the pre-specified acceptance criteria.
Pharmacokinetic parameters
The following PK parameters were calculated by non-compartmental methods using WinNonlin Enterprise (Version 5.0.1, Pharsight, Mountain View, CA, USA): maximal concentration (Cmax), time to Cmax (tmax), area under the concentration-time curve (AUC), apparent oral clearance (CL/F), apparent distribution volume at steady-state (Vss/F) and terminal half-life (t1/2). Six serial blood samples were collected after the first dose on Day 1 and 11 samples were collected through 336 h after the last dose (Day 14 or Day 15). In the cohorts that received a dose on Day 15 (100 and 300 mg), six serial blood samples were also collected on Day 14. Pre-dose blood samples for trough measurements were collected on Days 2, 3, 4, 7 and 10. PK data were analyzed for all randomized subjects who received at least one dose of evacetrapib. Dose proportionality of evacetrapib exposure on Day 14 was evaluated using a power model.[12]
Pharmacodynamic measurements
Serial samples for serum CETP activity were collected at the same times as for the measurement of PK parameters (above). Serum CETP activity was measured by fluorometric assay and expressed as the maximum inhibitable CETP activity with evacetrapib (Pacific Biomarkers, Seattle, WA, USA). CETP mass was measured in serum by a standard sandwich ELISA procedure (Polymedco, Courtlandt Manor, NY, USA) from samples collected at baseline and on Days 2, 7 and 14. Lipid analyses were performed at Covance Central Laboratory Services on a Roche Modular Analyzer (Roche Diagnostics, Indianapolis, IN, USA). Total cholesterol and triglycerides were measured by standard enzymatic methods. HDL-C was measured on a Roche Hitachi analyzer (Roche Diagnostics) after separation of LDL and VLDL by standard dextran sulfate precipitation and centrifugation. LDL-C was measured by a direct method (LDL-C Plus 2nd generation assay; Roche Diagnostics). Apolipoprotein (apo) concentrations were assayed by standard immunoturbidimetric methods on a Roche Modular Analyzer.
PD data were analyzed for all randomized subjects who received at least one study dose. Emax models were applied for the analysis of the relationships between dose of evacetrapib and changes in CETP activity and lipoproteins from baseline to 14 days, assuming a Hill coefficient of unity, and including baseline measurement as a covariate. Linear models were used for the analysis of total cholesterol and triglycerides. For the analysis of the relationships between plasma evacetrapib exposure and HDL or LDL change from baseline on Day 14, data were fit to an Emax model of the form
where Emax is the maximal response, AUC is the area under the plasma concentration vs time curve at steady-state for evacetrapib, and EAUC50 is the AUC producing 50% of the maximal response.
Safety parameters
Safety monitoring included assessments of vital signs, serum and plasma electrolyes, plasma renin activity, aldosterone and plasma and salivary cortisol and adverse event reporting. In addition to conventional blood pressure, blood pressure was monitored via ambulatory blood pressure monitoring (ABPM, Spacelabs Medical, Issaquah, WA, USA). Subjects were acclimated to wearing the ABPM device for approximately 12 h before the start of baseline measurements on Day 1. On Day 14, study drug (evacetrapib or placebo) was administered approximately 1 h after the start of ABPM. The device recorded ambulatory blood pressure and pulse every 20 min during the daytime and every 30 min throughout the night time. The first hour of data before study drug administration was excluded from data analysis, providing a 24-h measurement. To maximize detection of effects on blood pressure, a separate cohort received investigator and subject-blinded placebo and evacetrapib at 600 mg in a random treatment sequence over two periods with 24-h ABPM at baseline and on Day 14 for each period. Safety data were analyzed for all enrolled subjects. Adverse events were summarized by Medical Dictionary for Regulatory Acitivities (MedDRA 11.0) preferred term. ABPM measurements were analyzed as the mean value of the measurements for systolic blood pressure (SBP) and diastolic blood pressure (DBP) over the 24-h period following dosing of study drug.
