

Abstract
Summary
Background
Facial port-wine stains (PWSs) are usually isolated findings; however, when associated with cerebral and ocular vascular malformations they form part of the classical triad of Sturge–Weber syndrome (SWS).
Objectives
To evaluate the associations between the phenotype of facial PWS and the diagnosis of SWS in a cohort with a high rate of SWS.
Methods
Records were reviewed of all 192 children with a facial PWS seen in 2011–13. Adverse outcome measures were clinical (seizures, abnormal neurodevelopment, glaucoma) and radiological [abnormal magnetic resonance imaging (MRI)], modelled by multivariate logistic regression.
Results
The best predictor of adverse outcomes was a PWS involving any part of the forehead, delineated at its inferior border by a line joining the outer canthus of the eye to the top of the ear, and including the upper eyelid. This involves all three divisions of the trigeminal nerve, but corresponds well to the embryonic vascular development of the face. Bilateral distribution was not an independently significant phenotypic feature. Abnormal MRI was a better predictor of all clinical adverse outcome measures than PWS distribution; however, for practical reasons guidelines based on clinical phenotype are proposed.
Conclusions
Facial PWS distribution appears to follow the embryonic vasculature of the face, rather than the trigeminal nerve. We propose that children with a PWS on any part of the ‘forehead’ should have an urgent ophthalmology review and a brain MRI. A prospective study has been established to test the validity of these guidelines.
What’s already known about this topic?
Facial port-wine stains (PWSs) are common, but are rarely associated with Sturge–Weber syndrome (SWS).
Early diagnosis of SWS is important to reduce ophthalmological and neural complications.
Bilateral and ophthalmic division trigeminal nerve PWSs are thought to confer higher risk of SWS.
What does this study add?
The strongest predictor of SWS was found using a new classification of PWS based on the vascular embryological distribution and not the neural innervation of the face.
We propose new guidelines for investigation of children with facial PWS based on this new classification of phenotype.
The original description of Sturge–Weber syndrome (SWS) by William Sturge in 1879 was of a triad of extensive facial, scalp and truncal capillary malformation (port-wine stain, PWS), contralateral focal seizures suggested to be due to an ipsilateral abnormality on the surface of the brain, and ipsilateral intraocular vascular malformation with glaucoma.1 Kalischer confirmed the presence of an ipsilateral leptomeningeal vascular malformation on pathological examination in 1901,2 and in 1922 Weber described the radiological appearances of ipsilateral cerebral atrophy and the characteristic intravascular calcification in a patient with extensive bilateral facial, truncal and upper-limb PWS.3 Over the years the definition of SWS has expanded to include cases without cutaneous lesions,4 and variable distributions of brain and ophthalmological lesions.5,6 Neurological features have been described in detail since the advent of computed tomography and magnetic resonance imaging (MRI), revealing angiomatosis (a capillary–venous malformation)7,8 of the leptomeninges, atrophy and calcification of the affected cerebral hemisphere, absence of superficial cortical veins and/or dilated deep-draining veins, and vascular malformations involving the choroid plexus.5,9–11 Recent advances in functional imaging have demonstrated decreased cerebral perfusion and network connectivity in affected areas.12–14 Clinical neurological problems include seizures (often but not always contralateral focal motor seizures), neurodevelopmental delay, headache and stroke-like episodes.15 The ophthalmological features in SWS are enlarged venous vessels affecting the conjunctiva, episclera, retina and/or choroids, associated with glaucoma, retinal detachment and choroidal haemorrhage.1,16,17
Therapeutic studies have shown that early diagnosis and treatment of SWS may reduce ensuing complications, based on a new understanding of the pathogenesis of disease progression. The typical MRI findings of atrophy and calcification are now considered to be the consequence of chronic cortical hypoxaemia due to vascular stasis and decreased perfusion in the cortex underlying the leptomeningeal angioma.6,14 Imaging studies during the characteristic stroke-like episodes have suggested decreased perfusion of affected areas.18–20 This new understanding has led to the administration of prophylactic aspirin,21,22 and although randomized controlled trials are lacking, current data show reduced occurrence of stroke-like episodes and seizures.21 The potential benefits of diagnostic MRI and use of prophylactic aspirin need to be considered in relation to the potential adverse effects in the individual patient by the clinician. Given the severity of seizures and the association with acute neurological deficit, there is a rationale for the diagnosis of brain involvement in asymptomatic infants and children. Furthermore, as seizure activity correlates with poorer cognitive prognosis,23,24 some authors recommend prophylactic antiepileptic treatments.25 Early treatment of glaucoma is known to be critical in preserving visual function, and prompt diagnosis in SWS is important as glaucoma can be present from birth. Early identification of patients with facial PWS who may be at risk of SWS is therefore crucial in instigating early clinical and radiological investigation, and, where appropriate, prophylactic therapy.
