

Abstract
Seizures are a common manifestation of acute neurologic insults in neonates and are often resistant to the standard antiepileptic drugs that are efficacious in children and adults. The paucity of evidence-based treatment guidelines, coupled with a rudimentary understanding of disease pathogenesis, has made the current treatment of neonatal seizures empiric and often ineffective, highlighting the need for novel therapies. Key developmental differences in γ-aminobutyric acid (GABA)ergic neurotransmission between the immature and mature brain, and trauma-induced alterations in the function of the cation-chloride cotransporters (CCCs) NKCC1 and KCC2, probably contribute to the poor efficacy of standard antiepileptic drugs used in the treatment of neonatal seizures. Although CCCs are attractive drug targets, bumetanide and other existing CCC inhibitors are suboptimal because of pharmacokinetic constraints and lack of target specificity. Newer approaches including isoform-specific NKCC1 inhibitors with increased central nervous system penetration, and direct and indirect strategies to enhance KCC2-mediated neuronal chloride extrusion, might allow therapeutic modulation of the GABAergic system for neonatal seizure treatment.

Keywords: KCC2, NKCC1, Cation-chloride cotransporters, Neonate, Bumetanide
Seizures occur more often during the neonatal period than at any other time during life, with a reported incidence in postindustrial societies of up to 3.5 per 1,000 live births, and higher in preterm infants.1,2 Neonatal seizures are most commonly caused by perinatal asphyxia, and other causes include intracranial hemorrhage, cerebral infarction, and central nervous system (CNS) infection, such as meningitis.1
Neonatal seizures portend severe neurologic dysfunction later in life, with survivors experiencing higher rates of postneonatal epilepsy and motor and cognitive deficits.3,4 Rodent models have revealed that seizures early in development alter synaptic organization and plasticity, and may prime cortical neurons to increased seizure susceptibility.5 Therefore, the prompt diagnosis and successful treatment of seizures early in life is necessary for improving long-term neurologic outcomes.
Standard antiepileptic drugs (AEDs) that enhance γ-aminobutyric acid (GABA)ergic transmission by directly targeting GABAA receptors (GABAARs), such as phenobarbital and benzodiazepines, are less effective in suppressing seizures in neonates than in adults.6,7 This is not surprising, because the signaling mechanisms and pharmacologic properties of neurons in the immature brain are significantly different from those in the adult.8 It is notable that many neonatal seizures are clinically occult, going unnoticed without electroencephalography (EEG) monitoring.9 In neonates, GABAAR-enhancing AEDs can also elicit so-called “electroclinical uncoupling,” whereby the overt clinical manifestations of seizures (e.g., convulsions) are inhibited, but electrographic seizure activity is either unaffected or exacerbated.10 Clearly, new therapeutic strategies are needed.
Recently, there has been an increasing interest in the indirect modulation of GABAergic responses based on a strategy of targeting the plasmalemmal ion transporters responsible for the generation and maintenance of the ion gradients that drive GABAAR-mediated currents.11,12 This approach is motivated by the fact that short-term and long-term changes in the functional properties of ion transporters have a major influence on GABAergic signaling during neuronal maturation and following trauma and seizures in adults and neonates (Fig. 1).11,12 GABAAR-mediated currents are driven by Cl− and 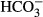 electrochemical gradients in postsynaptic neurons.13 Key molecules in neuronal homeostasis of Cl− include members of the SLC12 cation-chloride cotransporters (CCCs), especially the Na-K-2Cl cotransporter NKCC1 and the K-Cl cotransporter KCC2.13,14 As secondary active transporters, the CCCs derive their energy for active transport of Cl− from the transmembrane gradients of Na+ (for NKCC1) or K+ (for KCC2), which are generated by the plasmalemmal Na-K ATPase. Intraneuronal regulation of
electrochemical gradients in postsynaptic neurons.13 Key molecules in neuronal homeostasis of Cl− include members of the SLC12 cation-chloride cotransporters (CCCs), especially the Na-K-2Cl cotransporter NKCC1 and the K-Cl cotransporter KCC2.13,14 As secondary active transporters, the CCCs derive their energy for active transport of Cl− from the transmembrane gradients of Na+ (for NKCC1) or K+ (for KCC2), which are generated by the plasmalemmal Na-K ATPase. Intraneuronal regulation of 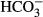 is based on secondary active acid-base transporters and carbonic anhydrases.13,15
is based on secondary active acid-base transporters and carbonic anhydrases.13,15
Figure 1.
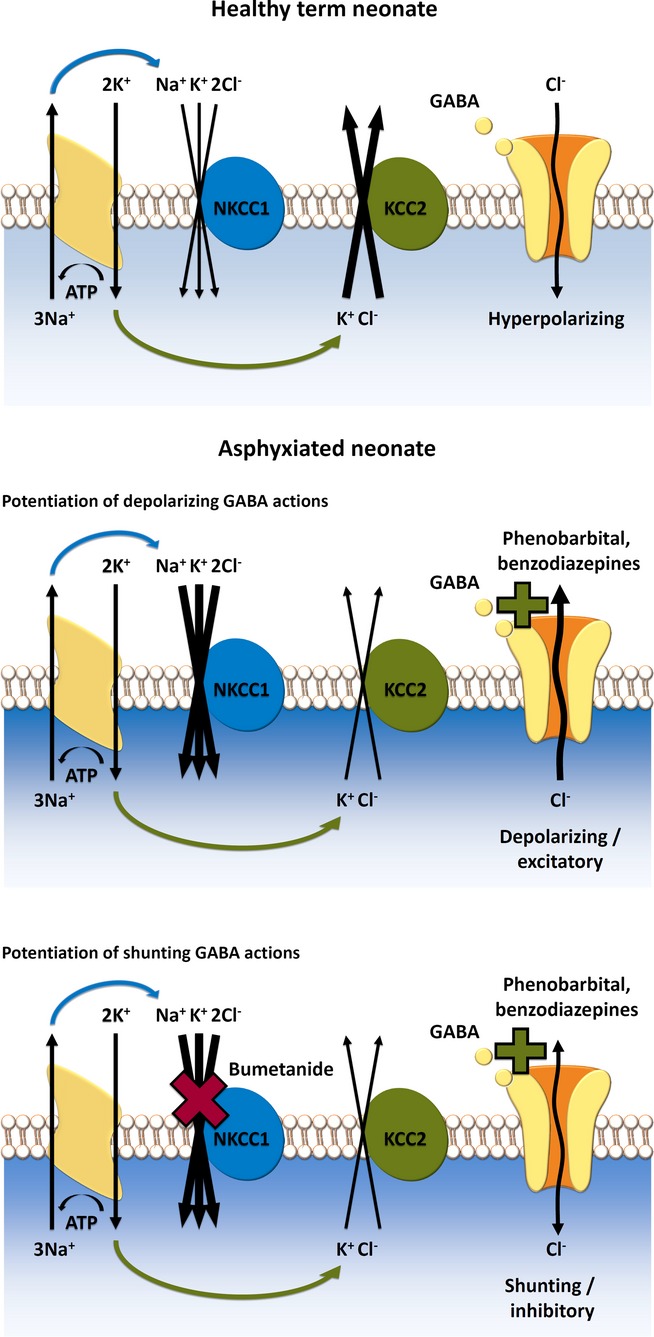
Hypothetical model of pharmacologic targeting of NKCC1 in postasphyxia neurons. NKCC1 and KCC2 are secondary active transporters that, under physiological conditions, mediate neuronal Cl− uptake and extrusion, respectively. Net ion transport by NKCC1 and KCC2 is fueled by the Na+ and K+ chemical gradients generated by the Na–K ATPase. The ion stoichiometry renders NKCC1 and KCC2 electrically silent (electroneutral). (Upper panel) In healthy cortical neurons of human neonates, Cl− extrusion via KCC2 is likely to be more efficient than uptake via NKCC1, which promotes a postsynaptic hyperpolarizing current triggered by GABAergic signaling. (Middle) After neuronal trauma caused by birth asphyxia, functional up-regulation of NKCC1 takes place, and the direction of the Cl− current is reversed which leads to depolarizing GABA responses. Under these conditions, application of positive modulators of GABAARs (phenobarbital, benzodiazepines) can lead to aggravation of seizures promoted by the depolarizing if not directly excitatory Cl− current. (Bottom) Pharmacologic block of NKCC1 by bumetanide attenuates or abolishes the depolarizing GABA response, and subsequent application of positive modulators of GABAARs will lead to effective shunting inhibition, which clamps the membrane potential close to its resting level, thereby preventing action-potential generation in the postsynaptic neuron. (For further details, see text.)