Results
Demographics
The study enrolled 76 subjects (52 (68%) African-Americans; 22 (29%) Caucasians; 2 (3%) Asians), with a mean (± standard deviation (SD)) age of 36.8 (±9.6) years (range 21–60 years). The majority of subjects were men (69/76; 91%). The mean BMI was 26.4 ± 3.1 kg/m2.
Pharmacokinetics
Figure 1 shows plasma concentration-time curves for evacetrapib (10, 100, 300 and 600 mg) after administration on Day 1 (a) and after the last day of dosing (b). Peak plasma concentrations of evacetrapib were typically observed approximately 4 h after dosing and did not appear to change between the first and last dose. Steady state appeared to be achieved after approximately 10 days of dosing. Table 1 shows PK parameters calculated by non-compartmental methods after multiple doses of evacetrapib. The mean terminal half-life ranged from approximately 24 to 44 h between doses of 10 and 600 mg, with no clear dependency on dose. Plasma exposure to evacetrapib increased in a less than dose-proportional manner. After 14 daily doses, AUCt,ss for evacetrapib 600 mg was approximately threefold higher than for evacetrapib 100 mg. Based on trough concentrations, the accumulation ratio during multiple dosing between Days 1 and 14 ranged 2–2.5. Mean apparent oral clearance of evacetrapib at steady state ranged 17.5–43.6 l/h and mean apparent steady-state volume of distribution ranged 543–1740 l. After a 2-week (i.e. 336 h) washout period, levels of evacetrapib for each dose group decreased to near the lower limit of detection. In the crossover cohort that received either placebo or evacetrapib 600 mg/day for 14 days, all subjects had undetectable levels of evacetrapib after a 3-week washout (data not shown).
Figure 1.

Plasma concentration (arithmetic means) curve vs time for evacetrapib following first dose (a) and last dose (b) of evacetrapib.
Table 1.
Pharmacokinetic parameters of evacetrapib after multiple doses
| Parameter | Steady-state values | |||
|---|---|---|---|---|
| 10 mg | 100 mg | 300 mg | 600 mg | |
| n | 12 | 11 | 10 | 25 |
| Cmax (ng/ml) | 41.6 (28) | 377 (34) | 744 (49) | 1330 (36) |
| tmaxa (h) | 4.02 (4.00–6.02) | 4.02 (4.00–8.00) | 4.00 (3.98–6.03) | 4.02 (0.00–6.08) |
| t1/2b (h) | 23.6 (14.4–43.2) | 44c (25.9–54.4) | 37.1 (26.8–51.3) | 37.4d (22.3–63.2) |
| AUCt,SS (ng · h/ml) | 572 (21) | 4610 (30) | 7570 (44) | 13 700e (34) |
| CL/F (l/h) | 17.5 (21) | 21.7 (30) | 39.6 (44) | 43.6e (34) |
| VSS/F (l) | 543 (29) | 998c (38) | 1350 (53) | 1740d (55) |
AUCt,SS, area under the concentration vs time curve over dosing interval at steady state; AUC(0-∞), area under the concentration vs time curve from 0 to infinite time; CL/F, apparent oral clearance; Cmax, maximum plasma concentration; tmax, time to Cmax; t1/2, terminal half-life; Vss/F, apparent volume of distribution at steady state after extravascular administration. Results are expressed as geometric means (coefficient of variation, %) except for amedians (range) and bgeometric means (range). cn = 10. dn = 11. Combined values from the initial 600-mg dose escalation cohort and the additional 600-mg cohort used to further evaluate blood pressure. Fewer PK samples were drawn from the additional 600-mg cohort, which prevented the evaluation of the terminal elimination rate for this cohort. Thus, t1/2,ss and Vss/F could not be reported for the additional 600-mg cohort. en = 24.
Pharmacodynamics
Cholesteryl ester transfer protein activity and mass
CETP activity decreased in a dose-dependent manner with multiple ascending doses of evacetrapib. Figure 2 shows mean CETP activity change from baseline vs time for Days 1 (a) and 14 (b) across each dose cohort of evacetrapib. Maximal reduction in CETP activity occurred approximately 6 h after administration, corresponding to evacetrapib tmax. At the highest dose tested (600 mg), the trough inhibition of CETP activity was 91.2% (Table 2). After a 2-week (i.e. 336 h) washout period, CETP activity for each dose group returned to baseline levels. CETP mass increased in a dose-dependent fashion over the dosing period, then returned to baseline approximately 2 weeks after the last dose of evacetrapib (data not shown).