Facial PWSs are a far more common occurrence than SWS, with an incidence of approximately 1 in 30026–28 for the former and an estimated incidence of between 1 in 20 000 and 1 in 50 000 for the latter.
In infants true PWS should not be confused with a salmon patch (naevus simplex). Naevus simplex is a transitory functional capillary lesion occurring in about 40% of newborns, which presents most commonly as an irregularly bordered, symmetrical pink macule overlying the midline of the neck, the forehead or the upper eyelids.
SWS occurs sporadically with equal frequency in boys and girls. Early studies of the relationship between facial PWS phenotype and risk of SWS implicated PWS in the ophthalmic (V1) division of the trigeminal nerve, a bilateral distribution, and lesions affecting the upper eyelids.29,30 However, more recent studies comparing PWS phenotype with cerebral and ocular phenotype have shown that while bilaterality and V1 distribution appear to be a risk factor, more extensive PWS with additional V2 and/or V3 involvement of the trigeminal nerve contribute to the risk of SWS.31,32
The aim of this study was to correlate facial PWS phenotype with intracranial and intraocular abnormalities, to review the validity of the trigeminal nerve classification, and to propose guidelines for investigation of facial PWS that can be used in our population for future prospective studies.
Patients and methods
In total 192 children (97 female) with a facial PWS were seen sequentially between March 2011 and January 2013 in the paediatric dermatology and neurology departments at Great Ormond Street Hospital for Children. This cohort included both new patients and follow-up patients. As this is a tertiary referral centre the cohort included a much higher percentage of children with SWS than would be seen in the general population of children with PWS; however, we considered that this may improve the power of our statistical analysis of phenotypic associations. We were not aiming to document the incidence of SWS in a population of patients with PWS.
Hospital notes, radiological results (MRI) and professional high-resolution photographs of these patients were reviewed retrospectively. Clinical data collected were age, sex, cutaneous phenotype, presence of glaucoma, seizures and abnormal neurodevelopment, defined as any degree of motor or cognitive impairment on formal assessment. Facial PWSs were classified using the traditional method based on the sensory branches of the trigeminal nerve (V1, V2, V3) (Fig. 1) and whether they were in a unilateral or bilateral distribution. A second classification was also performed by dividing up the face into eight areas on each side, and indicating which were affected. These areas were the central forehead, lateral forehead, upper eyelid, lower eyelid, maxillary area, mandibular area, ear (where visible) and chin. Scalp involvement was not classified, as the majority of the photographs did not show this adequately.
Fig 1.
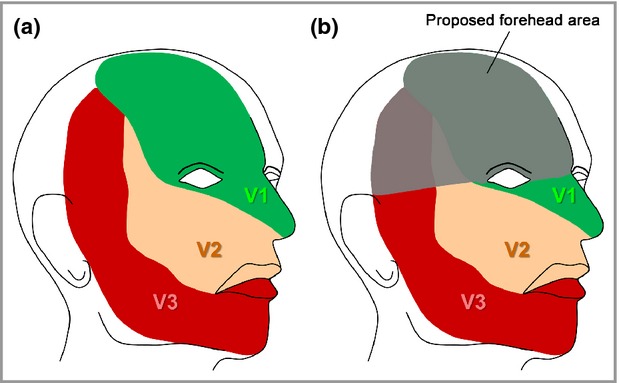
(a) Distribution of the three branches of the trigeminal nerve. (b) Distribution of the ‘forehead’, defined as any part of the forehead from the midline to an imaginary line between the outer canthus of the eye and the top of the ear including the upper eyelids. Figure adapted from Anatomy of the Human Body.36
Adverse outcome measures were (i) clinical: seizures, abnormal neurodevelopment and glaucoma; and (ii) radiological: abnormal MRI. Absence of adverse outcome measures was recorded only where definite negative clinical information was documented, rather than by assumption (for example absence of glaucoma was recorded only where a normal ophthalmological examination had occurred). Statistical analysis of the association between the cutaneous phenotype variables and outcome measures was by multivariate logistic regression. For calculation of odds ratios for the effect of involvement of the forehead on outcome measures, 1 was added to each group to correct for a value of 0 in one cell.