During early development, neurons harbor a low functional expression of the main neuron-specific Cl− extruder KCC2, and GABAARs mediate a net efflux of Cl− ions down an electrochemical gradient, which is generated by Cl− accumulation via NKCC1.13 This results in depolarizing, and sometimes even excitatory actions of GABA, which can trigger release of conventional neurotransmitters and neuroactive peptides. During neuronal maturation, the expression and functionality of KCC2 are progressively increased, which sets the basis for the generation of classical hyperpolarizing inhibitory postsynaptic potentials (IPSPs).16 The efficacy of hyperpolarizing IPSPs is determined by the number of open GABAARs and functionality of CCCs, which generate the driving force (Vm − EIPSP) and thus the polarity of GABAAR-mediated currents. For example, experimental knockdown of KCC2 in mature cortical principal and cerebellar Purkinje neurons abolishes the driving force for hyperpolarizing GABAAR-mediated signaling.16,17 In addition to the effects on membrane potential, opening of GABAARs increases the conductance of the target neuron, which has an inhibitory action by short-circuiting or “shunting” of simultaneous excitatory signals.13 Blocking NKCC1 enhances the efficacy of GABA-mediated shunting inhibition in neurons that would otherwise sustain strongly depolarizing or even excitatory GABA responses (Fig. 1).
In rodent cortical neurons, the “developmental EGABA shift” from depolarizing to hyperpolarizing takes place postnatally,14 and the lack of efficacy of drugs such as phenobarbital in human neonates has often been attributed to the lack of KCC2 expression in the cortical structures of the newborn.18,19 This kind of rodent-to-human extrapolation ignores the fact that in terms of cortical development, the human neonate is much more advanced than the rodent, which is born at a stage corresponding roughly to the second half of gestation in humans.20,21 The cortical EEG of neonatal rodents and preterm infants is discontinuous and dominated by discrete events known as “delta brushes” or “spontaneous activity transients”; in humans these events disappear already at or soon after term birth.8,22 This profound change in the overall properties of the human EEG is associated with a steep prenatal increase in KCC2 expression (see also Fig. 2).23
Figure 2.
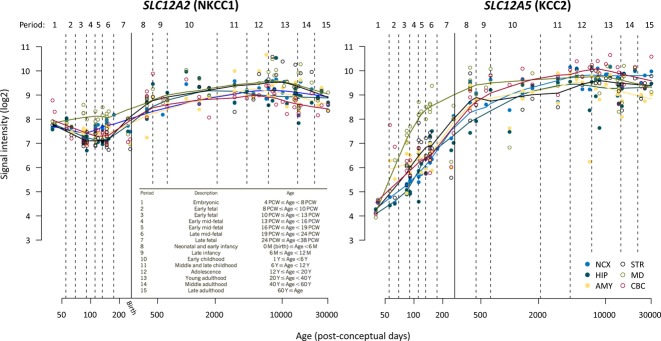
Expression of SLC12A2 (NKCC1) and SLC12A5 (KCC2) transcripts during human brain development. Line plots show the log2-transformed NKCC1 and KCC2 exon array signal intensity from the early fetal period to late adulthood. The solid line with arrow between periods 7 and 8 separates prenatal from postnatal periods. NCX, neocortex; HIP, hippocampus; AMY, amygdala; STR, striatum; MD, mediodorsal nucleus of the thalamus; CBC, cerebellar cortex; PCW, postconceptional week; M, month; Y, year. Data reproduced with permission from http://hbatlas.org; see Kang et al.114
Descriptions of the developmental expression patterns of NKCC1 in the rodent cortex appear discrepant. Plotkin et al.24 first reported a developmental peak in NKCC1 expression around the first postnatal week in the rat forebrain, with down-regulation of NKCC1 messenger RNA (mRNA) and protein after this time point. In contrast, no down-regulation of NKCC1 mRNA was observed in the rat cortex by Clayton et al.26, who suggested that the loss of NKCC1 expression observed by Plotkin et al. may actually reflect changes in the C-terminal splicing of NKCC1. Two ubiquitously expressed splice variants of NKCC1 have been characterized in mouse and human.25,27 The mRNA of the shorter of the two variants NKCC1b which is produced by splicing out exon 21, constitutes up to ∼80% of the total NKCC1 transcript in the adult human brain.27 It is not unlikely that the reported “developmental down-regulation” of NKCC1 protein in the human cortex,19 reflects the use of an NKCC1 rabbit antibody (Chemicon International28) raised against a 22 amino acid sequence near the C-terminus of rat NKCC1; a sequence that is absent from human NKCC1b as it strongly overlaps with exon 21. Use of such an antibody is expected to result in failure of detecting the major NKCC1 splice variant in the adult brain. Indeed, in the human cortex, no down-regulation, but rather progressive up-regulation of NKCC1 transcripts across the entire life-span is evident (Fig. 2).29 Such data are not, however, sufficient to yield information about the functional expression of NKCC1, as the subcellular expression pattern of NKCC1 determines its physiologic actions.30 Electrophysiological work on NKCC1 knockout (KO) animals has shown that this transporter modulates GABAergic signaling at the axon initial segment of adult neocortical and hippocampal principal neurons.30 Unfortunately, the lack of specific NKCC1 antibodies has complicated the interpretation of immunochemical studies on the subcellular distribution of NKCC1.14
The low level of KCC2 activity is likely to contribute to the poor anticonvulsant action of phenobarbital and other GABAAR-enhancing drugs in newborn rodents, but does not necessarily provide a robust explanation as to why these compounds have limited efficacy in human neonates. Two major points should be considered here. (1) In order to maintain effective IPSPs under in vivo conditions, the efficacy of Cl− extrusion has to be sufficient to keep the reversal potential of currents carried by Cl− at a level more negative than the action potential threshold despite the large intracellular Cl− loads generated by synaptic transmission, especially, during seizures.31,32 In addition to being possibly attributable to different density and subunit composition of GABAARs, the lack of efficacy of GABAAR-enhancing AEDs in the human neonate may reflect the limited capacity (in other words, the small physiologic “safety factor” [cf. Ref. 33]) of Cl− extrusion in immature neurons. (2) The rapid functional up-regulation of NKCC1, shown to take place in response to neonatal hypoxia-ischemia,34 hypoxia-induced neonatal seizures,35 as well as hypoxic-ischemic and mechanical cellular trauma,36,37 is bound to cause an additional cellular Cl− load that would render GABAergic inhibition less effective, if not frankly excitatory.32 Thus, in addition to seizures, birth asphyxia, which often is accompanied by brain injury, is already in itself likely to induce fast functional up-regulation of NKCC1.
Given the therapeutic implications of the above observations and controversies, the aims of this review are to (1) evaluate the CCCs as putative drug targets in the treatment of neonatal seizures, with a primary but not exclusive focus on NKCC1; (2) examine the potential therapeutic utility of currently available CCC-targeting drugs, especially bumetanide; and (3) describe novel approaches of targeting the CCCs that, given the limitations of bumetanide, might be more efficacious modulators of Cl− transport in the CNS.
Cation-Chloride Cotransporters as CNS Drug Targets
Seven members (SLC12A1-7) of the SLC12A gene family encode for secondary active Cl− transporters. These include the Na-Cl cotransporter NCC, the Na-K-2Cl cotransporters, NKCC isoforms 1–2, and the K-Cl cotransporters, KCC isoforms 1–4.14 Of these, the expression of KCC2 is restricted to central nervous system (CNS) neurons.16,38 While NKCC1 is expressed ubiquitously,39 NCC and NKCC2 are not expressed in brain tissue,12,26 and the CNS expression of KCC1 and KCC4 is limited.14 Although widely expressed in the CNS, the exact physiological role of KCC3 remains unclear.14,17 Of interest, Andermann syndrome, a severe autosomal recessive Mendelian form of peripheral neuropathy associated with agenesis of the corpus callosum is caused by loss-of function mutations in KCC3,40 revealing the necessity of this KCC isoform for both CNS and peripheral nervous system (PNS) development. The indispensability of KCC2 to CNS function is underscored by the fact that mice with complete loss of this protein die at birth.41
KCC2
Compared to the other mammalian KCCs, KCC2 (encoded by SLC12A5) is unique in that it is expressed exclusively in CNS neurons. KCC2 has attracted attention as a prime target for the pharmacologic control of neuronal Cl− concentration, and consequently GABAergic responses. What is typically ignored, however, is that KCC2, independently of its ion transport function, is also a critical structural protein for the formation and function of dendritic spines in cortical neurons.42–44 Thus, effects correlated solely with alterations in KCC2 expression levels cannot be attributed directly to changes in Cl− homeostasis. Genetic deficits in KCC2, or its orthologs, result in increased excitability in organisms as diverse as Caenorhabditis elegans, Drosophila, and mice,45 but it is unclear to what extent this is attributable to the structural versus transport roles of KCC2. These observations suggest that elevating KCC2 protein levels by, for example, gene therapy might not be a useful strategy. Specifically enhancing the transport activity of KCC2 may be more relevant therapeutically.
Neuronal Cl− extrusion mediated by KCC2 is prone to fast activity-dependent modulation through posttranslational modifications, including protein phosphorylation and calpain-mediated cleavage.45 These nontranscriptional regulatory pathways have significant consequences for the efficacy of synaptic inhibition mediated by GABAARs, with implications for neurologic diseases. Recent work has demonstrated that KCC2-mediated Cl− extrusion is rapidly enhanced in response to neonatal seizures via recruitment of the available intracellular KCC2 pool to the plasma membrane.46 Such an intrinsic “antiepileptic” mechanism suggests a putative therapeutic strategy whereby pharmacologic kinetic activation of KCC2 might be utilized to help suppress neonatal seizures.