Figure 2.

Change in plasma cholesteryl ester transfer protein (CETP) activity (arithmetic means ± standard deviation) following first dose (a) and last dose (b) of evacetrapib.
Table 2.
Changes in pharmacodynamic parameters at trough level (predose) on day 14
| Dose | n | Mean % change from baseline (90% CI) | Mean % change vs placebo (90 % CI) |
|---|---|---|---|
| CETP activity | |||
| Placebo | 19 | 1.48 (−7.39, 10.35) | |
| 10 mg | 12 | −14.60 (−22.33, −6.87) | −16.08 (−23.23, −8.94)*** |
| 100 mg | 12 | −64.54 (−73.08, −56.00) | −66.02 (−78.93, −53.11)*** |
| 300 mg | 10 | −84.26 (−91.34, −77.18) | −85.74 (−97.43, −74.05)*** |
| 600 mg | 19 | −91.18 (−99.71, −82.66) | −92.66 (−104.92, −80.41)*** |
| CETP mass | |||
| Placebo | 19 | −0.40 (−12.63, 11.84) | |
| 10 mg | 12 | 20.18 (9.92, 30.45) | 20.58 (11.28, 29.88)*** |
| 100 mg | 12 | 95.77 (82.14, 109.41) | 96.17 (76.37, 115.97)*** |
| 300 mg | 10 | 131.72 (122.10, 141.34) | 132.12 (115.85, 148.38)*** |
| 600 mg | 19 | 145.34 (133.29, 157.39) | 145.73 (128.99, 162.48)*** |
| Total cholesterol | |||
| Placebo | 26 | −0.65 (−8.14, 6.83) | |
| 10 mg | 12 | −4.92 (−15.93, 6.10) | −4.26 (−17.58, 9.06) |
| 100 mg | 12 | 6.83 (−4.18, 17.85) | 7.49 (−5.83, 20.81) |
| 300 mg | 10 | 15.60 (3.53, 27.67) | 16.25 (2.05, 30.46) |
| 600 mg | 25 | 15.48 (7.85, 23.11) | 16.13 (5.44, 26.82)* |
| HDL-C | |||
| Placebo | 19 | −6.33 (−14.31, 1.64) | |
| 10 mg | 12 | 11.69 (4.39, 18.99) | 18.02 (9.76, 26.28)*** |
| 100 mg | 12 | 59.28 (51.54, 67.02) | 65.61 (53.34, 77.88)*** |
| 300 mg | 10 | 75.22 (68.87, 81.58) | 81.56 (70.90, 92.22)*** |
| 600 mg | 19 | 80.50 (73.05, 87.95) | 86.83 (75.96, 97.71)*** |
| LDL-C | |||
| Placebo | 19 | 1.65 (−3.43, 6.73) | |
| 10 mg | 12 | −11.94 (−18.31, −5.57) | −13.59 (−21.83, −5.35)** |
| 100 mg | 12 | −24.47 (−27.68, −21.26) | −26.13 (−32.39, −19.86)*** |
| 300 mg | 10 | −26.39 (−29.92, −22.85) | −28.04 (−34.18, −21.90)*** |
| 600 mg | 19 | −26.91 (−30.71, −23.11) | −28.56 (−34.76, −22.37)*** |
| Triglycerides | |||
| Placebo | 26 | −18.42 (−27.92, −8.93) | |
| 10 mg | 12 | −12.92 (−26.89, 1.06) | 5.51 (−11.39, 22.40) |
| 100 mg | 12 | −3.75 (−17.73, 10.23) | 14.67 (−2.22, 31.57) |
| 300 mg | 10 | −2.10 (−17.41, 13.21) | 16.32 (−1.69, 34.34) |
| 600 mg | 25 | −22.08 (−31.76, −12.40) | −3.66 (−17.22, 9.91) |
| Apo AI | |||
| Placebo | 12 | −11.76 (−18.51, −5.01) | |