Results
A cohort of 192 patients with facial PWS was seen between March 2011 and January 2013. The mean age at the time of analysis was 8·3 years (SEM 0·4) and the mean patient follow-up period was 8 years. Seventy-five patients had an ophthalmological examination, of whom 55 were diagnosed with glaucoma. One hundred and twenty-one patients had an MRI of the brain, of which 90 were abnormal. Forty-nine of 162 patients for whom there were reliable data had seizures, and 59 of 143 had neurodevelopmental delay. As a result of these investigations 104 children were diagnosed with SWS, using the definition of any facial PWS plus either MRI abnormalities or glaucoma. The frequencies of the original classification of V1–3 are shown in Table 1.
Table 1.
Frequency of phenotypic distribution of facial port-wine stain (PWS) using the traditional trigeminal nerve classification, with which no clear associations were found with SWS, in 171 patients
| Trigeminal nerve distribution of PWS | Patients, n (%) |
|---|---|
| V1 alone | 25 (14·6) |
| V2 alone | 21 (12·3) |
| V3 alone | 9 (5·3) |
| V1 and V2 | 63 (36·8) |
| V1 and V3 | 2 (1·2) |
| V2 and V3 | 10 (5·8) |
| V1, V2 and V3 | 41 (24·0) |
Strikingly, there was no significant association between the trigeminal nerve distribution and an abnormal MRI. Associations between the eight facial areas from the second classification and abnormal MRI were then tested, and independently significant areas were found to be the central forehead and lateral forehead. Upper-eyelid involvement and bilateral distribution were initially significant but became nonsignificant when combined with the lateral forehead and central forehead. We therefore defined a new area that was the best predictor of an abnormal MRI, which equates to the ‘forehead’. This is delineated at the lateral and inferior margins by a line joining the outer canthus of the eye to the top of the ear, and including the upper eyelid. This area covers parts of the distribution of all three branches of the trigeminal nerve (Fig. 1b).
Once the forehead area was identified as the relevant area this was used to model the association with clinical outcome measures. Of the 103 children in this cohort with involvement of the forehead, 83 had SWS, and 20 had only the facial PWS. However, this figure cannot be extrapolated to the total population as our cohort is selected for more severely affected patients. For patients with any involvement of this area the odds ratio of neurodevelopmental abnormality was 24·7 [95% confidence interval (CI) 3·2–188·8, P = 0·002], of seizures 15·8 (95% CI 2·1–120·5, P = 0·008) and of glaucoma 14·4 (95% CI 1·8–113·3, P = 0·011). Absolute numbers of those in each group are shown in Table 2, with Fisher’s exact P-values. Bilateral involvement was not independently significant in any of these models when an interaction variable was used. On closer examination of the data we found that only six cases had bilateral involvement that did not include the forehead on either side. None of these six had any clinical or radiological adverse outcome.
Table 2.
New clinical classification of PWS phenotype with respect to clinical outcomes, where both adequate data and photographs were available
| Forehead involved | Forehead not involved | Fisher’s exact P-value | |
|---|---|---|---|
| Seizures | 36/111 | 0/33 | < 0·001 |
| Abnormal neurodevelopment | 42/93 | 0/30 | < 0·001 |
| Glaucoma | 45/92 | 0/15 | < 0·001 |
| Abnormal magnetic resonance imaging scan | 69/94 | 0/4 | 0·002 |
Interestingly, when abnormal MRI was included in the model for clinical outcome measures, this was a better predictor of all clinical outcome measures than the forehead distribution of PWS, with odds ratios for seizures of 80·3 (95% CI 9·0–714·6, P < 0·001), neurodevelopmental abnormalities 24·7 (95% CI 3·2–188·8, P = 0·002) and glaucoma 14·4 (95% CI 1·8–113·3, P = 0·011). However, guidelines based on clinical phenotyping are proposed for practical purposes so that MRI can be targeted to the highest-risk infants.