A possible way to manipulate CCC activity is by targeting their upstream regulatory molecules. For example, N-terminal threonine phosphorylation of NKCC1, required for the activity of this transporter, is catalyzed by the WNK/SPAK (with no lysine/Ste20-related proline alanine-rich kinase) kinase cascade. This is in contrast to the situation with KCC2, where activation of WNK kinases appears to have a suppressing effect on KCC2-mediated K-Cl cotransport.47 Specific manipulation of such kinase pathways with apparently opposing effects on KCC2 versus NKCC1 may pose as a novel pharmacotherapeutic strategy.45 Here, the fast membrane recycling of KCC248 could be exploited in generating KCC2-activating drugs that would act to modulate KCC2 membrane insertion and functionality.
A recent high-throughput screening identified the compound CLP257 and its carbamate prodrug derivative CLP290 as KCC2-activating molecules.49 The parent compound, CLP257, was reported to restore Cl− extrusion in adult neurons of the rat dorsal horn neurons incubated with brain-derived neurotrophic factor and in an experimental model of neuropathic pain. It remains unclear whether this was due to effects on KCC2 membrane trafficking or on the intrinsic transport kinetics of KCC2. Unlike the parent compound CLP257, which has a plasma half-life of <15 min, the prodrug CLP290 with a 5 h plasma half-life is more likely to be utile in vivo. However, its pharmacodynamic profile, including KCC2 specificity and mode of action, as well as its brain availability remain to be assessed.
One should also note that intense activation of KCC2-mediated K-Cl extrusion might lead to a proconvulsant effect triggered by the consequent increase of K+ concentration in the brain's extracellular space.50,51 This is likely to confound the use of KCC2 activators for anticonvulsant purposes, as they might potentiate KCC2-mediated extracellular K+ transients during ictal events. The KCC2-mediated excitatory effect depends on the presence of neuronal carbonic anhydrases,52 particularly on the isoform VII, which is absent from neonatal pyramidal neurons in rodents but shows a high level of expression in cortical structures of the human neonate.15 This species-specific difference may turn out to be of much significance when extrapolating results and concepts from work on rodent models of neonatal seizures, usually performed before the time point (around postnatal day [P] 10) when carbonic anhydrase expression commences in pyramidal neurons.15
NKCC1
NKCC1 (encoded by SLC12A2) has been observed in virtually all cell types studied, with highest expression on the basolateral membranes of secretory epithelia, including the kidney, and on the apical membrane of choroid plexus in the brain. In epithelial cells NKCC1 contributes to the vectorial transport of solute and water to drive fluid secretion.53 It is also expressed in endocrine and neuroendocrine cells,54,55 vascular endothelial cells,53 including those in the blood–brain barrier (BBB),56,57 as well as in neurons and glia.14 Given the wide expression of NKCC1, systemic administration of drugs that target it will influence a wide spectrum of tissues throughout the organism. The participation of NKCC1 in a variety of physiologic processes is well exemplified by the diverse phenotypes of NKCC1 KO mice, including impaired gastrointestinal function, infertility, and deafness arising from a sensory neuronal defect and disrupted epithelial secretion of endolymph in the inner ear.14
Bumetanide as a Potential Anticonvulsant Drug in Neonates: Dosage and Pharmacokinetics
Bumetanide is currently the only available compound which, at low concentrations, inhibits NKCCs, without significantly affecting the function of KCCs. A number of studies have employed this drug in an attempt to suppress neonatal seizures in preclinical work on animal models (Table 1). In addition, two clinical trials (NCT01434225; NCT00830531) are being conducted on the efficacy of bumetanide in the treatment of neonatal seizures (see also Ref. 58). Therefore, we will devote much attention to the pharmacokinetics and pharmacodynamics of bumetanide.
Table 1.
Reported effects of bumetanide in in vivo animal models of neonatal seizures
| Dose (mg/kg i.p.) | Species | Age | Model | Reported effect | Behavioral analysis | EEG analysis | References |
|---|---|---|---|---|---|---|---|
| 0.1–0.2 | Rat | P9-12 | Kainate | Anticonvulsanta | No | Yes | Dzhala et al.19 |
| 0.15–0.3 | Rat | P10 | Hypoxia | No effect | Yes | Yes | Cleary et al.35 |
| 0.2–2.5 | Rat | P7, P18 | PTZ | No effect | Yes | No | Mares94 |
| 0.2–0.5 | Rat | P12 | PTZ | No effect | Yes | No | |
| 1 | Rat | P12 | PTZ | Anticonvulsant | Yes | No | |
| 2.5 | Rat | P12 | PTZ | Proconvulsant | Yes | No | |
| 0.5b | Rat | P11 | Kindled ADT | Anticonvulsant | Yes | Yes | Mazarati et al.93 |
| Rat | P14, P21 | Kindled ADT | No effect | Yes | Yes | ||
| 1.8 | Rat | P4-9 | Sevoflurane | Anticonvulsant | No | Yesc | Edwards et al.90 |
| 1.8 | Rat | P5-9 | Flurothyl | No effect | No | Yes | Minlebaev and Khazipov91 |
| 0.1 (daily P11-P60)d | Rat | – | Hyperthermia at P11 | Decreased susceptibility to PC-induced seizures at P60 | Yes | Yes | Koyama et al.96 |
| 2 (daily P1-5) | Mouse | – | Hyperthermia at P17 | Proconvulsant | Yes | No | Vargas et al.95 |
ADT, afterdischarge threshold; PC, pilocarpine; PTZ, pentylenetetrazole.
Anticonvulsant effect deduced from EEG power.
Administered already during kindling.
Under anesthesia.
Bumetanide was administered after hyperthermia.
Pharmacokinetic studies of bumetanide have been largely performed in the context of treatment for fluid overload in cardiopulmonary disease in adults and critically ill term and preterm neonates.58,59 Plasma half-lives of bumetanide are in the range of 2–7 h in neonates of up to 4 weeks of age,59,60 while shorter (1–2 h) values are typically seen in infants and adults.59,61 Although the longer half-life of bumetanide in neonates may be desirable from the standpoint of brain targeting, it may also exaggerate the undesired effects of bumetanide. As there is little difference in the bumetanide sensitivity between the ubiquitously expressed NKCC1 and the main kidney isoform NKCC2,62 targeting extrarenal NKCC1 with systemically administered bumetanide stimulates diuresis and has the potential side-effect of electrolyte abnormalities including hypokalemic metabolic alkalosis.58,63
Bumetanide and furosemide (a non-selective inhibitor of KCCs and NKCCs) are called “loop diuretics” because they are actively concentrated by organic anion transporters in the loop of Henle in the kidney's thick ascending limb,64,65 and their diuretic action is mediated by inhibition of both NKCC1 and the renal-specific NKCC2. In adult humans, 1 mg of orally administered bumetanide has a diuretic potency equivalent to approximately 40 mg of furosemide and this 1:40 dose equivalence ratio applies also to the intravenous (i.v.) administration route.66,67 The pediatric bumetanide doses (see Refs 59,68) appear to be based in part on this ratio of diuresis induction efficacy, as for example stated in an influential paper by Sullivan et al.69: “The maximal dose [of bumetanide] (0.10 mg/kg) was based on the usual pediatric dose of furosemide (1–2 mg/kg), reportedly 20–40-fold less potent than bumetanide in adults.” It appears that the choice of using a rather low dose in most rodent studies on bumetanide in neonatal seizures (see Table 1) has been guided by similar reasoning. In preclinical work on rats, it has largely gone unnoticed that the half-life of bumetanide in this species is very short: only about 10 min70,71 in adult and around 30 min35 in neonatal rats. Accordingly, bumetanide concentrations, which are high enough to exert pharmacologic actions outside the CNS in the rat (as indicated by diuresis), can be achieved only at high parenteral doses of ≥4 mg/kg.12,71 Likely due to less rapid elimination, the maximal diuretic effect of bumetanide in the adult human is reached at ∼0.1 mg/kg12 and in volume-overloaded critically ill infants at ∼0.04 mg/kg.60 As a reference, the doses used in the multicenter clinical trial NCT01434225 on bumetanide in neonatal seizures are in the range of 0.05–0.3 mg/kg i.v., delivered four times at 12 h intervals. It is important to note that effects of bumetanide in structures outside the CNS, such as the kidney, do not imply that the drug would have any direct effects on CNS neurons in rodents or humans.
Bumetanide as a Potential Anticonvulsant Drug in Neonates: Brain Availability
The question of whether bumetanide accumulates within the BBB-protected brain parenchyma at NKCC1-inhibiting concentrations after systemic administration has mostly been addressed indirectly, by assuming that the anticonvulsant and disease-modifying effects of bumetanide (for recent review, see Ref. 12) are indicative of the drug's actions in cortical and hippocampal neurons. However, such actions might be indirect, reflecting drug effects on other targets such as the endocrine system or the BBB, or effects on total brain extracellular volume or systemic electrolyte balance controlled by the kidney. Considerations based on the pharmacokinetic properties of bumetanide, as well as direct measurements of the drug's concentration in brain tissue, suggest that bumetanide is not likely to achieve pharmacologically relevant levels in cortical structures with the low doses used in most preclinical studies on neonatal seizures (see below), and possibly in the ongoing clinical trials referred to earlier on.