| 10 mg | 12 | 3.53 (−2.23, 9.30) | 15.29 (7.74, 22.84)** |
| 100 mg | 12 | 24.61 (20.07, 29.15) | 36.37 (27.66, 45.08)*** |
| 300 mg | 10 | 28.75 (23.70, 33.80) | 40.50 (31.66, 49.34)*** |
| 600 mg | 11 | 29.93 (24.51, 35.35) | 41.69 (32.70, 50.67)*** |
| Apo AII | |||
| Placebo | 12 | −5.59 (−10.57, −0.61) | |
| 10 mg | 12 | −1.30 (−5.34, 2.73) | 4.29 (−0.90, 9.48) |
| 100 mg | 12 | 8.85 (4.69, 13.02) | 14.44 (7.16, 21.72)** |
| 300 mg | 10 | 11.93 (7.75, 16.10) | 17.52 (10.86, 24.17)*** |
| 600 mg | 11 | 12.91 (8.06, 17.77) | 18.50 (11.71, 25.29)*** |
| Apo B | |||
| Placebo | 12 | 6.96 (1.27, 12.66) | |
| 10 mg | 12 | −8.09 (−13.41, −2.77) | −15.05 (−22.68, −7.43)** |
| 100 mg | 12 | −17.64 (−20.84, −14.43) | −24.60 (−31.28, −17.92)*** |
| 300 mg | 10 | −18.85 (−22.45, −15.25) | −25.81 (−32.59, −19.04)*** |
| 600 mg | 11 | −19.17 (−22.94, −15.41) | −26.14 (−32.97, −19.30)*** |
| Ratio of apo B/apo AI | |||
| Placebo | 12 | 19.76 (13.28, 26.24) | |
| 10 mg | 12 | −6.87 (−12.72, −1.02) | −26.63 (−34.88, −18.38)*** |
| 100 mg | 12 | −31.84 (−35.59, −28.09) | −51.60 (−59.36, −43.83)*** |
| 300 mg | 10 | −35.69 (−39.92, −31.46) | −55.45 (−63.26, −47.63)*** |
| 600 mg | 11 | −36.74 (−41.23, −32.25) | −56.50 (−64.39, −48.61)*** |
| Apo E | |||
| Placebo | 12 | 11.35 (−7.54, 30.24) | |
| 10 mg | 12 | 19.70 (5.10, 34.30) | 8.36 (−2.89, 19.60) |
| 100 mg | 12 | 55.81 (36.13, 75.50) | 44.47 (14.63, 74.30)* |
| 300 mg | 10 | 76.75 (62.23, 91.28) | 65.40 (40.39, 90.42)*** |
| 600 mg | 11 | 85.48 (65.47, 105.49) | 74.13 (47.95, 100.31)*** |
Apo, Apolipoprotein; CI, confidence interval; CETP, cholesteryl ester transfer protein. *P < 0.05; **P < 0.01; ***P < 0.001. n = number of subjects per dose group.
High-density lipoprotein and low-density lipoprotein cholesterol
Multiple daily doses of evacetrapib produced dose-dependent increases in HDL-C and decreases in LDL-C, which were significant at all doses examined (Table 2). At the highest dose tested (600 mg), evacetrapib increased HDL-C by 86.8% (P < 0.001 vs placebo) and decreased LDL by 28.6% (P < 0.001 vs placebo) from baseline to Day 14. After a 2-week washout period, HDL-C and LDL-C returned to levels at or near baseline for all dose groups. For the 600-mg dose group; the change (SD) from baseline to 2 weeks after the last dose was 7.3% (16.2) for HDL-C and −2.6% (20.0) for LDL-C.