Discussion
Recent groundbreaking research by Shirley et al.7 has identified the causative mutation underlying both Sturge–Weber syndrome and the majority of isolated PWSs. This is a somatic activating mutation in the gene GNAQ, which increases cell proliferation and inhibits apoptosis due to increased downstream signalling through the RAS effector pathways. The cell of origin affected by the mutation is not yet known, but it is likely that the mutation occurs earlier in development in SWS than in isolated PWS, thus affecting a more primitive progenitor with wider potential effects. As all the manifestations of SWS are thought to be related to abnormal vasculature, we hypothesized that the identified high-risk ‘forehead’ area might correspond to the vascular distribution of the face.
We therefore considered the adult arterial supply, the adult venous drainage and the embryological vasculature of the face. The central forehead in adult life is supplied by the supratrochlear and supraorbital arteries, which are branches of the internal carotid artery, whereas the lateral forehead is supplied by the superficial temporal artery, a branch of the external carotid. While this supply corresponds to the forehead area (with central and lateral areas often affected separately), the adult arterial supply to the cerebral cortex comes only from the internal carotid, and we therefore discounted adult arterial supply as a satisfactory explanation for the strong association between the forehead and cerebral involvement. Adult venous drainage is far more interconnected and could conceivably be a reasonable distribution for the observed pattern of facial PWS. The internal and external jugular veins (unlike their carotid artery counterparts) communicate at the level of the midneck, which could help to explain the importance of both the central and lateral forehead, and as the primary abnormality in the brain is venous, this seemed a plausible model. However, it was more difficult to see how the venous patterning could be related to PWS on other areas of the face, particularly the relatively common lesions with a sharp lower-edge cut-off joining the angle of the mouth to the bottom of the ear, which do not correspond to the venous drainage patterns.
We therefore considered the embryological origin of the face, which involves the fusion of placodes and the formation of the optic vesicles (Fig. 2a). Each placode brings its own developing vasculature from the neural crest, and the primitive vasculature therefore maps to the placode shaping. We found that the forehead corresponds to the frontonasal prominence plus the skin in the optic vesicle area. Crucially, these two structures are the only parts of the face that are formed by the migration of neural crest cells from the developing prosencephalon (forebrain) and anterior mesencephalon (midbrain), whereas the maxillary and mandibular prominences formed from the first branchial arch consist of neural crest cells from the posterior mesencephalon and rhombencephalon, respectively.33 As the cerebral cortex and the eye both develop from the forebrain, the co-occurrence of forehead involvement and neurological and ophthalmological abnormalities is strongly suggestive of a single mutation affecting the neural crest cells emanating from the forebrain region. In some support of this embryological theory is the notable similarity between the regions identified here and those identified as being affected by infantile haemangiomas.34,35 For haemangiomas clear patterns of distribution have been delineated, where the authors described an association of the nose and philtrum region with the midline forehead (in that publication so-called segment 4), suggesting that this midline forehead region was narrower than in previous publications, and described a temporal region with a lower border running horizontally from the outer canthus (segment 1).35 While it is possible that our forehead region corresponds to segments 4 and 1 together, we have not looked for associations between PWS in different regions of the face, as we were focused primarily on adverse outcomes. We do however have patients with wider central forehead involvement that does correspond to the more classical descriptions of the frontonasal prominence (Fig. 2b–d).
Fig 2.
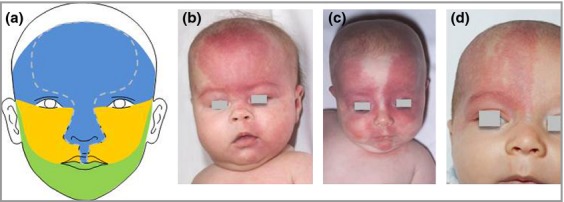
(a) Configuration of the facial placodes. The blue area represents the ‘forehead’, constituting a central frontonasal placode (marked by the dotted lines) and lateral optic vesicle areas. (b) Frontonasal prominence port-wine stain (PWS) – not to be confused with a salmon patch (naevus simplex). (c) PWS sparing the majority of the frontonasal prominence. (d) Unilateral PWS in the ‘forehead’, suggesting a mutation after division of vasculature into right and left.