A crucial part of CNS drug development is the assessment of the bioavailability of a candidate compound in the brain; and the ability of a compound to penetrate the BBB is often a major limiting factor.72 Only the unbound and nonionized (uncharged) form of bumetanide (or any other weak acid) is able to diffuse across plasma membranes, including those in the BBB. The degree of plasma protein binding of bumetanide is high, at ∼97–98% in rats and humans, including human neonates.73–77 Furthermore, bumetanide has a pKa = 3.6,78 and thus, the free (unbound to protein) plasma concentration (∼2–3% of total) is ∼99.98% ionized at plasma pH of 7.4 (see equation). Hence, the concentration of the free nonionized, BBB-permeable bumetanide species is several orders of magnitude lower than the total plasma concentration of the drug.
Accordingly, a study on the ability of clinically established drugs to penetrate the BBB reported the related loop diuretic furosemide as unable to cross the BBB largely due to its acidic pKa (<4).79
The bumetanide concentrations typically used in in vitro studies for effective CNS inhibition of NKCC1 are in the range of ∼2–10 μM.14 Given that bumetanide is likely to inhibit NKCC1 from an extracellular site by binding within the translocation cavity in the third transmembrane helix,80 its high ionization is not a problem for use in vitro, or for acting on targets in vivo that are not protected by the BBB (e.g., NKCC1 expressed on the luminal membrane of the cerebral vascular endothelium). However, because the bumetanide fraction unbound to plasma protein is only ∼2–3% of total, substantially higher serum concentrations are needed to achieve a transient half-maximal inhibition of NKCC1 in vivo compared to in vitro conditions, where plasma protein binding and the BBB do not limit neuronal target access. Such concentrations are reached only transiently in the blood, even after very high bumetanide doses, which are two orders of magnitude higher than those used in the neonatal and pediatric patient. Also, when evaluating the in vivo bumetanide dose-effect data, one should note that a given dose of bumetanide applied i.v., will, of course, lead to a higher concentration in brain capillaries than when applied via the intraperitoneal (i.p.) route.
In addition to the high degree of protein-binding and ionization of bumetanide in the blood, a number of its pharmacokinetic parameters indicate poor BBB penetration. The nonionized form of bumetanide is relatively lipophilic (log P ∼3)62,81,82; however, the experimentally determined distribution coefficient (log D7.4), which takes into account ionization at pH 7.4, is very low (between −0.11 and 0.14).82,83 The parallel artificial membrane permeability assay (PAMPA) score of bumetanide at pH 7.4 is also low (0.3).82 Accordingly, following i.v. administration of14 C-labeled bumetanide to healthy human volunteers, accumulation into red blood cells could not be detected, despite a marked diuretic effect.61 Another key factor restricting the entry of compounds into the CNS is a drug polar surface area (PSA) value higher than 80 Å2.72,84 The PSA of bumetanide is ∼123 Å2.81,82 For comparison, the log D7.4 of the commonly used CNS-acting AED phenytoin is 2.26, the PAMPA score at pH 7.4 is 5.1, and it has a PSA of 65 Å2.82 Indeed, 3H-labeled bumetanide has been reported to cross the rat BBB at a rate 100-fold slower than that predicted by passive diffusion.83 There is also some indication that bumetanide may be subject to active efflux transport from the brain via transport mechanisms of the BBB.85,86
Direct data on bumetanide brain accumulation obtained using liquid chromatography are in full accord with the poor BBB permeability predicted by the above physicochemical parameters. In neonatal rats, extremely low brain/plasma ratios with peak values of only 0.0036–0.0056 were observed during the first hour after i.p. administration of bumetanide at 0.15–0.3 mg/kg.35 Thus, only <1% of the total bumetanide in the plasma penetrated into the brain. Indeed, total brain concentrations achieved with this dose in the neonatal brain were only ∼1 ng/g brain tissue,35 which, assuming an average brain density of ∼1 g/ml, is equal to a negligible level of ∼3 nM. Notably, in neonatal rats which had experienced hypoxia-induced seizures, the brain concentrations achieved were within the same range (Fig. 3).35 Such total brain concentrations of bumetanide are orders of magnitude lower than the ∼200–300 nM half-maximal inhibitory concentration (IC50) of bumetanide for NKCC1.53,62
Figure 3.
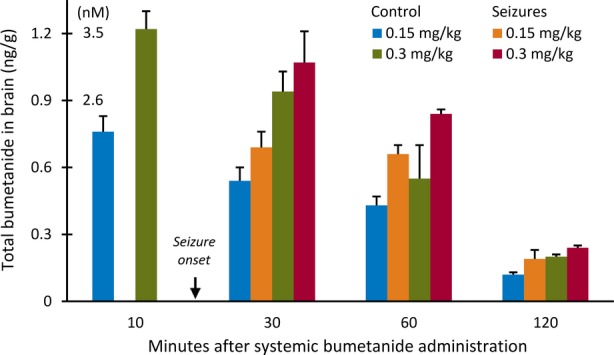
Total bumetanide concentrations in brain tissue of naive and hypoxia-exposed neonatal rats. Bumetanide concentrations of ∼2–3 nM (100-fold lower than the half-maximal NKCC1 inhibitory concentration) are reached in brain tissue of 10-day-old rats following intraperitoneal administration at 0.15–0.3 mg/kg. Notably, brain concentrations of bumetanide are similar in naive control animals and those which experienced hypoxia-induced seizures. Error bars indicate SEM. Data in the diagram derived from Cleary et al.35
In order to achieve pharmacologically relevant concentrations of bumetanide in the CNS, systemic doses much higher than those above are needed. Similarly to neonates, a very low brain/plasma concentration ratio of 0.0068 was measured for bumetanide 30 min after an i.p. dose of 0.3 mg/kg in adult rats.87 Only after increasing the systemically administered dose of bumetanide by nearly two orders of magnitude, could brain concentrations in the range of the half maximal inhibition constant of NKCC1 be briefly reached. Bumetanide at 10 mg/kg administered i.v. resulted in concentrations of ∼0.3 μM at 15 min and ∼0.2 μM at 30 min in hippocampi of adult rats.71 With a higher dose of 15 mg/kg (given i.p.) total bumetanide concentrations in the hippocampus ranged between 0.3 and 0.7 μM 30-60 min after the injection, with similar concentration values detected in the amygdala and the piriform cortex.70 In mice, a species with a longer bumetanide plasma half-life of ∼50 min, the lower dose 10 mg/kg (i.v.) yielded only marginally higher total brain levels of 0.75 μM at 30 min.71 In nephrectomized dogs, where the renal clearance of bumetanide does not limit its half-life, i.v. infusion of bumetanide (50 mg/kg) resulted only in submicromolar (0.7 μM) cerebrospinal fluid bumetanide concentrations, despite reaching extremely high total plasma concentrations (close to 300 μM) for several hours.88
Töpfer et al.71 examined the use of the cytochrome P450 inhibitor piperonyl butoxide with the aim of prolonging the elimination half-life of bumetanide and thus increasing the translational value of results based on rodent models. Using this approach in adult animals, the authors were able to increase the maximal hippocampal bumetanide levels (with a drug dose of 10 mg/kg i.v.) from 0.2–0.3 μM to 0.6 μM in rats and from 0.7–0.8 μM to 2 μM in mice. However, this modest increase did not lead to an anticonvulsant effect in the seizure models tested.
In light of bumetanide's poor BBB penetration, its effects on the CNS have often been a priori attributed in part to enhanced brain accumulation as a result of a seizure-induced breakdown of the BBB. The only study to investigate this in neonatal rats did not show any significant increase in brain accumulation of bumetanide (given at 0.2–0.3 mg/kg i.p.) following hypoxia-induced seizures.35 In line with this, after a relatively high dose of bumetanide (10 mg/kg i.v.), no difference in total hippocampal bumetanide levels were reported between nonkindled and fully kindled adult rats,71 or in total brain bumetanide levels between adult mice 24 h after pilocarpine status epilepticus or sham treatment.89
In summary, all the above data argue against the view that systemic administration of bumetanide at the low concentrations used in the majority of studies reporting anticonvulsant effects in rodent models of neonatal seizures (see Table 1) would achieve physiologically relevant levels in brain regions protected by the BBB.
Preclinical Observations with Bumetanide as an Anticonvulsant in Neonates
The preclinical in vitro and in vivo data on actions of NKCC1 inhibitors in seizure models have been recently reviewed in detail.12 With regard to neonatal seizures, no data exist for furosemide, whereas a number of studies have examined the anticonvulsant potential of bumetanide in vivo (Table 1).