The relationships between plasma exposure to evacetrapib and the change from baseline in HDL-C and LDL-C levels on Day 14 were further explored with an Emax model (Figure 3b). Baseline HDL-C had a significant impact on HDL-C change from baseline, whereas baseline LDL-C did not significantly impact its change from baseline. The maximum theoretical change from baseline for HDL-C and LDL-C after 14 days of dosing with evacetrapib were (model estimate, 90% confidence interval) 126% (107 to 144) and −28.2% (−34.7 to −22.2), respectively. The EAUC50 for the HDL-C model and LDL-C models were 4110 ng · h/ml (2410 to 6300) and 582 (194 to 1810), respectively, indicating that a maximal LDL-C change occurred at a lower exposure than for HDL-C change.
Figure 3.
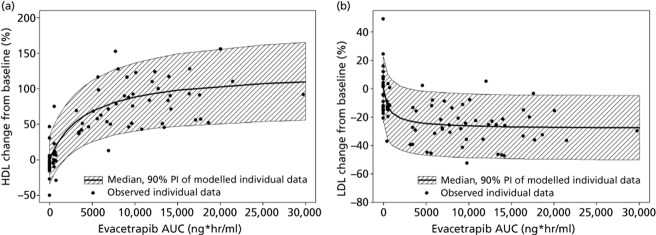
Relationship between evacetrapib exposure (AUC) and changes from baseline in HDL-C (a) and LDL-C (b) on Day 14 based on observed subject data and Emax model.
Apolipoproteins, total cholesterol and triglycerides
The effect of multiple daily dosing with evacetrapib on mean percent change from baseline to Day 14 in apo AI, apo AII, apo B, apo E, total cholesterol and triglycerides is shown in Table 2. Evacetrapib increased apo AI, apo AII, and apo E levels, and decreased apo B levels in a dose-dependent manner relative to placebo over the dose range of 10–600 mg. For 600 mg evacetrapib, increases in apo AI, apo AII and apo E vs placebo were 41.7, 18.5, and 74.1%, respectively, while decreases in apo B and apo B/AI ratio were 26.1 and 56.5%, respectively, at 600 mg. The increase in apo AI and decreases in apo B and ratio of apo B to apo AI were significant at all dose levels of evacetrapib. The increase in apo AII and apo E were significant at dose levels ≥100 mg of evacetrapib. There was a statistically significant mean % increase in total cholesterol with 600-mg evacetrapib compared with placebo. There were no statistically significant changes in triglycerides.
Safety findings
Evacetrapib was generally well tolerated in healthy subjects. Adverse events (AEs) were mild to moderate in severity and none had a dose-related pattern within a dose range of 10–600 mg. No clinically relevant changes in routine serum chemistries (including liver transaminases, complete blood counts and urinalysis) were observed with evacetrapib in the study. Four subjects reported rash-related AEs (i.e. with the terms ‘rash’ (2), drug eruption (1) and rash papular (1)) and discontinued the study. These four cases of rash were assessed by the investigator to be related to evacetrapib administration: two at the 300-mg dose level and two at the 600-mg dose level. One of the rashes in a subject receiving 600 mg was a drug eruption with recurrent fever that occurred after 8 days of evacetrapib. This was reported as a serious adverse event (SAE) because hospitalization was required for further evaluation. This event resolved after discontinuation of evacetrapib and treatment with oral corticosteroid therapy.
To exclude potential effects of evacetrapib on blood pressure, the additional cohort using crossover design was used to make within-subject comparisons of ABPM at the highest dose. No significant differences in either SBP or DBP were observed with evacetrapib 600 mg compared with placebo (Table 3). There was no correlation between exposure and blood pressure in this crossover amendment (Spearman's rank correlation coefficient = 0.00954; P = 0.964). There were no clinically relevant differences between evacetrapib and placebo in changes in the following laboratory measurements: plasma potassium, serum sodium, serum bicarbonate plasma renin activity or plasma cortisol or salivary cortisol (data not shown).
Table 3.