Anecdotally we tested our theory of vasculature-based classification of PWS using archived pictures of PWS on the limbs in our patient cohort. We found a striking visual correlation between the vascular but not the neural supply of both extremities and the distribution of PWS (examples in Fig. 3), strengthening our finding in the face. As there were only a few patients presenting with a PWS on their hands or feet, a separate study including a larger group of patients with PWS on their limbs is needed to examine the concept of vascular-based distribution of PWS on the extremities.
Fig 3.
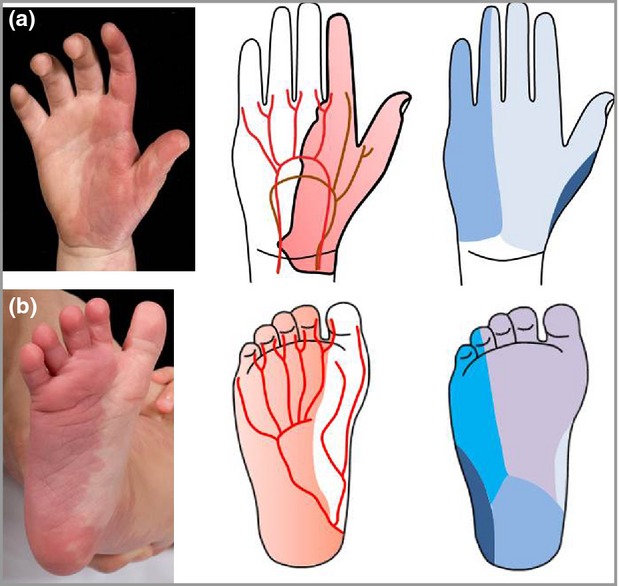
Comparison of port-wine stain with the vascular and neural distribution, showing similarity to the vascular distribution in (a) the palm and (b) the sole. Figure adapted from Anatomy of the Human Body.36
This study was not designed to assess the controversial issue of the optimal timing of brain MRI in the investigation of PWS; however, it is known that the features of SWS can be missed through early MRI.5 As our study demonstrates that an abnormal MRI is the best predictor of all adverse clinical outcomes for these patients, we propose that a gadolinium-enhanced brain MRI be done within the first 3 months of life, with the caveat that a negative result should not be considered conclusive if neurological symptoms develop (Fig. 4). In the context of clinical suspicion of SWS the MRI should be repeated at a later date if negative. In addition, it is important that the appropriate information be given to families surrounding the use of gadolinium enhancement in young children. The potential benefits of diagnostic MRI and use of prophylactic aspirin need to be considered in relation to the potential adverse effects in the individual patient by the clinician. Given the severity of seizures and the association with acute neurological deficit, there is a rationale for the diagnosis of brain involvement in asymptomatic infants and children. These guidelines will now be followed by a prospective study in our department to assess their validity, and ideally would also be assessed in secondary-care settings.
Fig 4.

Great Ormond Street Hospital management guidelines for children with facial port-wine stain (PWS) on the forehead. MRI, magnetic resonance imaging.
In conclusion, the distribution of facial PWS appears to follow the embryological vasculature of the face rather than the trigeminal nerve distribution, and this new classification improves the prediction of SWS based on facial PWS phenotype. In this cohort, only children with PWS involving the ‘forehead’ had any seizures, neurodevelopmental abnormalities, glaucoma or abnormal MRI of the brain, making this the most useful clinical feature. We propose that children with a PWS affecting any part of the forehead should have an ophthalmology review as early as possible, ideally on the first day of life, and a brain MRI with gadolinium contrast. Given the potential use of prophylactic aspirin therapy for those with abnormal MRI we propose that MRI should be performed ideally within the first 3 months, with the understanding that it may need to be repeated at a later date if reported as normal but with a clinical suspicion of SWS. Children with an abnormal MRI should have an electroencephalogram and regular neurological, neurodevelopmental and ophthalmological follow-up. A prospective study has been set up to test these new guidelines.
Acknowledgments
The authors would like to acknowledge the expert input of Professor Peter Scambler, UCL Institute of Child Health, regarding the embryonic vasculature.