Only a few studies have assessed the effect of bumetanide on electrographic seizures in vivo. Bumetanide had a suppressing effect on epileptiform activity induced by sevoflurane,90 but no effect was seen using a similar dose of bumetanide (1.8 mg/kg i.p.) in the flurothyl model.91 In the former study,90 bumetanide was administered 15 min before sevoflurane inhalation, whereas in the latter bumetanide was given 60 min before flurothyl,91 which may not be a trivial difference given the 30 min half-life of bumetanide in rats of this age (see above). In a paper on kainate-induced seizures, which generated much of the interest in bumetanide as an anticonvulsant in neonates, Dzhala et al.19 reported that bumetanide at an i.p. dose of 0.1–0.2 mg/kg, given 10 min after the kainate injection, led to a reduction in the power of EEG activity when compared to control animals. But, as pointed out by Vanhatalo et al.,92 mere changes in the spectral power of ictal EEG do not reliably define an anticonvulsant effect, especially in view of the low number of bumetanide-injected animals (n = 3) that were subject to EEG analysis in the study by Dzhala et al. A recent investigation on hypoxia-induced seizures35 failed to induce significant changes in EEG spectral power in rats of similar age and exposed to similar bumetanide concentrations as in the study by Dzhala et al. Using rapid hippocampal kindling, Mazarati et al. reported that bumetanide at 0.5 mg/kg, but not at 0.2 mg/kg, given during electrical kindling (see Ref. 12), increased afterdischarge threshold and shortened afterdischarge duration in P11 but not in P14 or P21 rats.93 In the P11 group, bumetanide at 0.5 mg/kg doubled the number of stimulations required to evoke the first full motor seizure as well as reduced the total number of motor seizures.93
Regarding the effects of bumetanide on behavioral seizures in neonatal rats, a modest reduction in acute hypoxia-induced seizures was observed in P10 rats using video-EEG after a bumetanide dose of 0.3 mg/kg given i.p. 15 min before seizure onset, but not after a dose of 0.15 or 0.5 mg/kg.35 In the pentylenetetrazole seizure model, bumetanide at 1 mg/kg i.p. had an anticonvulsant effect in rat pups at 12 days of age, but not at P7 or P18.94 Increasing the dose to 2.5 mg/kg was found to be proconvulsant but, again, only in the P12 group.94 Daily i.p. administration of 2 mg/kg of bumetanide during the first 5 days of life potentiated febrile seizures experimentally induced in the third postnatal week.95 In contrast, a daily low dose (0.1 mg/kg, i.p.) of bumetanide administered to rats after experimental febrile seizures at P11 decreased the susceptibility to pilocarpine-induced seizures at P60.96 Taken together, the preclinical in vivo evidence in support of bumetanide alone as an efficient neonatal anticonvulsant is not robust.
The use of bumetanide to enhance the effects of phenobarbital in neonatal seizure suppression was proposed by Staley et al.97 The underlying idea is elegant: blocking first the depolarizing driving force of GABA currents and Cl− accumulation using bumetanide, and thereafter increasing the GABA-induced conductance using phenobarbital or a benzodiazepine is expected to lead to a potent anticonvulsant effect. Also, as pointed out by Ben-Ari et al., the earlier administration of bumetanide may help to ameliorate the eventual down-regulation of KCC2 and possible seizure aggravation,32 which take place after an initial functional up-regulation of KCC2 by neonatal seizures.46 In clinical practice, where GABAAR enhancers must be used as first-line drugs, it is not possible to begin drug administration with bumetanide. Recent work in vivo using a rat model of hypoxia-induced neonatal seizures reported lower seizure burden obtained with a combination of phenobarbital and bumetanide, versus phenobarbital or bumetanide (0.15–0.3 mg/kg i.p.) alone.35 Interestingly, reduced cerebral damage and improved sensorimotor functions have been reported when a high dose of bumetanide (10 mg/kg i.p.) was used in conjunction with phenobarbital and hypothermia in a neonatal rat model of cerebral hypoxia-ischemia.98 However, the effects on seizures were not assessed in this study. In view of the already poor pharmacokinetic properties of bumetanide it is important to note that, as seen in rats, phenobarbital may increase the metabolic clearance of bumetanide.99
In sum, considering the fact that clinical trials are ongoing, the number of studies on bumetanide actions on seizures in vivo is surprisingly small and no data are available on large-animal models.
Neuroprotective Effects of Bumetanide Acting on BBB-Located NKCC1
The BBB endothelium luminal (blood-facing) Na+ and Cl− transporter systems generate up to a third of the brain interstitial fluid volume.57 During the early stages following ischemic injury, a common consequence of birth asphyxia, up-regulation of channels and transporters in the BBB facilitates net uptake of cations and water from the blood into the brain interstitial space across the yet intact BBB. This results in ionic edema and swelling of the brain parenchyma.56 NKCC1, highly expressed in the BBB and choroid plexus,57,100 is phosphorylated and functionally stimulated under ischemic conditions, thereby facilitating ionic edema in the early stages of brain ischemia.56 It is notable that cerebral edema and accompanying brain damage are strongly attenuated in NKCC1 KO mice.101 O'Donnell et al.57 reported that high doses of bumetanide in the range of 7–30 mg/kg (i.v.) ameliorate edema induced by middle cerebral artery occlusion without reperfusion in adult rats. The authors concluded that this effect is unlikely to involve inhibition of NKCC1 of the brain parenchyma but rather that of endothelial cells, due to the poor ability of bumetanide to permeate the BBB.57 Work by others indicates that bumetanide administered at a high dose (15 mg/kg, i.v.), prior to BBB breakdown, attenuates brain edema induced by traumatic brain injury in adult rats.102 Due to its luminal expression in BBB endothelial cells,57 NKCC1 is directly exposed to systemically administered bumetanide. Therefore, it is possible that bumetanide prevents BBB breakdown by inhibiting endothelial NKCC1 activity. Thus, a relevant question is whether targeting BBB-located NKCC1 with bumetanide may suppress hypoxia-ischemia–induced seizures by preventing the escalation of ionic edema into vasogenic edema and BBB disruption in the asphyxic neonate.
NKCC1 in the Hypothalamic-Pituitary-Adrenal Axis: A Possible Site of the Anticonvulsant Actions of Bumetanide?
Mounting evidence indicates that the mechanisms underlying seizure generation and stress are closely linked.103,104 Intriguingly, bumetanide (and furosemide) has been shown to exert anxiolytic effects.105 The mediator of the body's stress response, the hypothalamic-pituitary-adrenal (HPA) axis undergoes robust activation in response to neonatal hypoxia-ischemia.106,107 The major mediator of the hormonal and behavioral responses to stress is the neuropeptide corticotropin releasing hormone (CRH). CRH is an excitatory neuropeptide that is tightly coupled to seizure susceptibility in both the adult and neonatal brain, with the latter apparently more sensitive to CRH as exemplified by the fact that the lowest seizure triggering CRH concentrations are two orders of magnitude lower in neonatal rats.103 Significantly higher CRH concentrations have been reported in umbilical cord plasma of asphyxiated human neonates than in those that had experienced uncomplicated birth.106 Recent work has demonstrated that CRH application triggers N-methyl-d-aspartate receptor/calpain–dependent downstream mechanisms108 that have been also implicated in the activity-dependent down-regulation of KCC2.109
The release of CRH from neurosecretory nerve terminals of periventricular (PVN) neurons in the median eminence is tightly regulated by GABAergic inhibitory input onto PVN neurons, which limits the release of neuropeptides.104 Recent work by Maguire et al.110,111 on adult rodents demonstrated rapid down-regulation of KCC2 paralleled by up-regulation of NKCC1 and emergence of excitatory GABA action on CRH-releasing PVN neurons by acute stress and seizures. Presynaptic nerve terminals are known to express both GABAARs and NKCC1 but not KCC2.14 This highlights the activity of NKCC1 in the neurosecretory terminals as an important determinant of the nature of GABAergic control over CRH release. Such a role may be relevant, especially in neurons in which KCC2 has been down-regulated by seizures. The median eminence, where the neurosecretory terminals of PVN neurons are located, is one of the circumventricular organs of the brain, a group of discrete brain regions characterized by a “leaky” BBB with fenestrated endothelial cells lacking tight junctions. Because of this, neuroendocrine cells in the PVN, including CRH releasing neurons, are likely to be directly exposed to systemically administered bumetanide. An interesting hypothesis is that bumetanide might suppress seizures by inhibiting NKCC1 in the PVN neurons, thus limiting CRH release and HPA-axis activation.104 It is also important to note that dehydration and hypovolemia, which are likely to be associated with the renal effects of bumetanide, may have a suppressing action on transcription of CRH.112 Of interest, bumetanide administered at low concentrations (0.2 mg/kg i.p.) previously shown to result in brain levels that are two orders of magnitude lower than the half maximal inhibition constant for NKCC1 inhibition (see above), was demonstrated to block seizure-induced activation of the HPA axis and decrease susceptibility to future seizures in adult mice.111 An important next step is to investigate whether bumetanide attenuates hypoxia-ischemia- and seizure-induced activation of the HPA axis in neonatal animals.
Concluding Remarks
The work reviewed presently focuses on KCC2 and NKCC1 as potential drug targets in the treatment of neonatal seizures. Testing the hypothesis that transient pharmacologic activation of KCC2 would ameliorate neonatal seizures directly and/or increase the efficacy of GABA-enhancing AEDs, such as phenobarbital, is currently limited by the lack of well-characterized KCC2 activators. Although the first steps have recently been taken in this direction,49 the drugs' specificity to KCC2 should be confirmed. Notably, the putative KCC2 activators should not interfere with the role of KCC2 in promoting the maturation of dendritic spines and other neurodevelopmental processes (see above).