Effects of evacetrapib on 24-h ambulatory blood pressure
| Treatment group | n | Day 1 mean (mmHg) | Day 14 mean (mmHg) | LS mean change (mmHg) | Difference | Comparison with placebo | P-value |
|---|---|---|---|---|---|---|---|
| Systolic blood pressure | |||||||
| Placebo | 13 | 109.29 (105.02, 113.57) | 118.34 (115.54, 121.14) | 1.69 (−0.68, 4.07) | |||
| Evacetrapib 600 mg | 14 | 112.16 (109.12, 115.19) | 118.28 (113.78, 122.77) | −0.27 (−2.64, 2.11) | −1.96 | (−5.32, 1.40) | 0.327 |
| Diastolic blood pressure | |||||||
| Placebo | 13 | 67.55 (63.39, 71.71) | 74.56 (72.00, 77.12) | 2.88 (0.83, 4.93) | |||
| Evacetrapib 600 mg | 14 | 68.27 (64.82, 71.72) | 73.72 (70.26, 77.18) | 0.60 (−1.45, 2.65) | −2.28 | (−5.18, 0.62) | 0.191 |
CI, confidence interval; LS mean, least-squares mean; mmHg, millimetres of mercury; n, number. Results are expressed as means (90% CI).
Discussion
Here, we demonstrate significant dose-related inhibition of CETP activity, increases in HDL-C and decreases in LDL-C following multiple doses of the potent CETP inhibitor evacetrapib in healthy adults. After multiple doses, the highest dose of evacetrapib (600 mg) resulted in 93% inhibition of CETP activity, 87% increase in HDL-C and 29% decrease in LDL-C compared with placebo. The pharmacodynamic effects of evacetrapib in healthy adults are similar to those reported in the Phase 2 study of evacetrapib in dyslipidemic patients.[10]
The time course of CETP inhibition appears to be similar for evacetrapib and anacetrapib, with peak inhibition occurring 6 and 4 h after dosing, respectively, in both cases corresponding to Tmax values for plasma drug concentrations.[13] Less than dose-proportional increases in evacetrapib exposure were observed in healthy volunteers studied here, as well as in dyslipidemic subjects in the Phase 2 study.[14] The less than dose-proportional increases in exposure for both evacetrapib and anacetrapib likely results from their limited solubility in the gastrointestinal tract, which in turn limits the oral absorption of these molecules. The present study showed that complete washout of evacetrapib occurred within the 3-week washout period after 14 daily doses of 600 mg. This observation is consistent with the terminal half-life of evacetrapib. Anacetrapib has shown significant plasma concentrations 12 weeks after cessation of treatment in the Determining the EFficacy and tolerability of CETP INhibition with AnacEtrapib (DEFINE) study,[15] suggesting that evacetrapib and anacetrapib may differ in the way they are distributed and/or eliminated. The PD effects of evacetrapib, including inhibition of CETP activity and increases and decreases in HDL-C and LDL-C, respectively, return near baseline levels after a 2-week washout period.
The analyses between exposure of evacetrapib and changes in HDL-C and LDL-C on Day 14 indicate that the dose range that was tested in this study produced exposures that covered the range of minimal to maximal effect for these PD endpoints. Maximal effect on LDL-C appeared to be produced by a lower evacetrapib exposure than HDL-C. Further exploration of the relationships between evacetrapib exposure and duration of treatment and changes in HDL-C and LDL-C are discussed in a publication of the PK/PD analysis for the evacetrapib Phase 2 study.[14]
All doses of evacetrapib examined here (10–600 mg) produced a substantial and significant increase in apo AI and decrease in apo B levels, with consequent decrease of the apo B to apo AI ratio. Apo AII and apo E levels also significantly increased at doses of 100 mg or more, as previously reported.[10] Taken together, these observations are consistent with a reduced fractional catabolic rate of HDL following pharmacological CETP inhibition.[16] This is supported by data in homozygous CETP deficiency, which causes markedly delayed catabolism of apo AI and apo AII without affecting the production rates of these apos.[17] Increased levels of apo E have previously been reported in HDL of subjects with CETP deficiency and those treated with CETP inhibitors and has been associated with an increased ability of HDL particles to promote cholesterol efflux.[18]