References
- 1.Sturge WA. A case of partial epilepsy apparently due to a lesion of one of the vasomotor centres of the brain. Trans Clin Soc Lond. 1879;12:162–7. [Google Scholar]
- 2.Kalischer S. Ein Fall von Telangiektasie (Angiom) des Gesichts und der weichen Hirnhäute. Arch Psychiatr Nervenkrankheiten Berl. 1901;34:171–80. (in German) [Google Scholar]
- 3.Weber FP. Right-sided hemi-hypotrophy resulting from right-sided congenital spastic hemiplegia, with a morbid condition of the left side of the brain, revealed by radiograms. J Neurol Psychopathol. 1922;3:134–9. doi: 10.1136/jnnp.s1-3.10.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piram M, Lorette G, Sirinelli D, et al. Sturge-Weber syndrome in patients with facial port-wine stain. Pediatr Dermatol. 2012;29:32–7. doi: 10.1111/j.1525-1470.2011.01485.x. [DOI] [PubMed] [Google Scholar]
- 5.Adams ME, Aylett SE, Squier W, Chong W. A spectrum of unusual neuroimaging findings in patients with suspected Sturge-Weber syndrome. AJNR Am J Neuroradiol. 2009;30:276–81. doi: 10.3174/ajnr.A1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baselga E. Sturge-Weber syndrome. Semin Cutan Med Surg. 2004;23:87–98. doi: 10.1016/j.sder.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971–9. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu J, Yu Y, Juhász C, et al. MR susceptibility weighted imaging (SWI) complements conventional contrast enhanced T1 weighted MRI in characterizing brain abnormalities of Sturge-Weber syndrome. J Magn Reson Imaging. 2008;28:300–7. doi: 10.1002/jmri.21435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fogarasi A, Loddenkemper T, Mellado C, et al. Sturge-Weber syndrome: clinical and radiological correlates in 86 patients. Ideggyogy Sz. 2013;66:53–7. [PubMed] [Google Scholar]
- 10.Sperner J, Schmauser I, Bittner R, et al. MR-imaging findings in children with Sturge-Weber syndrome. Neuropediatrics. 1990;21:146–52. doi: 10.1055/s-2008-1071483. [DOI] [PubMed] [Google Scholar]
- 11.Stimac GK, Solomon MA, Newton TH. CT and MR of angiomatous malformations of the choroid plexus in patients with Sturge-Weber disease. AJNR Am J Neuroradiol. 1986;7:623–7. [PMC free article] [PubMed] [Google Scholar]
- 12.Jeong JW, Chugani HT, Behen ME, et al. Quantitative assessment of brain networks in children with Sturge-Weber syndrome using resting state functional magnetic resonance imaging (MRI) J Child Neurol. 2013;28:1448–55. doi: 10.1177/0883073812469296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeong JW, Chugani HT, Juhász C. Localization of function-specific segments of the primary motor pathway in children with Sturge-Weber syndrome: a multimodal imaging analysis. J Magn Reson Imaging. 2013;38:1152–61. doi: 10.1002/jmri.24076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miao Y, Juhász C, Wu J, et al. Clinical correlates of white matter blood flow perfusion changes in Sturge-Weber syndrome: a dynamic MR perfusion-weighted imaging study. AJNR Am J Neuroradiol. 2011;32:1280–5. doi: 10.3174/ajnr.A2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comi AM. Presentation, diagnosis, pathophysiology, and treatment of the neurological features of Sturge-Weber syndrome. Neurologist. 2011;17:179–84. doi: 10.1097/NRL.0b013e318220c5b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arora KS, Quigley HA, Comi AM, et al. Increased choroidal thickness in patients with Sturge-Weber syndrome. JAMA Ophthalmol. 2013;131:1216–19. doi: 10.1001/jamaophthalmol.2013.4044. [DOI] [PubMed] [Google Scholar]
- 17.Sullivan TJ, Clarke MP, Morin JD. The ocular manifestations of the Sturge-Weber syndrome. J Pediatr Ophthalmol Strabismus. 1992;29:349–56. doi: 10.3928/0191-3913-19921101-05. [DOI] [PubMed] [Google Scholar]