The major problems when using bumetanide to block NKCC1 are related to its poor pharmacokinetic properties, which largely limit the inhibition of NKCC1 in BBB-protected brain areas, as well as its side effects including but not limited to diuresis, hypokalemic alkalosis, and hearing loss;58,63 and the latter is likely to be potentiated by coadministration of antibiotics (such as aminoglycosides).68 Notably, a systemic alkalosis is likely to be harmful given that a very small pH increase (0.2 units or even less) in the neonate brain may trigger seizures.113
The above considerations related to the poor brain availability of bumetanide do not exclude inhibition of NKCC1 in hippocampal and neocortical neurons as another potentially useful approach in neonatal seizures, as exemplified by encouraging work on a combination of bumetanide with phenobarbital in an in vitro model of neonatal seizures.97 However, testing this more reliably in vivo requires specific NKCC1 inhibitors with improved CNS availability. Here, a strategy based on prodrugs where bumetanide is initially neutralized by an ester bond to facilitate CNS availability, may turn out to be useful.89
Acknowledgments
We thank Prof. Wolfgang Löscher, Drs. Tarek Z. Deeb, Jamie Maguire, and Patricia Seja for comments on an early version of the manuscript. The authors' original research work was supported by grants from the Letten Foundation, the Academy of Finland, the Sigrid Jusélius Foundation, the Jane and Aatos Erkko Foundation (K. Kaila), the National Institutes of Health, and the Manton Center for Orphan Disease Research (K. Kahle).
Disclosure
No conflicts of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting Information
References
- 1.van Rooij LG, Hellström-Westas L, De Vries LS. Treatment of neonatal seizures. Semin Fetal Neonatal Med. 2013;18:209–215. doi: 10.1016/j.siny.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Volpe J. Neurology of the newborn. 5th edn. Philadelphia, PA: Saunders; 2008. [Google Scholar]
- 3.Pisani F, Piccolo B, Cantalupo G, et al. Neonatal seizures and postneonatal epilepsy: a 7-y follow-up study. Pediatr Res. 2012;72:186–193. doi: 10.1038/pr.2012.66. [DOI] [PubMed] [Google Scholar]
- 4.Ronen GM, Buckley D, Penney S, et al. Long-term prognosis in children with neonatal seizures: a population-based study. Neurology. 2007;69:1816–1822. doi: 10.1212/01.wnl.0000279335.85797.2c. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Ari Y, Holmes GL. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006;5:1055–1063. doi: 10.1016/S1474-4422(06)70626-3. [DOI] [PubMed] [Google Scholar]
- 6.Painter MJ, Scher MS, Stein AD, et al. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. N Engl J Med. 1999;341:485–489. doi: 10.1056/NEJM199908123410704. [DOI] [PubMed] [Google Scholar]
- 7.Boylan GB, Rennie JM, Chorley G, et al. Second-line anticonvulsant treatment of neonatal seizures: a video-EEG monitoring study. Neurology. 2004;62:486–488. doi: 10.1212/01.wnl.0000106944.59990.e6. [DOI] [PubMed] [Google Scholar]
- 8.Vanhatalo S, Kaila K. Development of neonatal EEG activity: from phenomenology to physiology. Semin Fetal Neonatal Med. 2006;11:471–478. doi: 10.1016/j.siny.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Boylan GB, Stevenson NJ, Vanhatalo S. Monitoring neonatal seizures. Semin Fetal Neonatal Med. 2013;18:202–208. doi: 10.1016/j.siny.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 10.Connell J, Oozeer R, de Vries L, et al. Clinical and EEG response to anticonvulsants in neonatal seizures. Arch Dis Child. 1989;64:459–464. doi: 10.1136/adc.64.4_spec_no.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaila K, Ruusuvuori E, Seja P, et al. GABA actions and ionic plasticity in epilepsy. Curr Opin Neurobiol. 2014;26:34–41. doi: 10.1016/j.conb.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Löscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. 2013;69:62–74. doi: 10.1016/j.neuropharm.2012.05.045. [DOI] [PubMed] [Google Scholar]
- 13.Farrant M, Kaila K. The cellular, molecular and ionic basis of GABAA receptor signalling. Prog Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- 14.Blaesse P, Airaksinen MS, Rivera C, et al. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Ruusuvuori E, Huebner AK, Kirilkin I, et al. Neuronal carbonic anhydrase VII provides GABAergic excitatory drive to exacerbate febrile seizures. EMBO J. 2013;32:2275–2286. doi: 10.1038/emboj.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rivera C, Voipio J, Payne JA, et al. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 17.Seja P, Schonewille M, Spitzmaul G, et al. Raising cytosolic Cl− in cerebellar granule cells affects their excitability and vestibulo-ocular learning. EMBO J. 2012;31:1217–1230. doi: 10.1038/emboj.2011.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glykys J, Dzhala VI, Kuchibhotla KV, et al. Differences in cortical versus subcortical GABAergic signaling: a candidate mechanism of electroclinical uncoupling of neonatal seizures. Neuron. 2009;63:657–672. doi: 10.1016/j.neuron.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dzhala VI, Talos DM, Sdrulla DA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- 20.Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. 2001;105:7–17. doi: 10.1016/s0306-4522(01)00171-3. [DOI] [PubMed] [Google Scholar]
- 21.Erecinska M, Cherian S, Silver IA. Energy metabolism in mammalian brain during development. Prog Neurobiol. 2004;73:397–445. doi: 10.1016/j.pneurobio.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Lamblin MD, Andre M, Challamel MJ, et al. [Electroencephalography of the premature and term newborn. Maturational aspects and glossary] Neurophysiol Clin. 1999;29:123–219. doi: 10.1016/s0987-7053(99)80051-3. [DOI] [PubMed] [Google Scholar]
- 23.Vanhatalo S, Palva JM, Andersson S, et al. Slow endogenous activity transients and developmental expression of K+-Cl− cotransporter 2 in the immature human cortex. Eur J Neurosci. 2005;22:2799–2804. doi: 10.1111/j.1460-9568.2005.04459.x. [DOI] [PubMed] [Google Scholar]
- 24.Plotkin MD, Snyder EY, Hebert SC, et al. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol. 1997;33:781–795. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 25.Randall J, Thorne T, Delpire E. Partial cloning and characterization of Slc12a2: the gene encoding the secretory Na+-K+-2Cl− cotransporter. Am J Physiol. 1997;273:C1267–C1277. doi: 10.1152/ajpcell.1997.273.4.C1267. [DOI] [PubMed] [Google Scholar]
- 26.Clayton GH, Owens GC, Wolff JS, et al. Ontogeny of cation-Cl− cotransporter expression in rat neocortex. Dev Brain Res. 1998;109:281–292. doi: 10.1016/s0165-3806(98)00078-9. [DOI] [PubMed] [Google Scholar]
- 27.Vibat CRT, Holland MJ, Kang JJ, et al. Quantitation of Na+-K+-2Cl(−) cotransport splice variants in human tissues using kinetic polymerase chain reaction. Anal Biochem. 2001;298:218–230. doi: 10.1006/abio.2001.5398. [DOI] [PubMed] [Google Scholar]
- 28.Moore-Hoon ML, Turner RJ. Molecular and topological characterization of the rat parotid Na+-K+-2Cl− cotransporter1. Biochim Biophys Acta. 1998;1373:261–269. doi: 10.1016/s0005-2736(98)00112-6. [DOI] [PubMed] [Google Scholar]
- 29.Hyde TM, Lipska BK, Ali T, et al. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci. 2011;31:11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khirug S, Yamada J, Afzalov R, et al. GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J Neurosci. 2008;28:4635–4639. doi: 10.1523/JNEUROSCI.0908-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buzsaki G, Kaila K, Raichle M. Inhibition and brain work. Neuron. 2007;56:771–783. doi: 10.1016/j.neuron.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nardou R, Yamamoto S, Chazal G, et al. Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain. 2011;134:987–1002. doi: 10.1093/brain/awr041. [DOI] [PubMed] [Google Scholar]
- 33.Diamond J. Quantitative evolutionary design. J Physiol. 2002;542:337–345. doi: 10.1113/jphysiol.2002.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai Y, Tang J, Zhang JH. Role of Cl− in cerebral vascular tone and expression of Na+-K+-2Cl− co-transporter after neonatal hypoxia-ischemia. Can J Physiol Pharmacol. 2005;83:767–773. doi: 10.1139/y05-076. [DOI] [PubMed] [Google Scholar]
- 35.Cleary RT, Huynh T, Manning SM, et al. Bumetanide enhances phenobarbital efficacy in a rat model of hypoxic neonatal seizures. PLoS ONE. 2013;8:e57148. doi: 10.1371/journal.pone.0057148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foroutan S, Brillault J, Forbush B, et al. Moderate-to-severe ischemic conditions increase activity and phosphorylation of the cerebral microvascular endothelial cell Na+-K+-Cl− cotransporter. Am J Physiol Cell Physiol. 2005;289:C1492–C1501. doi: 10.1152/ajpcell.00257.2005. [DOI] [PubMed] [Google Scholar]