Safety findings from the present monotherapy studies align with safety results of the Phase 2 study, which showed that 12-week therapy with daily doses of evacetrapib up to 500 mg as monotherapy or 100 mg as combination therapy with a statin was well tolerated.[10] Neither the present studies nor the Phase 2 study demonstrated any liver, kidney or muscle safety concern. A recent study has shown that evacetrapib appears to be well tolerated in doses of up to 1200 mg for 10 days in healthy volunteers, with no evidence of QT interval prolongation.[19]
Consistent with the Phase 2 study, these results indicate that evacetrapib had no adverse effects on blood pressure. In the evacetrapib 600-mg cohort, no significant differences were observed between evacetrapib and placebo in 24-h ambulatory systolic or diastolic blood pressure monitoring. In addition, multiple daily doses of evacetrapib had no effect on aldosterone, suggesting there was no increase in mineralocorticoid levels.[10] Therefore, evacetrapib, like other CETP inhibitors examined in late-stage clinical testing, does not seem to exhibit the presumably off-target effects on aldosterone and blood pressure observed with torcetrapib.[3],[20] These findings are supported by a branched DNA analysis that showed that torcetrapib, but not evacetrapib, induced expression of genes regulating CYP11B1 (steroid 11 β-hydroxylase) and CYP11B2 (aldosterone synthase), leading to increases in in-vitro production of aldosterone and cortisol.[7],[8],[21]
The four cases of rash during multiple dosing with evacetrapib occurred with the middle to higher dose groups (two subjects on 300 mg and two subjects on 600 mg) and resolved after discontinuing therapy or after a short course of corticosteroid treatment. In the SAE case, the ensuing hospital work-up for drug eruption and fever lacked evidence of other organ involvement or infectious aetiology. Attribution of the SAE was confounded by prior administration of concomitant medications before onset of drug eruption and fever. One significant rash also was observed in an evacetrapib-treated patient in the Phase 2 trial, but the relationship to study drug was confounded by the concomitant administration of rosuvastatin and the delay in the onset of the rash relative to completion of study drug.[10]
The Phase 1 study discussed here was conducted in a small number of healthy adults and was limited to 15 days of daily dosing with evacetrapib, thus limiting extrapolation of our results to a dyslipidemic patient population. However, the Phase 2 study entailed daily administration of evacetrapib up to 90 days in 400 dyslipidemic patients and provides supporting evidence for continued development of evacetrapib.[10] Confirmation of a beneficial effect of evacetrapib on CV events in patients with high-risk vascular disease awaits completion of the ongoing CV outcomes study, Assessment of Clinical Effects of Cholesteryl Ester Transfer Protein Inhibition with Evacetrapib in Patients at a High-Risk for Vascular Outcomes (ACCELERATE (ClinicalTrials.gov identifier NCT01687998)).
Conclusion
Multiple doses of evacetrapib (10–600 mg) potently inhibited CETP activity, leading to substantial elevations in HDL-C and lowering of LDL-C. The PK and PD effects of evacetrapib returned to levels near or at baseline 2 weeks after the last dose in this multiple ascending dose study. Evacetrapib was well tolerated and was devoid of clinically relevant effects on blood pressure and mineralocorticoid levels.
Declarations
Conflict of interest
All authors are employees of Eli Lilly and Company and hold equity ownership and/or stock options in Eli Lilly and Company.
Funding
This study was funded by Eli Lilly and Company.
Acknowledgments
The authors thank Drs Malcolm Ian Mitchell, Jennie Lin Francis and Wei Zhang for helpful review of this manuscript.