- 18.Kumar KR, Hon K, Schultz D, et al. Transient changes on brain magnetic resonance imaging in a patient with Sturge-Weber syndrome presenting with hemiparesis. Neurologist. 2009;15:351–4. doi: 10.1097/NRL.0b013e3181940244. [DOI] [PubMed] [Google Scholar]
- 19.Maria BL, Neufeld JA, Rosainz LC, et al. Central nervous system structure and function in Sturge-Weber syndrome: evidence of neurologic and radiologic progression. J Child Neurol. 1998;13:606–18. doi: 10.1177/088307389801301204. [DOI] [PubMed] [Google Scholar]
- 20.Alkonyi B, Miao Y, Wu J, et al. A perfusion-metabolic mismatch in Sturge-Weber syndrome: a multimodality imaging study. Brain Dev. 2012;34:553–62. doi: 10.1016/j.braindev.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bay MJ, Kossoff EH, Lehmann CU, et al. Survey of aspirin use in Sturge-Weber syndrome. J Child Neurol. 2011;26:692–702. doi: 10.1177/0883073810388646. [DOI] [PubMed] [Google Scholar]
- 22.Lance EI, Sreenivasan AK, Zabel TA, et al. Aspirin use in Sturge-Weber syndrome: side effects and clinical outcomes. J Child Neurol. 2013;28:213–18. doi: 10.1177/0883073812463607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Behen ME, Juhász C, Wolfe-Christensen C, et al. Brain damage and IQ in unilateral Sturge-Weber syndrome: support for a ‘fresh start’ hypothesis. Epilepsy Behav. 2011;22:352–7. doi: 10.1016/j.yebeh.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raches D, Hiscock M, Chapieski L. Behavioral and academic problems in children with Sturge-Weber syndrome: differences between children with and without seizures. Epilepsy Behav. 2012;25:457–63. doi: 10.1016/j.yebeh.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 25.Comi AM. Sturge-Weber syndrome and epilepsy: an argument for aggressive seizure management in these patients. Expert Rev Neurother. 2007;7:951–6. doi: 10.1586/14737175.7.8.951. [DOI] [PubMed] [Google Scholar]
- 26.Alper JC, Holmes LB. The incidence and significance of birthmarks in a cohort of 4,641 newborns. Pediatr Dermatol. 1983;1:58–68. doi: 10.1111/j.1525-1470.1983.tb01093.x. [DOI] [PubMed] [Google Scholar]
- 27.Kanada KN, Merin MR, Munden A, Friedlander SF. A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161:240–5. doi: 10.1016/j.jpeds.2012.02.052. [DOI] [PubMed] [Google Scholar]
- 28.Tsai FJ, Tsai CH. Birthmarks and congenital skin lesions in Chinese newborns. J Formos Med Assoc. 1993;92:838–41. [PubMed] [Google Scholar]
- 29.Mazereeuw-Hautier J, Syed S, Harper JI. Bilateral facial capillary malformation associated with eye and brain abnormalities. Arch Dermatol. 2006;142:994–8. doi: 10.1001/archderm.142.8.994. [DOI] [PubMed] [Google Scholar]
- 30.Tallman B, Tan OT, Morelli JG, et al. Location of port-wine stains and the likelihood of ophthalmic and/or central nervous system complications. Pediatrics. 1991;87:323–7. [PubMed] [Google Scholar]
- 31.Ch’ng S, Tan ST. Facial port-wine stains – clinical stratification and risks of neuro-ocular involvement. J Plast Reconstr Aesthet Surg. 2008;61:889–93. doi: 10.1016/j.bjps.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Mehta M, Salas AH, Fay A. Trigeminal dermatome distribution in patients with glaucoma and facial port wine stain. Dermatology. 2009;219:219–24. doi: 10.1159/000235546. [DOI] [PubMed] [Google Scholar]
- 33.Odaci E, Schaitkin BM. Face embryology. Available at: http://emedicine.medscape.com/article/844962-overview (last accessed 11 August 2014)
- 34.Waner M, North PE, Scherer KA, et al. The nonrandom distribution of facial hemangiomas. Arch Dermatol. 2003;139:869–75. doi: 10.1001/archderm.139.7.869. [DOI] [PubMed] [Google Scholar]
- 35.Haggstrom AN, Lammer EJ, Schneider RA, et al. Patterns of infantile hemangiomas: new clues to hemangioma pathogenesis and embryonic facial development. Pediatrics. 2006;117:698–703. doi: 10.1542/peds.2005-1092. [DOI] [PubMed] [Google Scholar]
- 36.Gray H. Anatomy of the Human Body. 20th edn. Philadelphia: Lea & Febiger; 1918. [Google Scholar]