- 37.Pieraut S, Lucas O, Sangari S, et al. An autocrine neuronal interleukin-6 loop mediates chloride accumulation and NKCC1 phosphorylation in axotomized sensory neurons. J Neurosci. 2011;31:13516–13526. doi: 10.1523/JNEUROSCI.3382-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams JR, Sharp JW, Kumari VG, et al. The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J Biol Chem. 1999;274:12656–12664. doi: 10.1074/jbc.274.18.12656. [DOI] [PubMed] [Google Scholar]
- 39.Delpire E, Rauchman MI, Beier DR, et al. Molecular cloning and chromosome localization of a putative basolateral Na(+)-K(+)-2Cl− cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem. 1994;269:25677–25683. [PubMed] [Google Scholar]
- 40.Howard HC, Mount DB, Rochefort D, et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet. 2002;32:384–392. doi: 10.1038/ng1002. [DOI] [PubMed] [Google Scholar]
- 41.Hübner CA, Stein V, Hermans-Borgmeyer I, et al. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- 42.Fiumelli H, Briner A, Puskarjov M, et al. An ion transport-independent role for the cation-chloride cotransporter KCC2 in dendritic spinogenesis in vivo. Cereb Cortex. 2013;23:378–388. doi: 10.1093/cercor/bhs027. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Khirug S, Cai C, et al. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 44.Chamma I, Chevy Q, Poncer JC, et al. Role of the neuronal K-Cl co-transporter KCC2 in inhibitory and excitatory neurotransmission. Front Cell Neurosci. 2012;6:5. doi: 10.3389/fncel.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kahle KT, Deeb TZ, Puskarjov M, et al. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013;36:726–737. doi: 10.1016/j.tins.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khirug S, Ahmad F, Puskarjov M, et al. A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J Neurosci. 2010;30:12028–12035. doi: 10.1523/JNEUROSCI.3154-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kahle KT, Rinehart J, Lifton RP. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim Biophys Acta. 2010;1802:1150–1158. doi: 10.1016/j.bbadis.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee HHC, Walker JA, Williams JR, et al. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J Biol Chem. 2007;282:29777–29784. doi: 10.1074/jbc.M705053200. [DOI] [PubMed] [Google Scholar]
- 49.Gagnon M, Bergeron MJ, Lavertu G, et al. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat Med. 2013;19:1524–1528. doi: 10.1038/nm.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Viitanen T, Ruusuvuori E, Kaila K, et al. The K+-Cl cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol. 2010;588:1527–1540. doi: 10.1113/jphysiol.2009.181826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miles R, Blaesse P, Huberfeld G, et al. Chloride homeostasis and GABA signaling in temporal lobe epilepsy. In: Noebels JL, Avoli M, Rogawski MA, et al., editors. Jasper's basic mechanisms of the epilepsies. 4th ed. Oxford: Oxford University Press; 2012. pp. 581–590. [Google Scholar]
- 52.Kaila K, Lamsa K, Smirnov S, et al. Long-lasting GABA-mediated depolarization evoked by high- frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- 54.Haam J, Popescu IR, Morton LA, et al. GABA is excitatory in adult vasopressinergic neuroendocrine cells. J Neurosci. 2012;32:572–582. doi: 10.1523/JNEUROSCI.3826-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie Z, Currie KP, Cahill AL, et al. Role of Cl− co-transporters in the excitation produced by GABAA receptors in juvenile bovine adrenal chromaffin cells. J Neurophysiol. 2003;90:3828–3837. doi: 10.1152/jn.00617.2003. [DOI] [PubMed] [Google Scholar]
- 56.Kahle KT, Simard JM, Staley KJ, et al. Molecular mechanisms of ischemic cerebral edema: role of electroneutral ion transport. Physiology (Bethesda) 2009;24:257–265. doi: 10.1152/physiol.00015.2009. [DOI] [PubMed] [Google Scholar]
- 57.O'Donnell ME, Tran L, Lam TI, et al. Bumetanide inhibition of the blood–brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab. 2004;24:1046–1056. doi: 10.1097/01.WCB.0000130867.32663.90. [DOI] [PubMed] [Google Scholar]
- 58.Pressler RM, Mangum B. Newly emerging therapies for neonatal seizures. Semin Fetal Neonatal Med. 2013;18:216–223. doi: 10.1016/j.siny.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 59.Pacifici GM. Clinical pharmacology of the loop diuretics furosemide and bumetanide in neonates and infants. Paediatr Drugs. 2012;14:233–246. doi: 10.2165/11596620-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 60.Sullivan JE, Witte MK, Yamashita TS, et al. Analysis of the variability in the pharmacokinetics and pharmacodynamics of bumetanide in critically ill infants. Clin Pharmacol Ther. 1996;60:414–423. doi: 10.1016/S0009-9236(96)90198-8. [DOI] [PubMed] [Google Scholar]
- 61.Pentikäinen PJ, Penttilä A, Neuvonen PJ, et al. Fate of [14C]-bumetanide in man. Br J Clin Pharmacol. 1977;4:39–44. doi: 10.1111/j.1365-2125.1977.tb00664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hannaert P, Alvarez-Guerra M, Pirot D, et al. Rat NKCC2/NKCC1 cotransporter selectivity for loop diuretic drugs. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:193–199. doi: 10.1007/s00210-001-0521-y. [DOI] [PubMed] [Google Scholar]
- 63.Greenberg A. Diuretic complications. Am J Med Sci. 2000;319:10–24. [PubMed] [Google Scholar]
- 64.Hasannejad H, Takeda M, Taki K, et al. Interactions of human organic anion transporters with diuretics. J Pharmacol Exp Ther. 2004;308:1021–1029. doi: 10.1124/jpet.103.059139. [DOI] [PubMed] [Google Scholar]
- 65.Kobayashi Y, Ohbayashi M, Kohyama N, et al. Mouse organic anion transporter 2 and 3 (mOAT2/3[Slc22a7/8]) mediates the renal transport of bumetanide. Eur J Pharmacol. 2005;524:44–48. doi: 10.1016/j.ejphar.2005.09.054. [DOI] [PubMed] [Google Scholar]
- 66.Nappi JM. A retrospective evaluation of the efficacy of intravenous bumetanide and comparison of potency with furosemide. Pharm Pract (Granada) 2013;11:44–50. doi: 10.4321/s1886-36552013000100008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramsay LE, McInnes GT, Hettiarachchi J, et al. Bumetanide and frusemide: a comparison of dose-response curves in healthy men. Br J Clin Pharmacol. 1978;5:243–247. doi: 10.1111/j.1365-2125.1978.tb01631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Soul J. Novel medications for neonatal seizures: bumetanide and topiramate. J Pediatr Neurol. 2009;7:85–93. [Google Scholar]
- 69.Sullivan JE, Witte MK, Yamashita TS, et al. Pharmacokinetics of bumetanide in critically ill infants. Clin Pharmacol Ther. 1996;60:405–413. doi: 10.1016/S0009-9236(96)90197-6. [DOI] [PubMed] [Google Scholar]
- 70.Brandt C, Nozadze M, Heuchert N, et al. Disease-modifying effects of phenobarbital and the NKCC1 inhibitor bumetanide in the pilocarpine model of temporal lobe epilepsy. J Neurosci. 2010;30:8602–8612. doi: 10.1523/JNEUROSCI.0633-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Töpfer M, Töllner K, Brandt C, et al. Consequences of inhibition of bumetanide metabolism in rodents on brain penetration and effects of bumetanide in chronic models of epilepsy. Eur J Neurosci. 2014;39:673–687. doi: 10.1111/ejn.12424. [DOI] [PubMed] [Google Scholar]
- 72.Abbott NJ, Patabendige AA, Dolman DE, et al. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 73.Walker PC, Berry NS, Edwards DJ. Protein binding characteristics of bumetanide. Dev Pharmacol Ther. 1989;12:13–18. [PubMed] [Google Scholar]
- 74.Kim EJ, Lee MG. Pharmacokinetics and pharmacodynamics of intravenous bumetanide in mutant Nagase analbuminemic rats: importance of globulin binding for the pharmacodynamic effects. Biopharm Drug Dispos. 2001;22:147–156. doi: 10.1002/bdd.267. [DOI] [PubMed] [Google Scholar]
- 75.Turmen T, Thom P, Louridas AT, et al. Protein binding and bilirubin displacing properties of bumetanide and furosemide. J Clin Pharmacol. 1982;22:551–556. doi: 10.1002/j.1552-4604.1982.tb02648.x. [DOI] [PubMed] [Google Scholar]
- 76.Shim HJ, Lee MG, Lee MH. Factors influencing the protein binding of bumetanide using an equilibrium dialysis technique. J Clin Pharm Ther. 1991;16:467–476. doi: 10.1111/j.1365-2710.1991.tb00337.x. [DOI] [PubMed] [Google Scholar]
- 77.Oliveros M, Pham JT, John E, et al. The use of bumetanide for oliguric acute renal failure in preterm infants. Pediatr Crit Care Med. 2011;12:210–214. doi: 10.1097/PCC.0b013e3181e912a7. [DOI] [PubMed] [Google Scholar]