References
- Gordon T. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- deGoma EM. Clinical significance of high-density lipoprotein cholesterol in patients with low low-density lipoprotein cholesterol. J Am Coll Cardiol. 2008;51:49–55. doi: 10.1016/j.jacc.2007.07.086. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ. Evacetrapib. Curr Cardiol Rep. 2012;14:245–250. doi: 10.1007/s11886-012-0252-3. [DOI] [PubMed] [Google Scholar]
- Cannon CP. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- Barter PJ. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- Forrest MJ. Torcetrapib-induced blood pressure elevation is independent of CETP inhibition and is accompanied by increased circulating levels of aldosterone. Br J Pharmacol. 2008;154:1465–1473. doi: 10.1038/bjp.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G. Evacetrapib is a novel, potent, and selective inhibitor of cholesteryl ester transfer protein that elevates HDL cholesterol without inducing aldosterone or increasing blood pressure. J Lipid Res. 2011;52:2169–2176. doi: 10.1194/jlr.M018069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X. Torcetrapib induces aldosterone and cortisol production by an intracellular calcium-mediated mechanism independently of cholesteryl ester transfer protein (CETP) inhibition. Endocrinology. 2009;150:2211–2219. doi: 10.1210/en.2008-1512. [DOI] [PubMed] [Google Scholar]
- Schwartz GG. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: a randomized controlled trial. JAMA. 2011;306:2099–2109. doi: 10.1001/jama.2011.1649. [DOI] [PubMed] [Google Scholar]
- Ferrari P. The role of 11β-hydroxysteroid dehydrogenase type 2 in human hypertension. Biochim Biophys Acta. 2010;1802:1178–1187. doi: 10.1016/j.bbadis.2009.10.017. [DOI] [PubMed] [Google Scholar]
- Smith BP. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17:1278–1283. doi: 10.1023/a:1026451721686. [DOI] [PubMed] [Google Scholar]
- Krishna R. Multiple-dose pharmacodynamics and pharmacokinetics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Clin Pharmacol Ther. 2008;84:679–683. doi: 10.1038/clpt.2008.109. [DOI] [PubMed] [Google Scholar]
- Friedrich S. The pharmacokinetics and pharmacokinetic/pharmacodynamic relationships of evacetrapib administered as monotherapy or in combination with statins. CPT Pharmacometrics Syst Pharmacol. 2014;3:e94. doi: 10.1038/psp.2013.70. . doi: 10.1038/psp.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotto AM., Jr Evaluation of lipids, drug concentration, and safety parameters following cessation of treatment with the cholesteryl ester transfer protein inhibitor anacetrapib in patients with or at high risk for coronary heart disease. Am J Cardiol. 2014;113:76–83. doi: 10.1016/j.amjcard.2013.08.041. [DOI] [PubMed] [Google Scholar]
- Brousseau ME. Effects of cholesteryl ester transfer protein inhibition on high-density lipoprotein subspecies, apolipoprotein A-I metabolism, and fecal sterol excretion. Arterioscler Thromb Vasc Biol. 2005;25:1057–1064. doi: 10.1161/01.ATV.0000161928.16334.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikewaki K. Delayed catabolism of high density lipoprotein apolipoproteins A-I and A-II in human cholesteryl ester transfer protein deficiency. J Clin Invest. 1993;92:1650–1658. doi: 10.1172/JCI116750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall AR. The effects of cholesterol ester transfer protein inhibition on cholesterol efflux. Am J Cardiol. 2009;104(10 Suppl):39E–45E. doi: 10.1016/j.amjcard.2009.09.018. . doi: 10.1016/j.amjcard.2009.09.018. [DOI] [PubMed] [Google Scholar]
- Suico J. Evacetrapib at a supratherapeutic steady-state concentration does not prolong QT in a thorough QT/QTc study in healthy subjects. J Cardiovasc Pharmacol Ther. 2014;19:283–289. doi: 10.1177/1074248413510784. . doi: 10.1177/1074248413510784. [DOI] [PubMed] [Google Scholar]
- Johns DG. On- and off-target pharmacology of torcetrapib: current understanding and implications for the structure activity relationships (SAR), discovery and development of cholesteryl ester-transfer protein (CETP) inhibitors. Drugs. 2012;72:491–507. doi: 10.2165/11599310-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Burris TP. A novel method for analysis of nuclear receptor function at natural promoters: peroxisome proliferator-activated receptor gamma agonist actions on aP2 gene expression detected using branched DNA messenger RNA quantitation. Mol Endocrinol. 1999;13:410–417. doi: 10.1210/mend.13.3.0246. [DOI] [PubMed] [Google Scholar]