- 78.Song B, Kodukula K, Moos WH, et al. Evaluation of the pKa values and ionization sequence of bumetanide using 1H and 13C NMR and UV spectroscopy. Drug Dev Res. 2011;72:416–426. [Google Scholar]
- 79.Fischer H, Gottschlich R, Seelig A. Blood–brain barrier permeation: molecular parameters governing passive diffusion. J Membr Biol. 1998;165:201–211. doi: 10.1007/s002329900434. [DOI] [PubMed] [Google Scholar]
- 80.Somasekharan S, Tanis J, Forbush B. Loop diuretic and ion-binding residues revealed by scanning mutagenesis of transmembrane helix 3 (TM3) of Na-K-Cl cotransporter (NKCC1) J Biol Chem. 2012;287:17308–17317. doi: 10.1074/jbc.M112.356014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Linnankoski J, Makela JM, Ranta VP, et al. Computational prediction of oral drug absorption based on absorption rate constants in humans. J Med Chem. 2006;49:3674–3681. doi: 10.1021/jm051231p. [DOI] [PubMed] [Google Scholar]
- 82.Zhu C, Jiang L, Chen TM, et al. A comparative study of artificial membrane permeability assay for high throughput profiling of drug absorption potential. Eur J Med Chem. 2002;37:399–407. doi: 10.1016/s0223-5234(02)01360-0. [DOI] [PubMed] [Google Scholar]
- 83.Goda S, Thomas FC, Gaasch JA, et al. Blood–brain barrier permeability and transport of bumetanide; Program No.200.9/AAA23 Neuroscience Meeting Planner; San Diego, CA: Society for Neuroscience; 2007. [Google Scholar]
- 84.Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Uchida Y, Kamiie J, Ohtsuki S, et al. Multichannel liquid chromatography-tandem mass spectrometry cocktail method for comprehensive substrate characterization of multidrug resistance-associated protein 4 transporter. Pharm Res. 2007;24:2281–2296. doi: 10.1007/s11095-007-9453-7. [DOI] [PubMed] [Google Scholar]
- 86.Goda S, Thorsheim HR, Taskar KS, et al. Restricted brain distribution of bumetanide is mediated by active efflux transporters (rat Oatp & OAT3) at the blood–brain barrier. American Association of Pharmaceutical Scientists AAPS 2010; Published Meeting Abstracts. Number SA7187.2010. [Google Scholar]
- 87.Li Y, Cleary R, Kellogg M, et al. Sensitive isotope dilution liquid chromatography/tandem mass spectrometry method for quantitative analysis of bumetanide in serum and brain tissue. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:998–1002. doi: 10.1016/j.jchromb.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Javaheri S, Davis C, Rogers DH. Ionic composition of cisternal CSF in acute respiratory acidosis: lack of effect of large dose bumetanide. J Neurochem. 1993;61:1525–1529. doi: 10.1111/j.1471-4159.1993.tb13648.x. [DOI] [PubMed] [Google Scholar]
- 89.Töllner K, Brandt C, Töpfer M, et al. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann Neurol. 2014 doi: 10.1002/ana.24124. Feb 25 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 90.Edwards DA, Shah HP, Cao W, et al. Bumetanide alleviates epileptogenic and neurotoxic effects of sevoflurane in neonatal rat brain. Anesthesiology. 2010;112:567–575. doi: 10.1097/ALN.0b013e3181cf9138. [DOI] [PubMed] [Google Scholar]
- 91.Minlebaev M, Khazipov R. Antiepileptic effects of endogenous beta-hydroxybutyrate in suckling infant rats. Epilepsy Res. 2011;95:100–109. doi: 10.1016/j.eplepsyres.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 92.Vanhatalo S, Hellstrom-Westas L, De Vries LS. Bumetanide for neonatal seizures: based on evidence or enthusiasm? Epilepsia. 2009;50:1292–1293. doi: 10.1111/j.1528-1167.2008.01894.x. [DOI] [PubMed] [Google Scholar]
- 93.Mazarati A, Shin D, Sankar R. Bumetanide inhibits rapid kindling in neonatal rats. Epilepsia. 2009;50:2117–2122. doi: 10.1111/j.1528-1167.2009.02048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mares P. Age- and dose-specific anticonvulsant action of bumetanide in immature rats. Physiol Res. 2009;58:927–930. doi: 10.33549/physiolres.931897. [DOI] [PubMed] [Google Scholar]
- 95.Vargas E, Petrou S, Reid CA. Genetic and pharmacological modulation of giant depolarizing potentials in the neonatal hippocampus associates with increased seizure susceptibility. J Physiol. 2013;591:57–65. doi: 10.1113/jphysiol.2012.234674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Koyama R, Tao K, Sasaki T, et al. GABAergic excitation after febrile seizures induces ectopic granule cells and adult epilepsy. Nat Med. 2012;18:1271–1278. doi: 10.1038/nm.2850. [DOI] [PubMed] [Google Scholar]
- 97.Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63:222–235. doi: 10.1002/ana.21229. [DOI] [PubMed] [Google Scholar]
- 98.Liu Y, Shangguan Y, Barks JD, et al. Bumetanide augments the neuroprotective efficacy of phenobarbital plus hypothermia in a neonatal hypoxia-ischemia model. Pediatr Res. 2012;71:559–565. doi: 10.1038/pr.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Choi YM, Lee SH, Jang SH, et al. Effects of phenobarbital and 3-methylcholanthrene pretreatment on the pharmacokinetics and the pharmacodynamics of bumetanide in rats. Biopharm Drug Dispos. 1991;12:311–324. doi: 10.1002/bdd.2510120408. [DOI] [PubMed] [Google Scholar]
- 100.Ho HT, Dahlin A, Wang J. Expression profiling of solute carrier gene families at the blood-CSF barrier. Front Pharmacol. 2012;3:154. doi: 10.3389/fphar.2012.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen H, Luo J, Kintner DB, et al. Na+-dependent chloride transporter (NKCC1)-null mice exhibit less gray and white matter damage after focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:54–66. doi: 10.1038/sj.jcbfm.9600006. [DOI] [PubMed] [Google Scholar]
- 102.Lu KT, Wu CY, Cheng NC, et al. Inhibition of the Na+ -K+-2Cl− -cotransporter in choroid plexus attenuates traumatic brain injury-induced brain edema and neuronal damage. Eur J Pharmacol. 2006;548:99–105. doi: 10.1016/j.ejphar.2006.07.048. [DOI] [PubMed] [Google Scholar]
- 103.Baram TZ, Hatalski CG. Neuropeptide-mediated excitability: a key triggering mechanism for seizure generation in the developing brain. Trends Neurosci. 1998;21:471–476. doi: 10.1016/s0166-2236(98)01275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maguire J, Salpekar JA. Stress, seizures, and hypothalamic-pituitary-adrenal axis targets for the treatment of epilepsy. Epilepsy Behav. 2013;26:352–362. doi: 10.1016/j.yebeh.2012.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Krystal AD, Sutherland J, Hochman DW. Loop diuretics have anxiolytic effects in rat models of conditioned anxiety. PLoS ONE. 2012;7:e35417. doi: 10.1371/journal.pone.0035417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Okamoto E, Takagi T, Makino T, et al. Immunoreactive corticotropin-releasing hormone, adrenocorticotropin and cortisol in human plasma during pregnancy and delivery and postpartum. Horm Metab Res. 1989;21:566–572. doi: 10.1055/s-2007-1009289. [DOI] [PubMed] [Google Scholar]
- 107.Charles MS, Ostrowski RP, Manaenko A, et al. Role of the pituitary-adrenal axis in granulocyte-colony stimulating factor-induced neuroprotection against hypoxia-ischemia in neonatal rats. Neurobiol Dis. 2012;47:29–37. doi: 10.1016/j.nbd.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Andres AL, Regev L, Phi L, et al. NMDA receptor activation and calpain contribute to disruption of dendritic spines by the stress neuropeptide CRH. J Neurosci. 2013;33:16945–16960. doi: 10.1523/JNEUROSCI.1445-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Puskarjov M, Ahmad F, Kaila K, et al. Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J Neurosci. 2012;32:11356–11364. doi: 10.1523/JNEUROSCI.6265-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sarkar J, Wakefield S, MacKenzie G, et al. Neurosteroidogenesis is required for the physiological response to stress: role of neurosteroid-sensitive GABAA receptors. J Neurosci. 2011;31:18198–18210. doi: 10.1523/JNEUROSCI.2560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.O'Toole KK, Hooper A, Wakefield S, et al. Seizure-induced disinhibition of the HPA axis increases seizure susceptibility. Epilepsy Res. 2013;108:29–43. doi: 10.1016/j.eplepsyres.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Watts AG, Sanchez-Watts G. Interactions between heterotypic stressors and corticosterone reveal integrative mechanisms for controlling corticotropin-releasing hormone gene expression in the rat paraventricular nucleus. J Neurosci. 2002;22:6282–6289. doi: 10.1523/JNEUROSCI.22-14-06282.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Helmy MM, Ruusuvuori E, Watkins PV, et al. Acid extrusion via blood–brain barrier causes brain alkalosis and seizures after neonatal asphyxia. Brain. 2012;135:3311–3319. doi: 10.1093/brain/aws257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kang HJ, Kawasawa YI, Cheng F, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.